
Tumor Microenvironment in Melanoma—Characteristic and Clinical Implications


Abstract
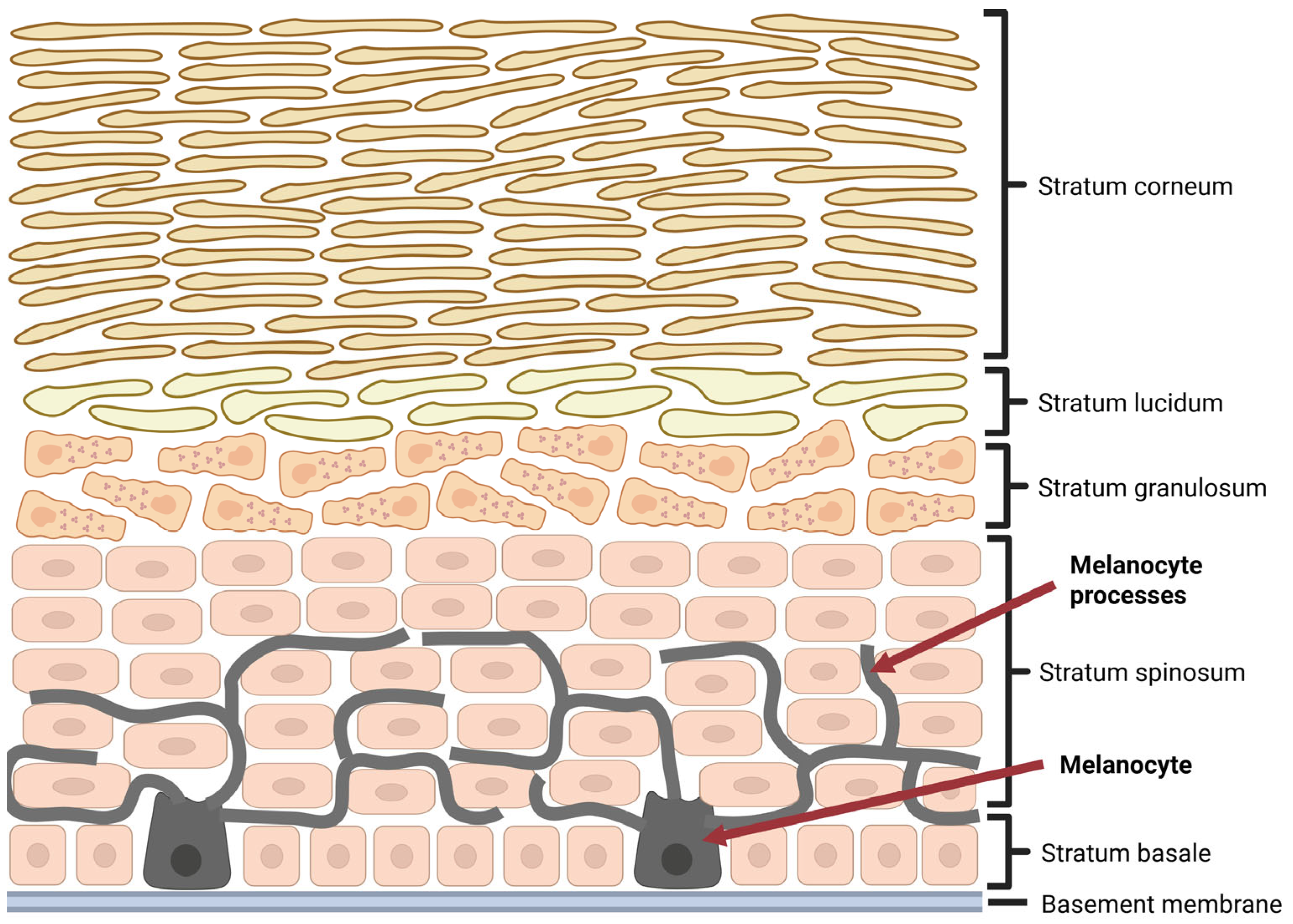
1. Introduction
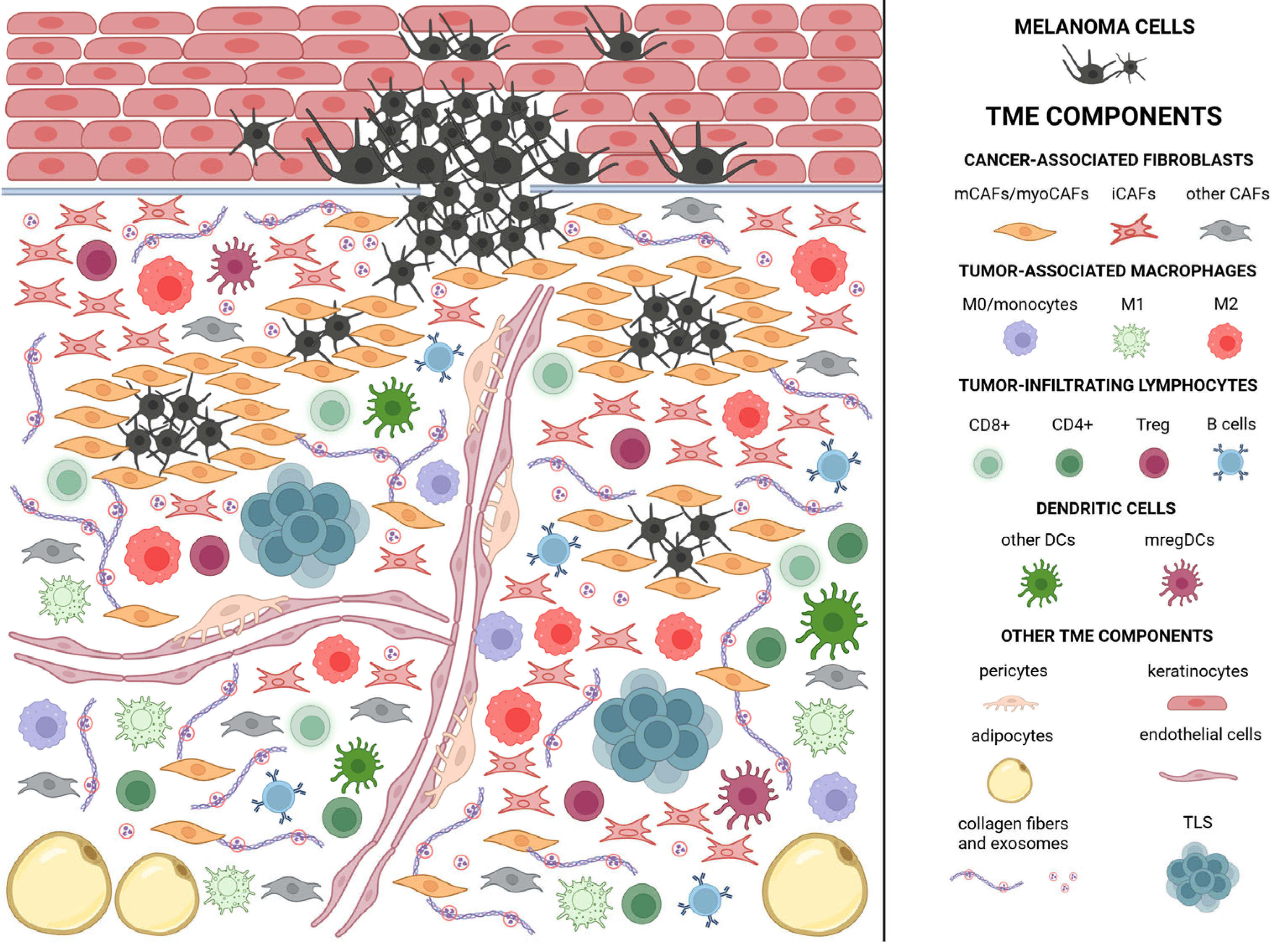
2. Melanoma Microenvironment—Characteristic and Clinical Implications
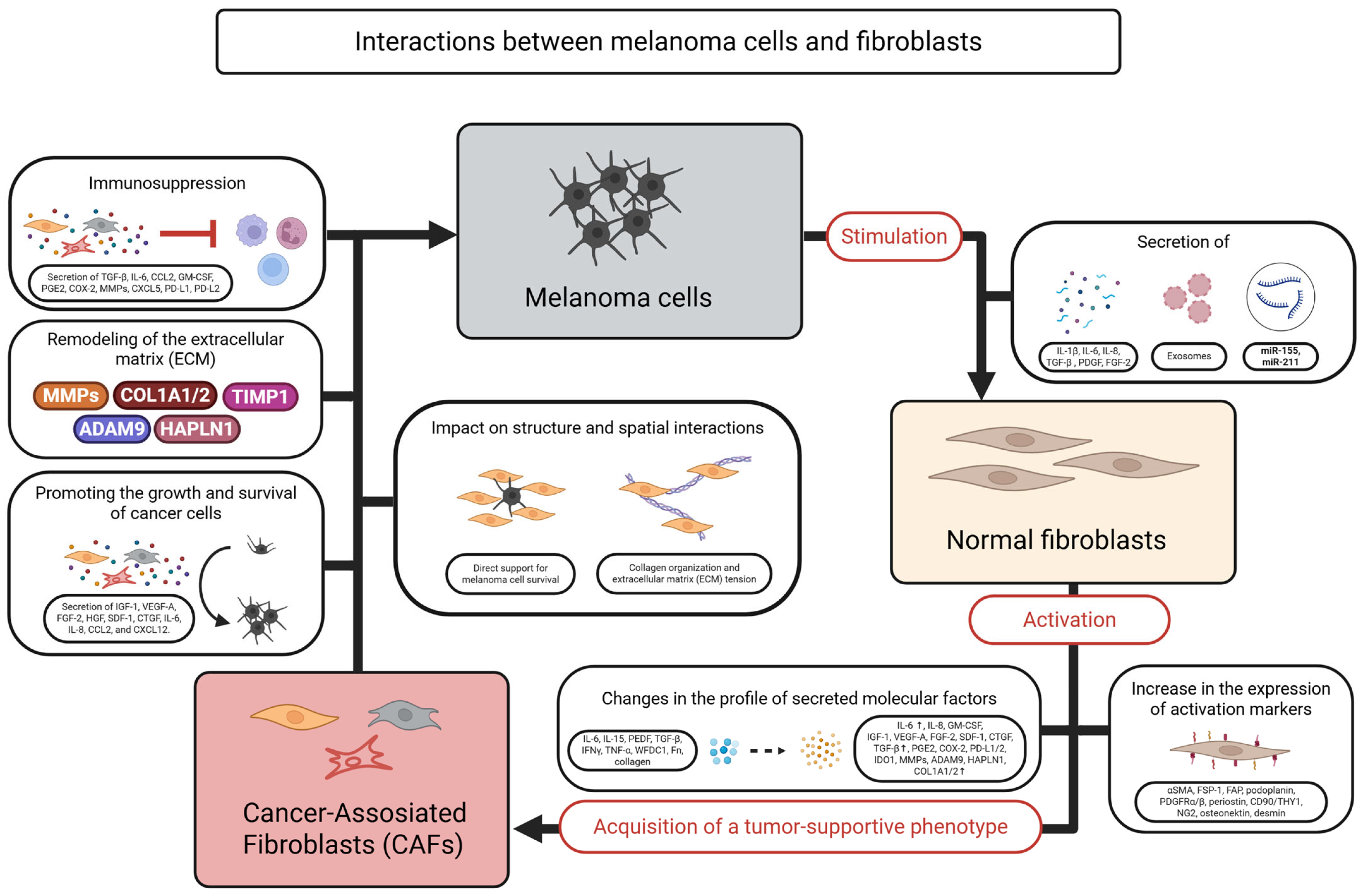
2.1. Cancer-Associated Fibroblasts/Melanoma-Associated Fibroblasts (CAFs)
2.2. Tumor-Associated Macrophages (TAMs)
2.3. Tumor-Infiltrating Lymphocytes (TILs)
2.3.1. B-Cells
2.3.2. CD4+ T-Cells
2.3.3. CD8+ T-Cells
2.3.4. Tregs
2.4. Dendritic Cells (DCs)
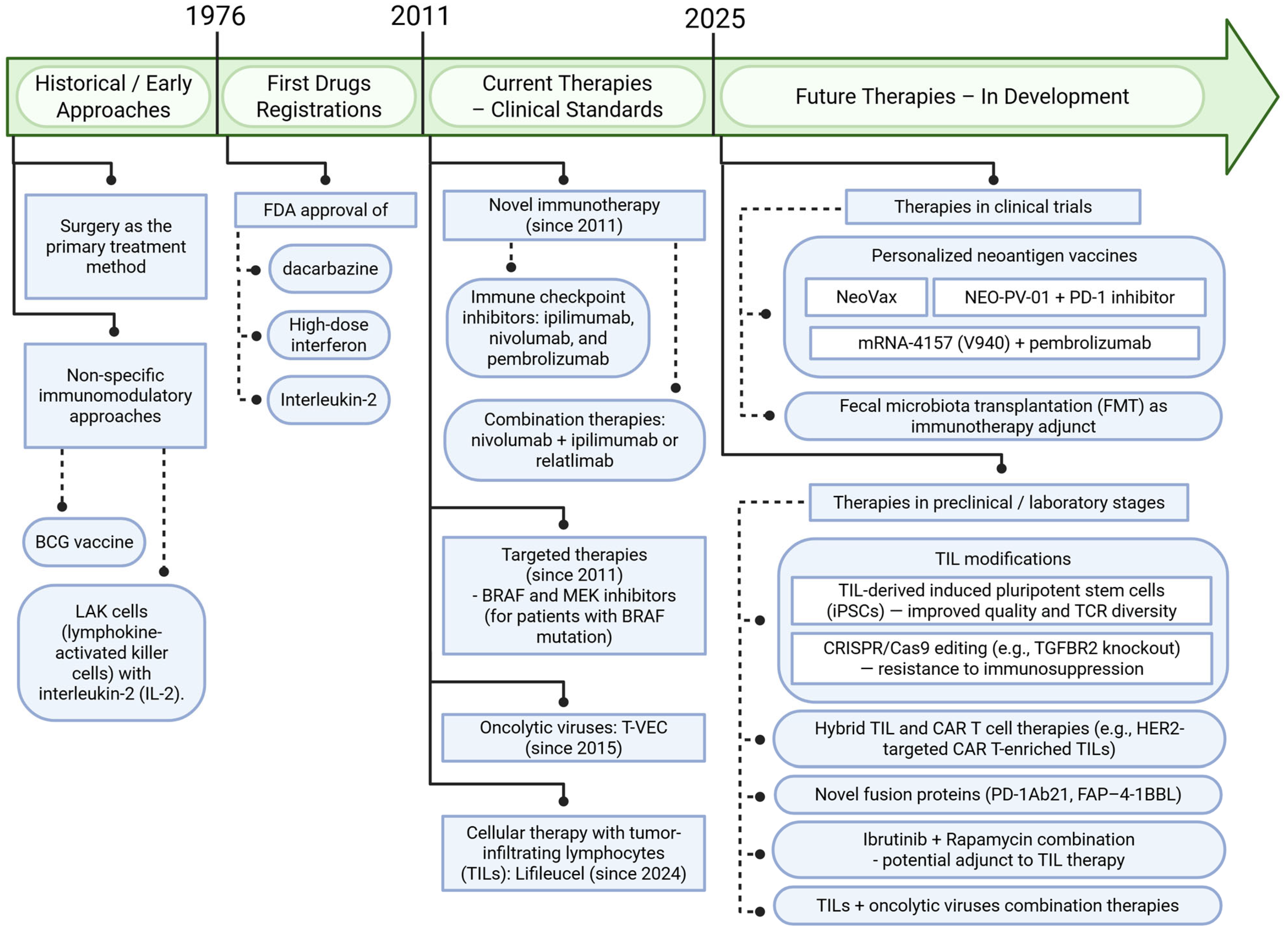
3. Therapeutic Implications and Future Perspectives
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTA2 | Actin alpha cardiac muscle 2 |
| ADAM9 | ADAM metallopeptidase domain 9 |
| ARID5A | AT-rich interactive domain 5A |
| αSMA | Alpha smooth muscle actin |
| Bcl-XL | B-cell lymphoma-extra large |
| BIRC3 | Baculoviral IAP repeat containing 3 |
| BDCA1 | Blood dendritic cell antigen 1 |
| BDCA3 | Blood dendritic cell antigen 3 |
| BTLA | B- and T-lymphocyte attenuator |
| CAFs | Cancer-associated fibroblasts |
| cDC | Conventional dendritic cell |
| cDC1 | Conventional dendritic cell type 1 |
| cDC2 | Conventional dendritic cell type 2 |
| CD103 | Cluster of differentiation 103 |
| CD107a | Cluster of differentiation 107a |
| CD11c | Cluster of differentiation 11c |
| CD141 | Cluster of differentiation 141 |
| CD19 | Cluster of differentiation 19 |
| CD25 | Cluster of differentiation 25 |
| CD5 | Cluster of differentiation 5 |
| CD80 | Cluster of differentiation 80 |
| CD90 | Cluster of differentiation 90 |
| CD206 | Cluster of differentiation 206 |
| CCR1 | C-C motif chemokine receptor 1 |
| CCR2 | C-C motif chemokine receptor 2 |
| CCR7 | C-C motif chemokine receptor 7 |
| CXCL16 | C-X-C motif chemokine ligand 16 |
| CXCL9 | C-X-C motif chemokine ligand 9 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| CXCR3 | C-X-C motif chemokine receptor 3 |
| CXCR5 | C-X-C motif chemokine receptor 5 |
| DNGR-1 | Dendritic cell natural killer receptor-1 |
| EOMES | Eomesodermin |
| FCεR1 | High-affinity immunoglobulin epsilon receptor 1 |
| FLT3L | Fms-like tyrosine kinase 3 ligand |
| FAP | Fibroblast-activation protein |
| FOXP3 | Forkhead box P3 |
| GNLY | Granulysin |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| ICOS-L | Inducible co-stimulator ligand |
| ILT7 | Immunoglobulin-like transcript 7 |
| IFN-γ | Interferon gamma |
| IFNγ | Interferon gamma |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IL-10 | Interleukin-10 |
| IL-15 | Interleukin-15 |
| IL-12 | Interleukin-12 |
| IL-17 | Interleukin-17 |
| ID2 | Inhibitor of DNA-binding 2 |
| IDO | Indoleamine 2,3-dioxygenase |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| IRF1 | Interferon regulatory factor 1 |
| IRF4 | Interferon regulatory factor 4 |
| IRF8 | Interferon regulatory factor 8 |
| IRF9 | Interferon regulatory factor 9 |
| KCNJ8 | Potassium channel, inwardly rectifying subfamily J member 8 |
| LAMP-1 | Lysosomal-associated membrane protein 1 |
| LAMP3 | Lysosomal-associated membrane protein 3 |
| LIPSTIC | Liposome-based short interfering RNA delivery technology for immunocellular targeting |
| MMP1 | Matrix metalloproteinase 1 |
| MMP3 | Matrix metalloproteinase 3 |
| MMPs | Matrix metalloproteinases |
| MRC1 | Mannose receptor C-type 1 |
| MARCKSL1 | Myristoylated alanine-rich C-kinase substrate-like 1 |
| MZB1 | Marginal zone B-cell marker 1 |
| NKG7 | Natural killer cell granule protein 7 |
| NK | Natural killer cells |
| OX40 | Ox40 receptor |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PD-L2 | Programmed cell death ligand 2 |
| PRDM1 | PR domain containing 1 |
| RELB | RelB proto-oncogene |
| RGS5 | Regulator of G-protein signaling 5 |
| RUNX3 | Runt-related transcription factor 3 |
| SDF-1 | Stromal-derived factor 1 |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| T-bet | T-box transcription factor |
| TCR | T-cell receptor |
| TILs | Tumor-infiltrating lymphocytes |
| TLR4 | Toll-like receptor 4 |
| TLR7 | Toll-like receptor 7 |
| TLR9 | Toll-like receptor 9 |
| TLS | Tumor lymphoid structures |
| TNF | Tumor necrosis factor |
| TNF-α | Tumor necrosis factor alpha |
| TME | Tumor microenvironment |
| TWIST1 | Twist family bHLH transcription factor 1 |
| TWIST2 | Twist family bHLH transcription factor 2 |
| XCL1 | C-X-C motif chemokine ligand 1 |
| XCL2 | C-X-C motif chemokine ligand 2 |
| XCR1 | C-X-C motif chemokine receptor 1 |
| ZEB2 | Zinc finger E-box binding homeobox 2 |
References
- Available online: https://www.who.int/uv/faq/skincancer/en/index1.html (accessed on 11 July 2025).
- Rutkowski, P.; Owczarek, W.; Nejc, D.; Jeziorski, A.; Wysocki, W.M.; Słowińska, M.; Dudzisz-Śledź, M.; Wiśniewski, P.; Koseła-Paterczyk, H.; Kiprian, D.; et al. Skin carcinomas. Oncol. Clin. Pract. 2018, 14, 129–147. [Google Scholar] [CrossRef]
- Khan, N.H.; Mir, M.; Qian, L.; Baloch, M.; Ali Khan, M.F.; Rehman, A.U.; Ngowi, E.E.; Wu, D.D.; Ji, X.Y. Skin cancer biology and barriers to treatment: Recent applications of polymeric micro/nanostructures. J. Adv. Res. 2022, 36, 223–247. [Google Scholar] [CrossRef]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2020, 10, 626129. [Google Scholar] [CrossRef]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Bello, D.M. Adjuvant immunotherapy for melanoma. J. Surg. Oncol. 2021, 123, 789–797. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Rutkowski, P.; Wysocki, P.J.; Nasierowska-Guttmejer, A.; Jeziorski, A.; Wysocki, W.M.; Kalinka, E.; Świtaj, T.; Kozak, K.; Kamińska-Winciorek, G.; Czarnecka, A.M.; et al. Cutaneous melanoma. Oncol. Clin. Pract. 2020, 16, 163–182. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.; Hölzel, D.; Coebergh, J.W.; Engel, J.; Group, M.M. Gender differences in melanoma survival: Female patients have a decreased risk of metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef]
- Murali, T.; Schwartz, M.; Reynolds, A.Z.; Luo, L.; Ridgeway, G.; Busam, K.J.; Cust, A.E.; Anton-Culver, H.; Gallagher, R.P.; Zanetti, R.; et al. Sex differences in melanoma survival-a GEM study. JNCI Cancer Spectr. 2025, 9, pkaf005. [Google Scholar] [CrossRef]
- Behbahani, S.; Maddukuri, S.; Cadwell, J.B.; Lambert, W.C.; Schwartz, R.A. Gender differences in cutaneous melanoma: Demographics, prognostic factors, and survival outcomes. Dermatol. Ther. 2020, 33, e14131. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Patel, T.N. Melanoma therapeutics: A literature review. J. Biomed. Res. 2022, 36, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Wierzbicka, J.; Zmijewski, M.A. Vitamin D in the skin physiology and pathology. Acta Biochim. Pol. 2016, 63, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Brozyna, A.A.; Hoffman, R.M.; Slominski, A.T. Relevance of Vitamin D in Melanoma Development, Progression and Therapy. Anticancer Res. 2020, 40, 473–489. [Google Scholar] [CrossRef]
- Domżalski, P.; Piotrowska, A.; Tuckey, R.C.; Zmijewski, M.A. Anticancer Activity of Vitamin D, Lumisterol and Selected Derivatives against Human Malignant Melanoma Cell Lines. Int. J. Mol. Sci. 2024, 25, 10914. [Google Scholar] [CrossRef]
- Craig, S.; Earnshaw, C.H.; Virós, A. Ultraviolet light and melanoma. J. Pathol. 2018, 244, 578–585. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The mitogen-activated protein kinase pathway in melanoma part I—Activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Haist, M.; Kuske, M.; Grabbe, S.; Bros, M. Immunomodulatory Properties of BRAF and MEK Inhibitors Used for Melanoma Therapy-Paradoxical ERK Activation and Beyond. Int. J. Mol. Sci. 2021, 22, 9890. [Google Scholar] [CrossRef]
- Villanueva, J.; Herlyn, M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, X.; Chen, Z.; Liu, C.; Wu, W.; Zhang, N.; Liu, Z.; Yang, L.; Jiang, Q.; Cheng, Q.; et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: New opportunities in cancer immunotherapy and advances in clinical trials. Mol. Cancer 2023, 22, 159. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Strnadová, K.; Pfeiferová, L.; Přikryl, P.; Dvořánková, B.; Vlčák, E.; Frýdlová, J.; Vokurka, M.; Novotný, J.; Šáchová, J.; Hradilová, M.; et al. Exosomes produced by melanoma cells significantly influence the biological properties of normal and cancer-associated fibroblasts. Histochem. Cell Biol. 2022, 157, 153–172. [Google Scholar] [CrossRef]
- Pizzurro, G.A.; Liu, C.; Bridges, K.; Alexander, A.F.; Huang, A.; Baskaran, J.P.; Ramseier, J.; Bosenberg, M.W.; Mak, M.; Miller-Jensen, K. 3D Model of the Early Melanoma Microenvironment Captures Macrophage Transition into a Tumor-Promoting Phenotype. Cancers 2021, 13, 4579. [Google Scholar] [CrossRef]
- Jeon, T.J.; Kim, O.H.; Kang, H.; Lee, H.J. Preadipocytes potentiate melanoma progression and M2 macrophage polarization in the tumor microenvironment. Biochem. Biophys. Res. Commun. 2024, 721, 150129. [Google Scholar] [CrossRef]
- Romano, V.; Belviso, I.; Venuta, A.; Ruocco, M.R.; Masone, S.; Aliotta, F.; Fiume, G.; Montagnani, S.; Avagliano, A.; Arcucci, A. Influence of Tumor Microenvironment and Fibroblast Population Plasticity on Melanoma Growth, Therapy Resistance and Immunoescape. Int. J. Mol. Sci. 2021, 22, 5283. [Google Scholar] [CrossRef]
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530. [Google Scholar] [CrossRef]
- Chhabra, Y.; Fane, M.E.; Pramod, S.; Hüser, L.; Zabransky, D.J.; Wang, V.; Dixit, A.; Zhao, R.; Kumah, E.; Brezka, M.L.; et al. Sex-dependent effects in the aged melanoma tumor microenvironment influence invasion and resistance to targeted therapy. Cell 2024, 187, 6016–6034.e25. [Google Scholar] [CrossRef]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Arcucci, A. Insights into Melanoma Fibroblast Populations and Therapeutic Strategy Perspectives: Friends or Foes? Curr. Med. Chem. 2022, 29, 6159–6168. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Forsthuber, A.; Aschenbrenner, B.; Korosec, A.; Jacob, T.; Annusver, K.; Krajic, N.; Kholodniuk, D.; Frech, S.; Zhu, S.; Purkhauser, K.; et al. Cancer-associated fibroblast subtypes modulate the tumor-immune microenvironment and are associated with skin cancer malignancy. Nat. Commun. 2024, 15, 9678. [Google Scholar] [CrossRef]
- Suh, J.; Kim, D.H.; Lee, Y.H.; Jang, J.H.; Surh, Y.J. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinog. 2020, 59, 1028–1040. [Google Scholar] [CrossRef]
- Piotrowska, A.; Nowak, J.I.; Wierzbicka, J.M.; Domżalski, P.; Górska-Arcisz, M.; Sądej, R.; Popiel, D.; Wieczorek, M.; Żmijewski, M.A. Fibroblast Growth Factor Receptor Inhibitors Decrease Proliferation of Melanoma Cell Lines and Their Activity Is Modulated by Vitamin D. Int. J. Mol. Sci. 2024, 25, 2505. [Google Scholar] [CrossRef]
- Piotrowska, A.; Zaucha, R.; Król, O.; Żmijewski, M.A. Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs. Int. J. Mol. Sci. 2023, 24, 8037. [Google Scholar] [CrossRef]
- Christofides, A.; Strauss, L.; Yeo, A.; Cao, C.; Charest, A.; Boussiotis, V.A. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 2022, 23, 1148–1156. [Google Scholar] [CrossRef]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: An effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Loyher, P.L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef]
- Pizzurro, G.A.; Bridges, K.; Jiang, X.; Vidyarthi, A.; Miller-Jensen, K.; Colegio, O.R. Functionally and Metabolically Divergent Melanoma-Associated Macrophages Originate from Common Bone-Marrow Precursors. Cancers 2023, 15, 3330. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, S.; Yang, X.; Ren, M.; Qi, Y.; Mao, Y.; Yang, C. In vitro study of cold atmospheric plasma-activated liquids inhibits malignant melanoma by affecting macrophage polarization through the ROS/JAK2/STAT1 pathway. Biomed. Pharmacother. 2024, 175, 116657. [Google Scholar] [CrossRef]
- Yin, J.; Forn-Cuní, G.; Surendran, A.M.; Lopes-Bastos, B.; Pouliopoulou, N.; Jager, M.J.; Le Dévédec, S.E.; Chen, Q.; Snaar-Jagalska, B.E. Lactate secreted by glycolytic conjunctival melanoma cells attracts and polarizes macrophages to drive angiogenesis in zebrafish xenografts. Angiogenesis 2024, 27, 703–717. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Vargas-Delgado, M.E.; von Amsberg, G.; Janning, M.; Loges, S. Influence of Androgens on Immunity to Self and Foreign: Effects on Immunity and Cancer. Front. Immunol. 2020, 11, 1184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Liu, Z.; Wu, T.; Li, S.; Zhang, Y.; Yin, X.; Yang, G.; Zhang, G. Tumor cell-induced platelet aggregation accelerates hematogenous metastasis of malignant melanoma by triggering macrophage recruitment. J. Exp. Clin. Cancer Res. 2023, 42, 277. [Google Scholar] [CrossRef] [PubMed]
- Barrio-Alonso, C.; Nieto-Valle, A.; García-Martínez, E.; Gutiérrez-Seijo, A.; Parra-Blanco, V.; Márquez-Rodas, I.; Avilés-Izquierdo, J.A.; Sánchez-Mateos, P.; Samaniego, R. Chemokine profiling of melanoma-macrophage crosstalk identifies CCL8 and CCL15 as prognostic factors in cutaneous melanoma. J. Pathol. 2024, 262, 495–504. [Google Scholar] [CrossRef]
- Brummel, K.; Eerkens, A.L.; de Bruyn, M.; Nijman, H.W. Tumour-infiltrating lymphocytes: From prognosis to treatment selection. Br. J. Cancer 2023, 128, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Herlyn, M.; Wagner, S.N. The role of tumor microenvironment in melanoma therapy resistance. Melanoma Manag. 2016, 3, 23–32. [Google Scholar] [CrossRef]
- Chatani, P.D.; Lowery, F.J.; Parikh, N.B.; Hitscherich, K.J.; Yossef, R.; Hill, V.; Gartner, J.J.; Paria, B.; Florentin, M.; Ray, S.; et al. Cell surface marker-based capture of neoantigen-reactive CD8. J. Immunother. Cancer 2023, 11, e006264. [Google Scholar] [CrossRef]
- Smithy, J.W.; Schoenfeld, A.J.; Betof Warner, A. The Clinical TIL Experience in Melanoma: Past, Present, Future. Transpl. Cell Ther. 2025, 31, S626–S634. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Lindner, K.; Boschert, T.; Meng, Z.; Rodriguez Ehrenfried, A.; De Roia, A.; Haltenhof, G.; Faenza, A.; Imperatore, F.; Bunse, L.; et al. Prediction of tumor-reactive T cell receptors from scRNA-seq data for personalized T cell therapy. Nat. Biotechnol. 2025, 43, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Bida, M.; Miya, T.V.; Hull, R.; Dlamini, Z. Tumor-infiltrating lymphocytes in melanoma: From prognostic assessment to therapeutic applications. Front. Immunol. 2024, 15, 1497522. [Google Scholar] [CrossRef]
- Mei, Z.; Liu, Y.; Liu, C.; Cui, A.; Liang, Z.; Wang, G.; Peng, H.; Cui, L.; Li, C. Tumour-infiltrating inflammation and prognosis in colorectal cancer: Systematic review and meta-analysis. Br. J. Cancer 2014, 110, 1595–1605. [Google Scholar] [CrossRef]
- Elomaa, H.; Ahtiainen, M.; Väyrynen, S.A.; Ogino, S.; Nowak, J.A.; Friman, M.; Helminen, O.; Wirta, E.V.; Seppälä, T.T.; Böhm, J.; et al. Prognostic significance of spatial and density analysis of T lymphocytes in colorectal cancer. Br. J. Cancer 2022, 127, 514–523. [Google Scholar] [CrossRef]
- Canzian, J.; Conforti, F.; Jacobs, F.; Benvenuti, C.; Gaudio, M.; Gerosa, R.; De Sanctis, R.; Zambelli, A. Sex-Related Differences in Immunotherapy Toxicities: Insights into Dimorphic Responses. Cancers 2025, 17, 1054. [Google Scholar] [CrossRef]
- Caraban, B.M.; Matei, E.; Cozaru, G.C.; Aşchie, M.; Deacu, M.; Enciu, M.; Bălţătescu, G.I.; Chisoi, A.; Dobrin, N.; Petcu, L.; et al. PD-L1, CD4+, and CD8+ Tumor-Infiltrating Lymphocytes (TILs) Expression Profiles in Melanoma Tumor Microenvironment Cells. J. Pers. Med. 2023, 13, 221. [Google Scholar] [CrossRef]
- Di Gennaro, P.; Gerlini, G.; Caporale, R.; Sestini, S.; Brandani, P.; Urso, C.; Pimpinelli, N.; Borgognoni, L. T regulatory cells mediate immunosuppresion by adenosine in peripheral blood, sentinel lymph node and TILs from melanoma patients. Cancer Lett. 2018, 417, 124–130. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, K.; Liu, R.; Song, Y.; Lv, Y.; Bi, P.; Yang, F.; Li, S.; Zhao, J.; Li, X.; et al. Single-cell transcriptome sequencing of B-cell heterogeneity and tertiary lymphoid structure predicts breast cancer prognosis and neoadjuvant therapy efficacy. Clin. Transl. Med. 2023, 13, e1346. [Google Scholar] [CrossRef]
- Coppola, D.; Nebozhyn, M.; Khalil, F.; Dai, H.; Yeatman, T.; Loboda, A.; Mulé, J.J. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 2011, 179, 37–45. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Montauti, E.; Oh, D.Y.; Fong, L. CD4. Trends Cancer 2024, 10, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Aptsiauri, N.; Del Campo, A.; Maleno, I.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F.; Garcia-Lora, A. HLA and melanoma: Multiple alterations in HLA class I and II expression in human melanoma cell lines from ESTDAB cell bank. Cancer Immunol. Immunother. 2009, 58, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Draghi, A.; Presti, M.; Jensen, A.W.P.; Chamberlain, C.A.; Albieri, B.; Rasmussen, A.K.; Andersen, M.H.; Crowther, M.D.; Svane, I.M.; Donia, M. Uncoupling CD4+ TIL-Mediated Tumor Killing from JAK-Signaling in Melanoma. Clin. Cancer Res. 2023, 29, 3937–3947. [Google Scholar] [CrossRef]
- Bevington, S.L.; Cauchy, P.; Withers, D.R.; Lane, P.J.; Cockerill, P.N. T Cell Receptor and Cytokine Signaling Can Function at Different Stages to Establish and Maintain Transcriptional Memory and Enable T Helper Cell Differentiation. Front. Immunol. 2017, 8, 204. [Google Scholar] [CrossRef]
- Andreu-Sanz, D.; Kobold, S. Role and Potential of Different T Helper Cell Subsets in Adoptive Cell Therapy. Cancers 2023, 15, 1650. [Google Scholar] [CrossRef]
- Jacenik, D.; Karagiannidis, I.; Beswick, E.J. Th2 cells inhibit growth of colon and pancreas cancers by promoting anti-tumorigenic responses from macrophages and eosinophils. Br. J. Cancer 2023, 128, 387–397. [Google Scholar] [CrossRef]
- Li, C.; Yu, X.; Han, X.; Lian, C.; Wang, Z.; Shao, S.; Shao, F.; Wang, H.; Ma, S.; Liu, J. Innate immune cells in tumor microenvironment: A new frontier in cancer immunotherapy. iScience 2024, 27, 110750. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L. Cytotoxic CD4. Immunity 2021, 54, 2701–2711. [Google Scholar] [CrossRef]
- Li, C.; Phoon, Y.P.; Karlinsey, K.; Tian, Y.F.; Thapaliya, S.; Thongkum, A.; Qu, L.; Matz, A.J.; Cameron, M.; Cameron, C.; et al. A high OXPHOS CD8 T cell subset is predictive of immunotherapy resistance in melanoma patients. J. Exp. Med. 2022, 219, e20202084. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Dolton, G.; Rius, C.; Wall, A.; Szomolay, B.; Bianchi, V.; Galloway, S.A.E.; Hasan, M.S.; Morin, T.; Caillaud, M.E.; Thomas, H.L.; et al. Targeting of multiple tumor-associated antigens by individual T cell receptors during successful cancer immunotherapy. Cell 2023, 186, 3333–3349.e27. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, P.; Gerlini, G.; Urso, C.; Sestini, S.; Brandani, P.; Pimpinelli, N.; Borgognoni, L. CD4. Clin. Exp. Metastasis 2016, 33, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Serafin, P.K.; Popęda, M.; Bulak, K.; Zwara, A.; Galikowska-Bogut, B.; Przychodzka, A.; Mika, A.; Śledziński, T.; Stanisławowski, M.; Jendernalik, K.; et al. Knock-out of CD73 delays the onset of HR-negative breast cancer by reprogramming lipid metabolism and is associated with increased tumor mutational burden. Mol. Metab. 2024, 89, 102035. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- Laffont, S.; Seillet, C.; Guéry, J.C. Estrogen Receptor-Dependent Regulation of Dendritic Cell Development and Function. Front. Immunol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Sosa Cuevas, E.; Saas, P.; Aspord, C. Dendritic Cell Subsets in Melanoma: Pathophysiology, Clinical Prognosis and Therapeutic Exploitation. Cancers 2023, 15, 2206. [Google Scholar] [CrossRef]
- Chudnovskiy, A.; Castro, T.B.R.; Nakandakari-Higa, S.; Cui, A.; Lin, C.H.; Sade-Feldman, M.; Phillips, B.K.; Pae, J.; Mesin, L.; Bortolatto, J.; et al. Proximity-dependent labeling identifies dendritic cells that drive the tumor-specific CD4. Sci. Immunol. 2024, 9, eadq8843. [Google Scholar] [CrossRef] [PubMed]
- Gerlini, G.; Susini, P.; Sestini, S.; Brandani, P.; Giannotti, V.; Borgognoni, L. Langerhans Cells in Sentinel Lymph Nodes from Melanoma Patients. Cancers 2024, 16, 1890. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Ferrari, G.; Gazzurelli, L.; Bugatti, M.; Facchetti, F.; Vermi, W. Plasmacytoid dendritic cells at the forefront of anti-cancer immunity: Rewiring strategies for tumor microenvironment remodeling. J. Exp. Clin. Cancer Res. 2024, 43, 196. [Google Scholar] [CrossRef] [PubMed]
- Amon, L.; Seichter, A.; Vurnek, D.; Heger, L.; Lächele, L.; Tochoedo, N.R.; Kaszubowski, T.; Hatscher, L.; Baranska, A.; Tchitashvili, G.; et al. Clec12A, CD301b, and FcγRIIB/III define the heterogeneity of murine DC2s and DC3s. Cell Rep. 2024, 43, 113949. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Hara, I.; Gleave, M.E.; Eto, H. Protection of androgen-dependent human prostate cancer cells from oxidative stress-induced DNA damage by overexpression of clusterin and its modulation by androgen. Prostate 2004, 61, 318–323. [Google Scholar] [CrossRef]
- López Malizia, A.; Merlotti, A.; Bonte, P.E.; Sager, M.; Arribas De Sandoval, Y.; Goudot, C.; Erra Díaz, F.; Pereyra-Gerber, P.; Ceballos, A.; Amigorena, S.; et al. Clusterin protects mature dendritic cells from reactive oxygen species mediated cell death. Oncoimmunology 2024, 13, 2294564. [Google Scholar] [CrossRef]
- Lee, C.; Collichio, F.; Ollila, D.; Moschos, S. Historical review of melanoma treatment and outcomes. Clin. Dermatol. 2013, 31, 141–147. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Lim, S.Y.; Shklovskaya, E.; Lee, J.H.; Pedersen, B.; Stewart, A.; Ming, Z.; Irvine, M.; Shivalingam, B.; Saw, R.P.M.; Menzies, A.M.; et al. The molecular and functional landscape of resistance to immune checkpoint blockade in melanoma. Nat. Commun. 2023, 14, 1516. [Google Scholar] [CrossRef]
- Zou, Y.; Yaguchi, T. Programmed cell death-1 blockade therapy in melanoma: Resistance mechanisms and combination strategies. Exp. Dermatol. 2023, 32, 264–275. [Google Scholar] [CrossRef]
- Kawashima, S.; Togashi, Y. Resistance to immune checkpoint inhibitors and the tumor microenvironment. Exp. Dermatol. 2023, 32, 240–249. [Google Scholar] [CrossRef]
- Thornton, J.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Shirley, C.A.; Ahmad, N. Mechanisms of Immunotherapy Resistance in Cutaneous Melanoma: Recognizing a Shapeshifter. Front. Oncol. 2022, 12, 880876. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Agarwala, S.S.; Messersmith, H.; Alluri, K.C.; Ascierto, P.A.; Atkins, M.B.; Bollin, K.; Chacon, M.; Davis, N.; Faries, M.B.; et al. Systemic Therapy for Melanoma: ASCO Guideline Update. J. Clin. Oncol. 2023, 41, 4794–4820. [Google Scholar] [CrossRef] [PubMed]
- Farma, J.M.; Olszanski, A.J.; Messina, J.L.; Sondak, V.K. Annals of Surgical Oncology Practice Guidelines Series: Adjuvant and Neoadjuvant Therapy for Melanoma. Ann. Surg. Oncol. 2025, 32, 3–11. [Google Scholar] [CrossRef]
- Amaral, T.; Ottaviano, M.; Arance, A.; Blank, C.; Chiarion-Sileni, V.; Donia, M.; Dummer, R.; Garbe, C.; Gershenwald, J.E.; Gogas, H.; et al. Cutaneous melanoma: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2025, 36, 10–30. [Google Scholar] [CrossRef] [PubMed]
- Boutros, A.; Croce, E.; Ferrari, M.; Gili, R.; Massaro, G.; Marconcini, R.; Arecco, L.; Tanda, E.T.; Spagnolo, F. The treatment of advanced melanoma: Current approaches and new challenges. Crit. Rev. Oncol. Hematol. 2024, 196, 104276. [Google Scholar] [CrossRef]
- Kwong, M.L.M.; Yang, J.C. Lifileucel: FDA-approved T-cell therapy for melanoma. Oncologist 2024, 29, 648–650. [Google Scholar] [CrossRef]
- Natarelli, N.; Aleman, S.J.; Mark, I.M.; Tran, J.T.; Kwak, S.; Botto, E.; Aflatooni, S.; Diaz, M.J.; Lipner, S.R. A Review of Current and Pipeline Drugs for Treatment of Melanoma. Pharmaceuticals 2024, 17, 214. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Fujimura, T.; Aiba, S. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules 2020, 10, 1087. [Google Scholar] [CrossRef]
- Bernard, N.J. Stromal regulation of the tumor. Nat. Immunol. 2024, 25, 1509. [Google Scholar] [CrossRef] [PubMed]
- Badillo, O.; Helfridsson, L.; Niemi, J.; Hellström, M. Exploring dendritic cell subtypes in cancer immunotherapy: Unraveling the role of mature regulatory dendritic cells. Upsala J. Med. Sci. 2024, 129, e10627. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Brazel, D.; DeRogatis, J.; Valerin, J.B.G.; Whiteson, K.; Chow, W.A.; Tinoco, R.; Moyers, J.T. The cure from within? a review of the microbiome and diet in melanoma. Cancer Metastasis Rev. 2022, 41, 261–280. [Google Scholar] [CrossRef]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Oh, B.; Boyle, F.; Pavlakis, N.; Clarke, S.; Eade, T.; Hruby, G.; Lamoury, G.; Carroll, S.; Morgia, M.; Kneebone, A.; et al. The Gut Microbiome and Cancer Immunotherapy: Can We Use the Gut Microbiome as a Predictive Biomarker for Clinical Response in Cancer Immunotherapy? Cancers 2021, 13, 4824. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Q.; He, L.; Sun, X. The critical role of tumor microbiome in cancer immunotherapy. Cancer Biol. Ther. 2024, 25, 2301801. [Google Scholar] [CrossRef]
- Li, Y.; Cong, Y.; Jia, M.; He, Q.; Zhong, H.; Zhao, Y.; Li, H.; Yan, M.; You, J.; Liu, J.; et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 2021, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Shi, J.; Fang, Y.; Zhao, Y.; Xu, A.; Li, N. Developing innovative strategies of tumor-infiltrating lymphocyte therapy for tumor treatment. Oncol. Rep. 2024, 51, 85. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.R.; Maeda, T.; Tamaoki, N.; Good, M.L.; Kishton, R.J.; Paria, B.C.; Yu, Z.; Bosch-Marce, M.; Bedanova, N.M.; Liu, C.; et al. Reprogramming of Tumor-reactive Tumor-infiltrating Lymphocytes to Human-induced Pluripotent Stem Cells. Cancer Res. Commun. 2023, 3, 917–932. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Iwabuchi, K.; Fusaki, N.; Ito, F. Generation of Induced Pluripotent Stem Cells from Human Melanoma Tumor-infiltrating Lymphocytes. J. Vis. Exp. 2016, 117, 54375. [Google Scholar] [CrossRef]
- Feist, M.; Zhu, Z.; Dai, E.; Ma, C.; Liu, Z.; Giehl, E.; Ravindranathan, R.; Kowalsky, S.J.; Obermajer, N.; Kammula, U.S.; et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2021, 28, 98–111. [Google Scholar] [CrossRef]
- Santos, J.M.; Heiniö, C.; Cervera-Carrascon, V.; Quixabeira, D.C.A.; Siurala, M.; Havunen, R.; Butzow, R.; Zafar, S.; de Gruijl, T.; Lassus, H.; et al. Oncolytic adenovirus shapes the ovarian tumor microenvironment for potent tumor-infiltrating lymphocyte tumor reactivity. J. Immunother. Cancer 2020, 8, e000188. [Google Scholar] [CrossRef]
- Ye, K.; Li, F.; Wang, R.; Cen, T.; Liu, S.; Zhao, Z.; Li, R.; Xu, L.; Zhang, G.; Xu, Z.; et al. An armed oncolytic virus enhances the efficacy of tumor-infiltrating lymphocyte therapy by converting tumors to artificial antigen-presenting cells in situ. Mol. Ther. 2022, 30, 3658–3676. [Google Scholar] [CrossRef]
- Yang, H.; Berezowska, S.; Dorn, P.; Zens, P.; Chen, P.; Peng, R.W.; Marti, T.M.; Kocher, G.J.; Schmid, R.A.; Hall, S.R.R. Tumor-infiltrating lymphocytes are functionally inactivated by CD90+ stromal cells and reactivated by combined Ibrutinib and Rapamycin in human pleural mesothelioma. Theranostics 2022, 12, 167–185. [Google Scholar] [CrossRef]
- Trüb, M.; Uhlenbrock, F.; Claus, C.; Herzig, P.; Thelen, M.; Karanikas, V.; Bacac, M.; Amann, M.; Albrecht, R.; Ferrara-Koller, C.; et al. Fibroblast activation protein-targeted-4-1BB ligand agonist amplifies effector functions of intratumoral T cells in human cancer. J. Immunother. Cancer 2020, 8, e000238. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Shokrollahi Barough, M.; Momeni-Varposhti, Z.; Ghanavatinejad, A.; Zarehzadeh Mehrabadi, A.; Sadeghi, B.; Falak, R. Pentoxifylline changes the balance of immune cell population in breast tumor-infiltrating lymphocytes. Med. Oncol. 2023, 40, 168. [Google Scholar] [CrossRef] [PubMed]
- Fix, S.M.; Forget, M.A.; Sakellariou-Thompson, D.; Wang, Y.; Griffiths, T.M.; Lee, M.; Haymaker, C.L.; Dominguez, A.L.; Basar, R.; Reyes, C.; et al. CRISPR-mediated TGFBR2 knockout renders human ovarian cancer tumor-infiltrating lymphocytes resistant to TGF-β signaling. J. Immunother. Cancer 2022, 10, e003750. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.M.V.; Riise, R.; Saellström, S.; Karlsson, J.; Alsén, S.; Bucher, V.; Hemminki, A.E.; Olofsson Bagge, R.; Ny, L.; Nilsson, L.M.; et al. Treatment with Anti-HER2 Chimeric Antigen Receptor Tumor-Infiltrating Lymphocytes (CAR-TILs) Is Safe and Associated with Antitumor Efficacy in Mice and Companion Dogs. Cancers 2023, 15, 648. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Truong, N.T.H.; Gardam, B.; Yu, W.; Ebert, L.M.; Johnson, A.; Yeo, E.C.F.; Wittwer, N.L.; Tapia Rico, G.; Logan, J.; et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J. Immunother. Cancer 2024, 12, e008659. [Google Scholar] [CrossRef]
- Rojas-Quintero, J.; Díaz, M.P.; Palmar, J.; Galan-Freyle, N.J.; Morillo, V.; Escalona, D.; González-Torres, H.J.; Torres, W.; Navarro-Quiroz, E.; Rivera-Porras, D.; et al. Car T Cells in Solid Tumors: Overcoming Obstacles. Int. J. Mol. Sci. 2024, 25, 4170. [Google Scholar] [CrossRef]
- Edeline, J.; Houot, R.; Marabelle, A.; Alcantara, M. CAR-T cells and BiTEs in solid tumors: Challenges and perspectives. J. Hematol. Oncol. 2021, 14, 65. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomised, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyzed Feature | CAFs | Refs. | TAMs | Ref. | TILs | Refs. | DCs | Refs. |
|---|---|---|---|---|---|---|---|---|
| Origin | Resident fibroblasts, BM-MSCs, EMT, EndMT, MMT, adipocytes, pericytes | [25] | TRMs—derived from yolk sac and fetal liver BM-derived monocytes—from myeloid progenitors in the bone marrow | [40,41,42,43] | Recruited from the population of circulating lymphocytes | [50] | CDP-derived, MDP-derived, lymph node-derived, epidermis-derived | [79,81,82,83,85] |
| Activation mechanisms | Exosomes secreted by cancer cells, IL-1β, IL-6, IL-8, TGF-β, PDGF, FGF-2, miR-211/155 | [34] | Lactate, exosomes secreted by cancer cells, TCIPA (CCL2, IL-1α/β, SDF-1), CCL8, CCL15 | [28,29,47,48] | IFN-γ/IL-2/IL-4/IL-6/IL-21/TGF-β, antigen presentation by DC, interaction with MHC-I/MHC-II | [54,60,65,72] | XCL1, CCL5, FLT3L, TLR7, TLR9, tissue damage, contact with T-cells, CD40–CD40L interaction, CTLA-4/PD-1 blockade, IL-27 | [79,82,83,84,87] |
| Markers | mCAFs: COL1A1, COL1A2, COL3A1, LUM, POSTN, TNC; iCAFs: MMP1, MMP3, IL6, CXCL8, IDO1; myoCAFs: ACTA2, COL1A1, RGS5, KCNJ8, MCAM; other markers: FSP-1, vimentin, FAP, podoplanin, PDGFR α/β, decorin, osteonectin, desmin, CD90/THY1, NG2 | [34,36] | M1: TNF-α, IL-1β, IL-6, iNOS, CD80; M2: Arg1, Chi3l3, MRC1, CD206 | [44] | T: CD3; Th: CD4; Treg: FOXP3, CD25; cytotoxic: CD4, EOMES, TBET, RUNX3/CD8 degranulation: GNLY, NKG7, CD107a; tumor-reactive profile: CD39/CD103, PD-1, TIGIT, CXCL13; B: CD20, CD23/CD69/CXCR4/TCL1A/MKI67 (subtype dependent); plasma cells: PRDM1, XBP1, MZB1, SSR4 | [51,54,59,60,65,67,72] | cDC1: XCR1, DNGR-1, CLEC9A, CD103, BDCA3, CD141 cDC2: BDCA1, CD1c, CD11c, CD5, FCεR1, CCR2, BTLA LCs: CD83, IDO1 pDCs: BDCA-2, ILT7, TRAIL, Granzyme B, ICOS-L, IDO, PD-L1 mregDCs: LAMP3, CCR7, CD83, BIRC3, MARCKSL1, PD-L1, CD200, Clusterin DC3s: FcγRIIB/III | [79,81,82,83,84,85,87] |
| Functions | ECM remodeling, physical barrier for immune cells, production of immunosuppressive cytokines, promotion of cancer cell migration and invasiveness | [5,34,36] | Phagocytosis and antigen presentation, promotion of angiogenesis (VEGF, TGF-β, and IL-10), potential support of primary tumor development (TRMs) and metastasis (M2), facilitation of immunosuppression (PD-L1) | [40,42,45,47,48] | B: a crucial component of TLS, antigen processing, antibody production; T: cytotoxicity (cytotoxic CD4+/CD8+), recruitment and stimulation of immune cells (Th), induction of tolerance, adenosine secretion(Treg) | [54,59,60,65,70,72] | Antigen uptake, presentation, cross-presentation, migration, chemokine/cytokine secretion, T-cell activation, immune regulation, cytotoxicity (TRAIL/granzyme B), Treg induction, costimulatory/inhibitory molecule expression | [79,81,82,83,84,87] |
| Subpopulations | mCAFs, iCAFs, myoCAFs, ucCAFs | [36] | M0, M1, M2, F4/80^high, F4/80^low | [43,44] | B:Naive, CXCR4+, Follicular, Germinal center, Plasma; T: Th1, Th2, Th17, (n/i/o/x)Tregs, cytotoxic CD4+/CD8+ | [51,54,59,60,65,70,72] | cDC1, cDC2, pDC, LCs, mregDC, DC3 | [79,82,83,84,85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sikorski, H.; Żmijewski, M.A.; Piotrowska, A. Tumor Microenvironment in Melanoma—Characteristic and Clinical Implications. Int. J. Mol. Sci. 2025, 26, 6778. https://doi.org/10.3390/ijms26146778
Sikorski H, Żmijewski MA, Piotrowska A. Tumor Microenvironment in Melanoma—Characteristic and Clinical Implications. International Journal of Molecular Sciences. 2025; 26(14):6778. https://doi.org/10.3390/ijms26146778
Chicago/Turabian StyleSikorski, Hubert, Michał Aleksander Żmijewski, and Anna Piotrowska. 2025. "Tumor Microenvironment in Melanoma—Characteristic and Clinical Implications" International Journal of Molecular Sciences 26, no. 14: 6778. https://doi.org/10.3390/ijms26146778
APA StyleSikorski, H., Żmijewski, M. A., & Piotrowska, A. (2025). Tumor Microenvironment in Melanoma—Characteristic and Clinical Implications. International Journal of Molecular Sciences, 26(14), 6778. https://doi.org/10.3390/ijms26146778