
A New GlyT2 Variant Associated with Hyperekplexia

 , , , , , , and
, , , , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Patient Information
2.2. Genetic Analysis
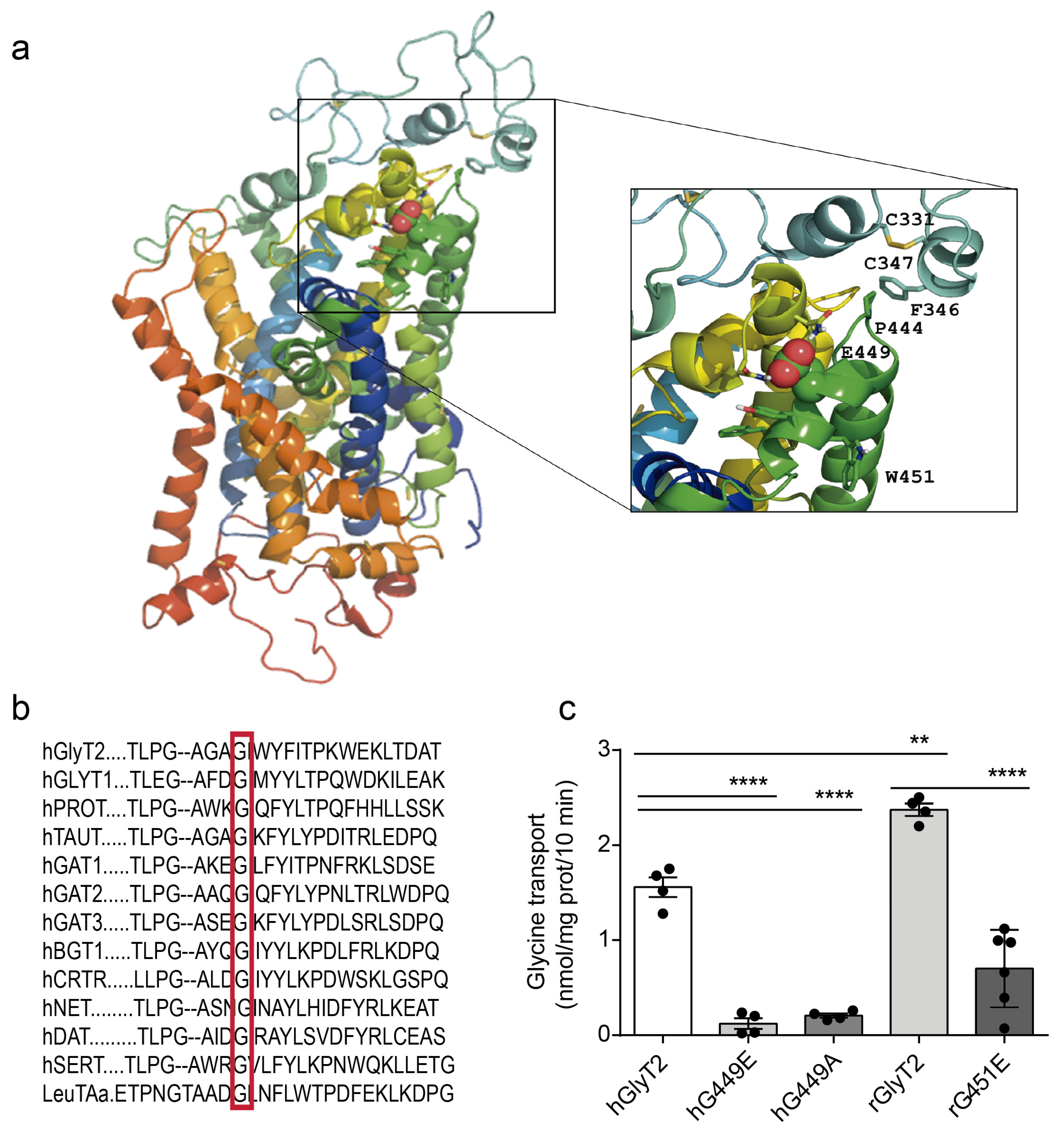
2.3. Mutagenesis and Functional Characterization
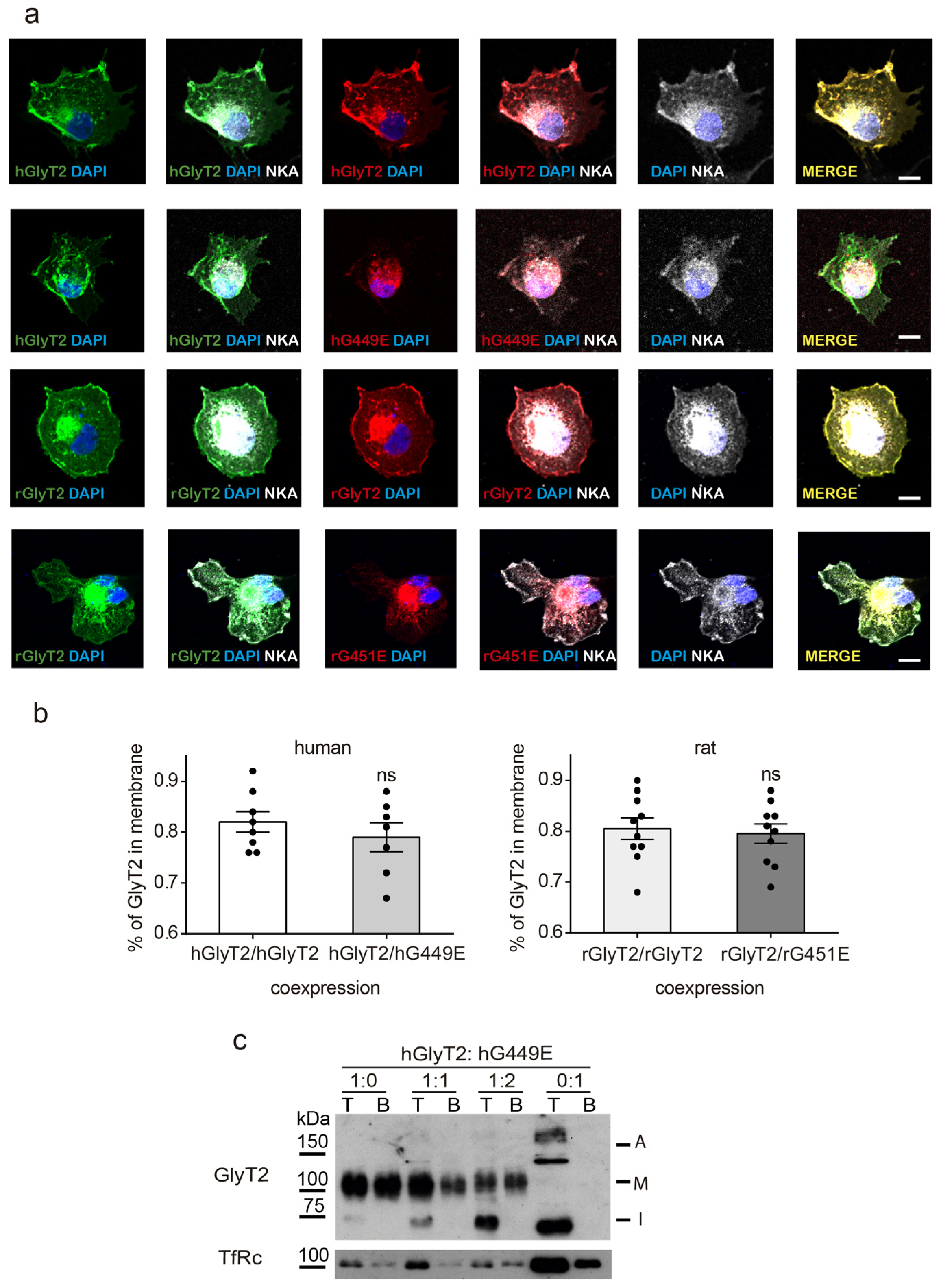
2.4. Transporter Expression
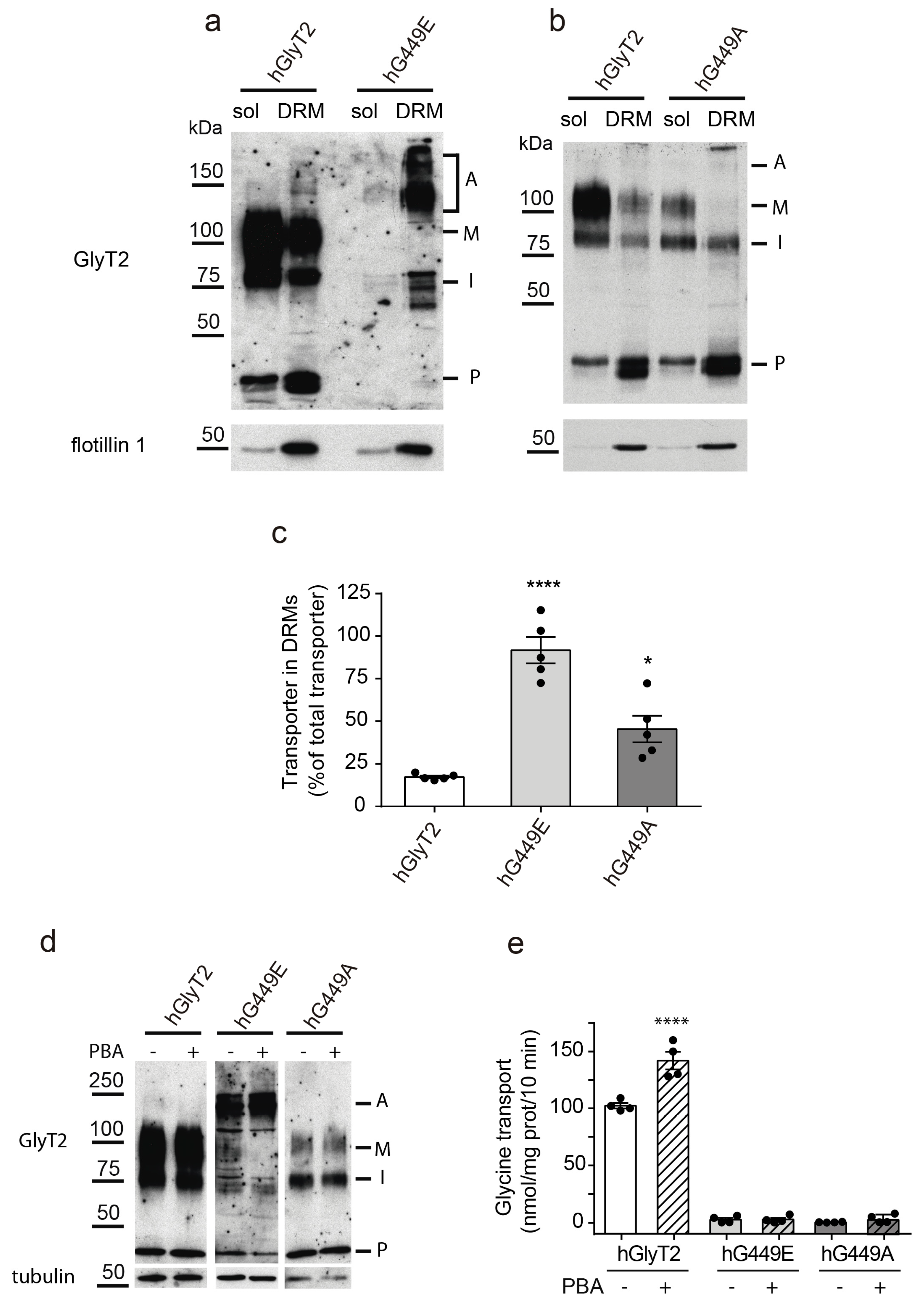
2.5. Lipid Raft Expression
2.6. Wild-Type–Mutant Coexpression
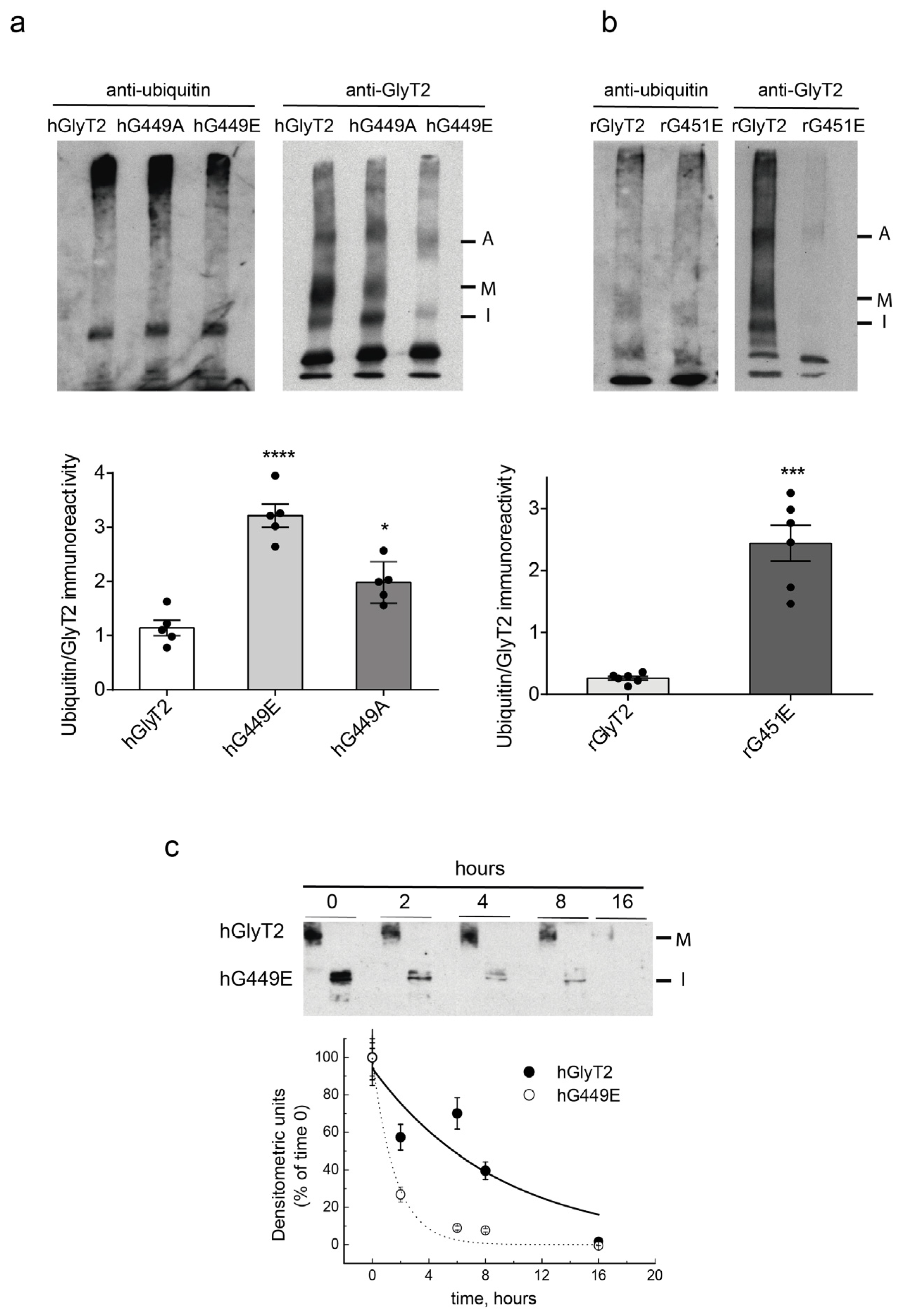
2.7. Mutant Transporter Degradation
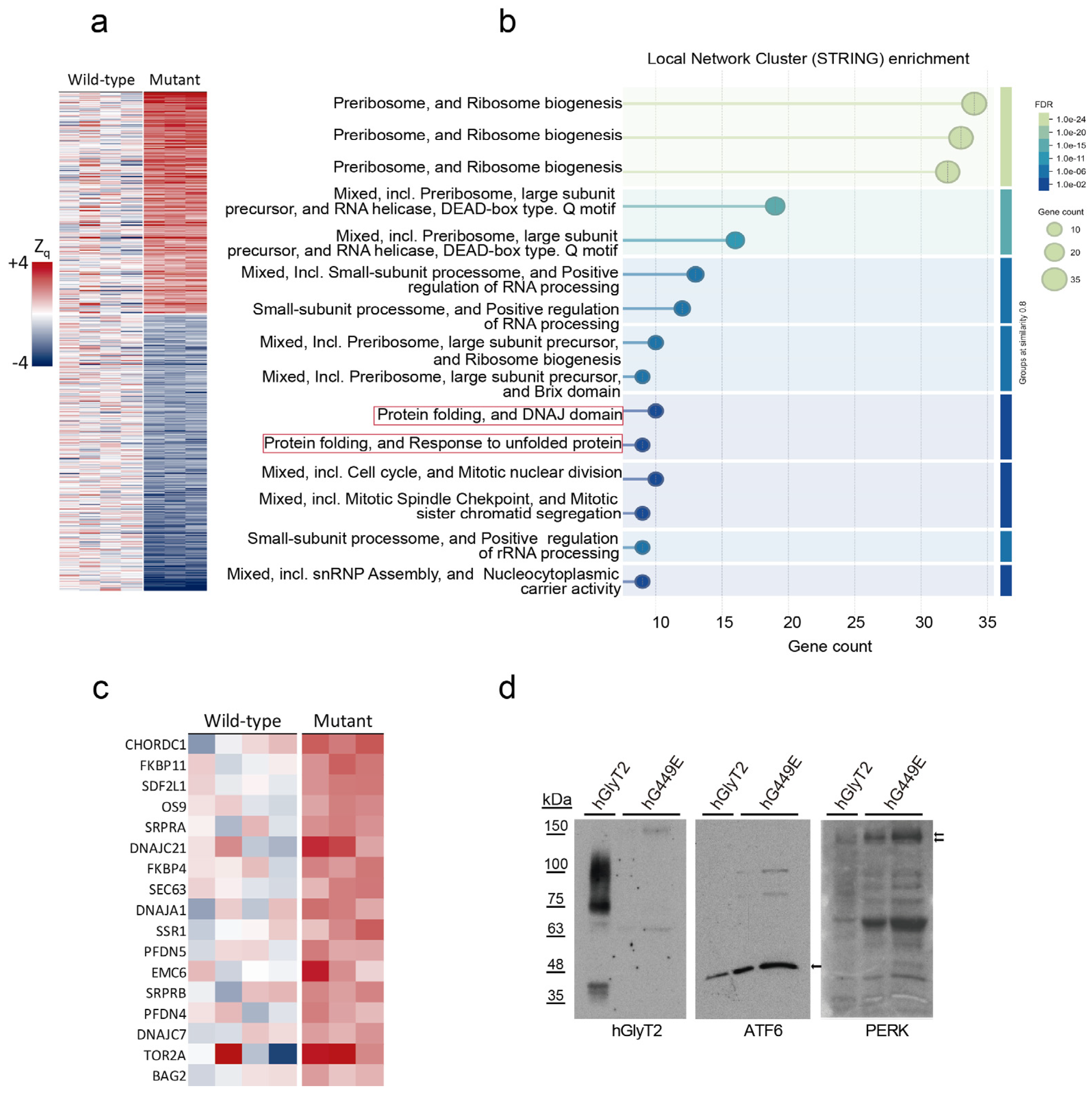
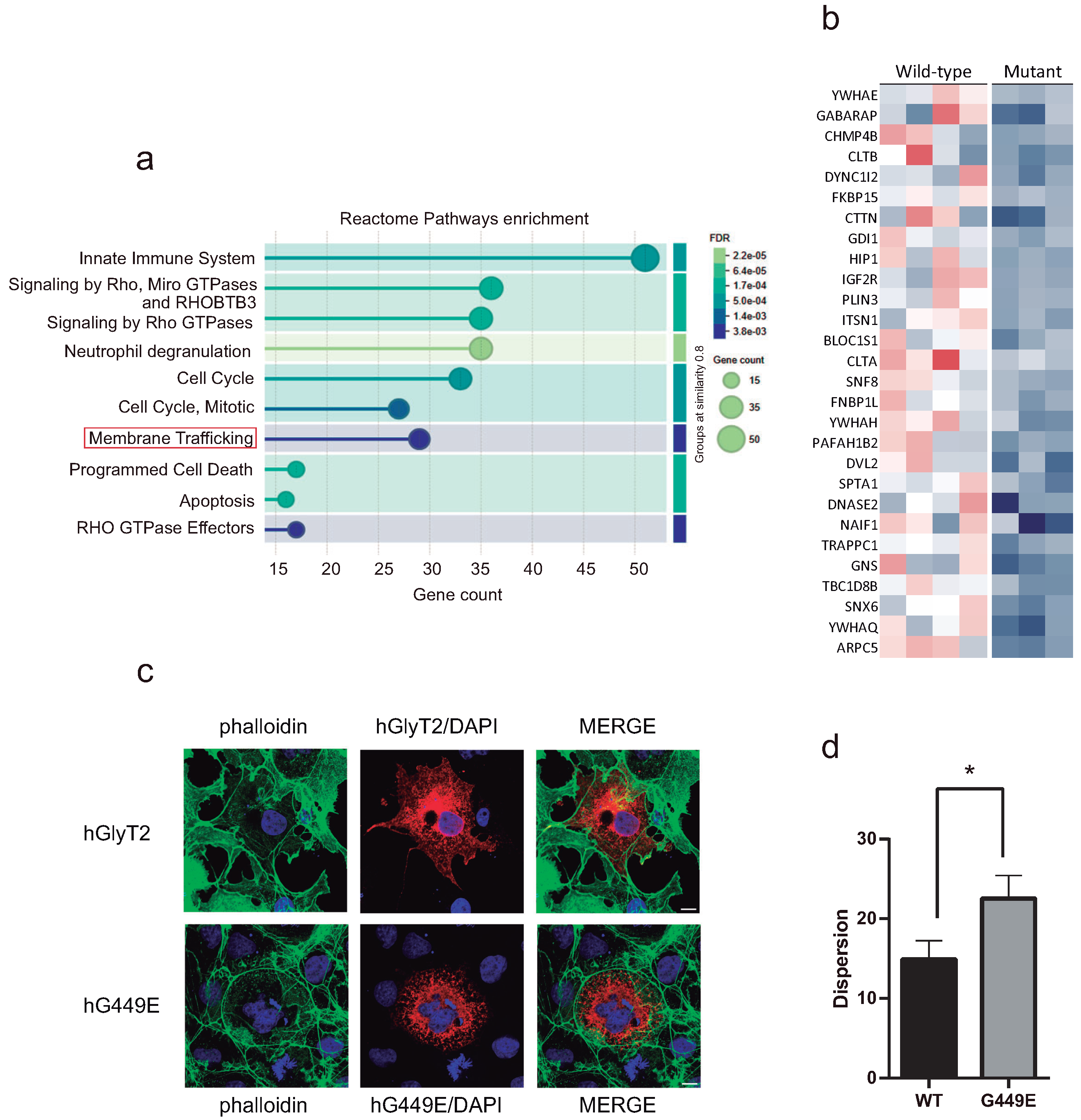
2.8. Proteomic Analysis
3. Discussion
4. Materials and Methods
4.1. Molecular Genetic Analysis of Human GlyT2 Gene (SLC6A5)
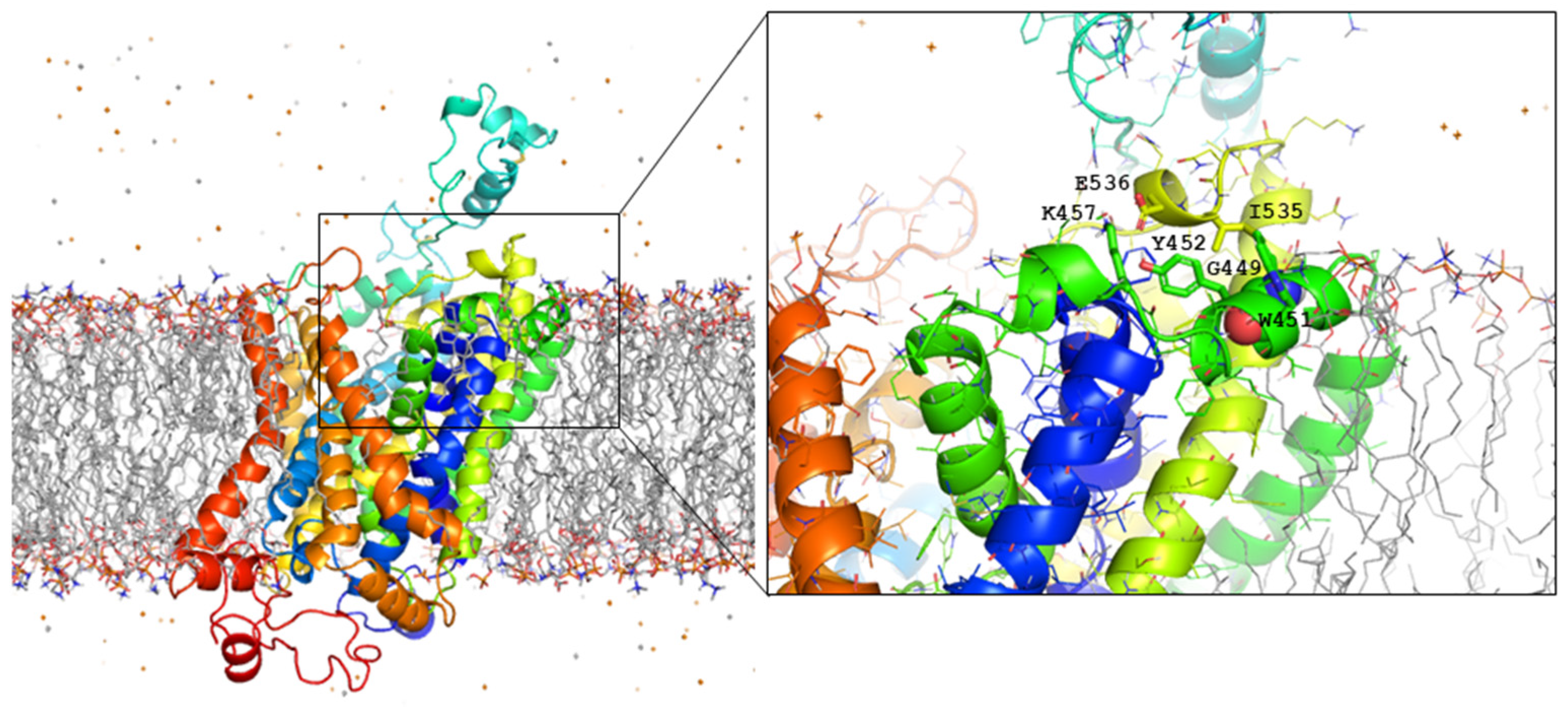
4.2. In Silico Analysis and Molecular Modeling
4.3. GlyT2 Mutagenesis and Transporter Expression
4.4. Transport Assays
4.5. Surface Biotinylation
4.6. Immunofluorescent Staining of Cultured Cells
4.7. Electrophoresis and Western Blotting
4.8. Carbohydrate Modification
4.9. Immunoprecipitation Assays
4.10. Isolation of Detergent-Resistant Membranes
4.11. Ubiquitination Assay
4.12. Proteomics
4.12.1. Preparation of Protein Extracts and Protein Digestion
4.12.2. Isobaric Labeling of Tryptic Peptides
4.12.3. Liquid Chromatography–Tandem Mass Spectrometry Analysis
4.12.4. Peptide and Protein Identification
4.12.5. Statistical Assessment of Protein Abundance Changes
4.13. Statistical Analysis and Data Representation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNX | calnexin |
| COPII | coatomer protein II |
| DRM | detergent-resistant membrane |
| ERAD | ER-associated degradation |
| GlyT | glycine transporter |
| GlyR | glycine receptor |
| GLR | glycine receptor subunit |
| LeuTAa | leucine transporter from A. aeolicus |
| NAG | N-arachidonoyl glycine |
| PBA | 4-phenylbutyric acid |
| SLC | solute carrier |
| TM | transmembrane domain |
References
- Saini, A.G.; Pandey, S. Hyperekplexia and Other Startle Syndromes. J. Neurol. Sci. 2020, 416, 117051. [Google Scholar] [CrossRef]
- Bakker, M.J.; van Dijk, J.G.; van den Maagdenberg, A.M.; Tijssen, M.A. Startle Syndromes. Lancet Neurol. 2006, 5, 513–524. [Google Scholar] [CrossRef]
- Foster, E.; Wildner, H.; Tudeau, L.; Haueter, S.; Ralvenius, W.T.; Jegen, M.; Johannssen, H.; Hosli, L.; Haenraets, K.; Ghanem, A.; et al. Targeted Ablation, Silencing, and Activation Establish Glycinergic Dorsal Horn Neurons as Key Components of a Spinal Gate for Pain and Itch. Neuron 2015, 85, 1289–1304. [Google Scholar] [CrossRef] [PubMed]
- Legendre, P. The Glycinergic Inhibitory Synapse. Cell. Mol. Life Sci. 2001, 58, 760–793. [Google Scholar] [CrossRef]
- Marques, B.L.; Oliveira-Lima, O.C.; Carvalho, G.A.; de Almeida Chiarelli, R.; Ribeiro, R.I.; Parreira, R.C.; da Madeira Freitas, E.M.; Resende, R.R.; Klempin, F.; Ulrich, H.; et al. Neurobiology of Glycine Transporters: From Molecules to Behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Gomeza, J.; Ohno, K.; Hulsmann, S.; Armsen, W.; Eulenburg, V.; Richter, D.W.; Laube, B.; Betz, H. Deletion of the Mouse Glycine Transporter 2 Results in a Hyperekplexia Phenotype and Postnatal Lethality. Neuron 2003, 40, 797–806. [Google Scholar] [CrossRef]
- Hartenstein, B.; Schenkel, J.; Kuhse, J.; Besenbeck, B.; Kling, C.; Becker, C.M.; Betz, H.; Weiher, H. Low Level Expression of Glycine Receptor Beta Subunit Transgene Is Sufficient for Phenotype Correction in Spastic Mice. EMBO J. 1996, 15, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- López-Corcuera, B.; Arribas-González, E.; Aragón, C. Hyperekplexia-Associated Mutations in the Neuronal Glycine Transporter 2. Neurochem. Int. 2019, 123, 95–100. [Google Scholar] [CrossRef]
- Rees, M.I.; Harvey, K.; Pearce, B.R.; Chung, S.K.; Duguid, I.C.; Thomas, P.; Beatty, S.; Graham, G.E.; Armstrong, L.; Shiang, R.; et al. Mutations in the Gene Encoding GlyT2 (SLC6A5) Define a Presynaptic Component of Human Startle Disease. Nat. Genet. 2006, 38, 801–806. [Google Scholar] [CrossRef]
- Dreissen, Y.E.; Tijssen, M.A. The Startle Syndromes: Physiology and Treatment. Epilepsia 2012, 53 (Suppl. S7), 3–11. [Google Scholar] [CrossRef]
- Thomas, R.H.; Chung, S.-K.; Wood, S.E.; Cushion, T.D.; Drew, C.J.G.; Hammond, C.L.; Vanbellinghen, J.-F.; Mullins, J.G.L.; Rees, M.I. Genotype-Phenotype Correlations in Hyperekplexia: Apnoeas, Learning Difficulties and Speech Delay. Brain 2013, 136, 3085–3095. [Google Scholar] [CrossRef]
- Freissmuth, M.; Stockner, T.; Sucic, S. SLC6 Transporter Folding Diseases and Pharmacochaperoning. In Targeting Trafficking in Drug Development; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl−-Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.R.; Joseph, D.; Höfner, G.; Dakua, A.; Athreya, A.; Wanner, K.T.; Kanner, B.I.; Penmatsa, A. Cryo-EM Structure of GABA Transporter 1 Reveals Substrate Recognition and Transport Mechanism. Nat. Struct. Mol. Biol. 2023, 30, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.-C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin Transporter-Ibogaine Complexes Illuminate Mechanisms of Inhibition and Transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Shahsavar, A.; Stohler, P.; Bourenkov, G.; Zimmermann, I.; Siegrist, M.; Guba, W.; Pinard, E.; Sinning, S.; Seeger, M.A.; Schneider, T.R.; et al. Structural Insights into the Inhibition of Glycine Reuptake. Nature 2021, 591, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Gonzalez, E.; Alonso-Torres, P.; Aragon, C.; Lopez-Corcuera, B. Calnexin-Assisted Biogenesis of the Neuronal Glycine Transporter 2 (GlyT2). PLoS ONE 2013, 8, e63230. [Google Scholar] [CrossRef]
- Arribas-Gonzalez, E.; de Juan-Sanz, J.; Aragon, C.; Lopez-Corcuera, B. Molecular Basis of the Dominant Negative Effect of a Glycine Transporter 2 Mutation Associated with Hyperekplexia. J. Biol. Chem. 2015, 290, 2150–2165. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Kawalia, A.; Sprute, R.; Karakaya, M.; Malenica, A.; Herkenrath, P.; Nürnberg, P.; Motameny, S.; Thiele, H.; Cirak, S. Novel Mutations in SLC6A5 with Benign Course in Hyperekplexia. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004465. [Google Scholar] [CrossRef]
- Kitzenmaier, A.; Schaefer, N.; Kasaragod, V.B.; Polster, T.; Hantschmann, R.; Schindelin, H.; Villmann, C. The P429L Loss of Function Mutation of the Human Glycine Transporter 2 Associated with Hyperekplexia. Eur. J. Neurosci. 2019, 50, 3906–3920. [Google Scholar] [CrossRef]
- de la Rocha-Muñoz, A.; Melgarejo, E.; Aragón, C.; López-Corcuera, B. Rescue of Two Trafficking-Defective Variants of the Neuronal Glycine Transporter GlyT2 Associated to Hyperekplexia. Neuropharmacology 2021, 189, 108543. [Google Scholar] [CrossRef]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human Non-Synonymous SNPs: Server and Survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef] [PubMed]
- Benito-Munoz, C.; Perona, A.; Abia, D.; Dos Santos, H.G.; Nunez, E.; Aragon, C.; Lopez-Corcuera, B. Modification of a Putative Third Sodium Site in the Glycine Transporter GlyT2 Influences the Chloride Dependence of Substrate Transport. Front. Mol. Neurosci. 2018, 11, 347. [Google Scholar] [CrossRef]
- Gimenez, C.; Perez-Siles, G.; Martinez-Villarreal, J.; Arribas-Gonzalez, E.; Jimenez, E.; Nunez, E.; de Juan-Sanz, J.; Fernandez-Sanchez, E.; Garcia-Tardon, N.; Ibanez, I.; et al. A Novel Dominant Hyperekplexia Mutation Y705C Alters Trafficking and Biochemical Properties of the Presynaptic Glycine Transporter GlyT2. J. Biol. Chem. 2012, 287, 28986–29002. [Google Scholar] [CrossRef] [PubMed]
- Taner, S.B.; Pando, M.J.; Roberts, A.; Schellekens, J.; Marsh, S.G.E.; Malmberg, K.-J.; Parham, P.; Brodsky, F.M. Interactions of NK Cell Receptor KIR3DL1*004 with Chaperones and Conformation-Specific Antibody Reveal a Functional Folded State as Well as Predominant Intracellular Retention. J. Immunol. 2011, 186, 62–72. [Google Scholar] [CrossRef]
- de Juan-Sanz, J.; Nunez, E.; Zafra, F.; Berrocal, M.; Corbacho, I.; Ibanez, I.; Arribas-Gonzalez, E.; Marcos, D.; Lopez-Corcuera, B.; Mata, A.M.; et al. Presynaptic Control of Glycine Transporter 2 (GlyT2) by Physical and Functional Association with Plasma Membrane Ca2+-ATPase (PMCA) and Na+-Ca2+ Exchanger (NCX). J. Biol. Chem. 2014, 289, 34308–34324. [Google Scholar] [CrossRef] [PubMed]
- de Juan-Sanz, J.; Nunez, E.; Villarejo-Lopez, L.; Perez-Hernandez, D.; Rodriguez-Fraticelli, A.E.; Lopez-Corcuera, B.; Vazquez, J.; Aragon, C. Na+/K+-ATPase Is a New Interacting Partner for the Neuronal Glycine Transporter GlyT2 That Downregulates Its Expression in Vitro and in Vivo. J. Neurosci. 2013, 33, 14269–14281. [Google Scholar] [CrossRef] [PubMed]
- Nunez, E.; Alonso-Torres, P.; Fornes, A.; Aragon, C.; Lopez-Corcuera, B. The Neuronal Glycine Transporter GLYT2 Associates with Membrane Rafts: Functional Modulation by Lipid Environment. J. Neurochem. 2008, 105, 2080–2090. [Google Scholar] [CrossRef]
- García-García, E.; Brown, E.J.; Rosales, C. Transmembrane Mutations to FcgammaRIIA Alter Its Association with Lipid Rafts: Implications for Receptor Signaling. J. Immunol. 2007, 178, 3048–3058. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-Pi Interactions in Structural Biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef]
- Svistunov, V.O.; Ehrmann, K.J.; Lencer, W.I.; Schmieder, S.S. Sorting of Complex Sphingolipids within the Cellular Endomembrane Systems. Front. Cell Dev. Biol. 2024, 12, 1490870. [Google Scholar] [CrossRef] [PubMed]
- Piniella, D.; Canseco, A.; Vidal, S.; Xiol, C.; Díaz de Bustamante, A.; Martí-Carrera, I.; Armstrong, J.; Bastolla, U.; Zafra, F. Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1. Int. J. Mol. Sci. 2023, 24, 955. [Google Scholar] [CrossRef] [PubMed]
- Kasture, A.; El-Kasaby, A.; Szöllősi, D.; Asjad, H.M.M.; Grimm, A.; Stockner, T.; Hummel, T.; Freissmuth, M.; Sucic, S. Functional Rescue of a Misfolded Drosophila Melanogaster Dopamine Transporter Mutant Associated with a Sleepless Phenotype by Pharmacological Chaperones. J. Biol. Chem. 2016, 291, 20876–20890. [Google Scholar] [CrossRef]
- Bartholomaus, I.; Milan-Lobo, L.; Nicke, A.; Dutertre, S.; Hastrup, H.; Jha, A.; Gether, U.; Sitte, H.H.; Betz, H.; Eulenburg, V. Glycine Transporter Dimers: Evidence for Occurrence in the Plasma Membrane. J. Biol. Chem. 2008, 283, 10978–10991. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Zhang, Y.-W. Cross-Linking and Functional Analyses for Dimerization of a Cysteine Mutant of Glycine Transporter 1. Int. J. Mol. Sci. 2022, 23, 16157. [Google Scholar] [CrossRef] [PubMed]
- Sitte, H.H.; Freissmuth, M. Oligomer Formation by Na+-Cl−-Coupled Neurotransmitter Transporters. Eur. J. Pharmacol. 2003, 479, 229–236. [Google Scholar] [CrossRef]
- de Juan-Sanz, J.; Zafra, F.; López-Corcuera, B.; Aragón, C. Endocytosis of the Neuronal Glycine Transporter GLYT2: Role of Membrane Rafts and Protein Kinase C-Dependent Ubiquitination. Traffic 2011, 12, 1850–1867. [Google Scholar] [CrossRef]
- de Juan-Sanz, J.; Nunez, E.; Lopez-Corcuera, B.; Aragon, C. Constitutive Endocytosis and Turnover of the Neuronal Glycine Transporter GlyT2 Is Dependent on Ubiquitination of a C-Terminal Lysine Cluster. PLoS ONE 2013, 8, e58863. [Google Scholar] [CrossRef]
- Gregersen, N.; Bross, P.; Jørgensen, M.M.; Corydon, T.J.; Andresen, B.S. Defective Folding and Rapid Degradation of Mutant Proteins Is a Common Disease Mechanism in Genetic Disorders. J. Inherit. Metab. Dis. 2000, 23, 441–447. [Google Scholar] [CrossRef]
- Hetz, C.; Mollereau, B. Disturbance of Endoplasmic Reticulum Proteostasis in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Z.; Zhao, Y.; Zhao, C.; Shi, M.; Dong, X.; Zhang, J.; Tan, L.; Zhang, L.; Zhao, Y. TRAPPC1 Is Essential for the Maintenance and Differentiation of Common Myeloid Progenitors in Mice. EMBO Rep. 2023, 24, e55503. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, M.; Castillo, C.; Francesconi, A. Quantitative Profiling of Brain Lipid Raft Proteome in a Mouse Model of Fragile X Syndrome. PLoS ONE 2015, 10, e0121464. [Google Scholar] [CrossRef]
- Chadwick, W.; Brenneman, R.; Martin, B.; Maudsley, S. Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease. Int. J. Alzheimers Dis. 2010, 2010, 604792. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, R.; Tian, D.; Krishna, S.; Pollak, M.R.; Greka, A.; Schlöndorff, J. SNF8, a Member of the ESCRT-II Complex, Interacts with TRPC6 and Enhances Its Channel Activity. BMC Cell Biol. 2012, 13, 33. [Google Scholar] [CrossRef]
- Belvitch, P.; Rizzo, A.N.; Dudek, S.M. Cortactin in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2278–2280. [Google Scholar] [CrossRef]
- Ledoux, B.; Zanin, N.; Yang, J.; Mercier, V.; Coster, C.; Dupont-Gillain, C.; Alsteens, D.; Morsomme, P.; Renard, H.-F. Plasma Membrane Nanodeformations Promote Actin Polymerization through CIP4/CDC42 Recruitment and Regulate Type II IFN Signaling. Sci. Adv. 2023, 9, eade1660. [Google Scholar] [CrossRef]
- Geerlings, A.; Nunez, E.; Lopez-Corcuera, B.; Aragon, C. Calcium- and Syntaxin 1-Mediated Trafficking of the Neuronal Glycine Transporter GLYT2. J. Biol. Chem. 2001, 276, 17584–17590. [Google Scholar] [CrossRef]
- Sommer, T.; Wolf, D.H. Endoplasmic Reticulum Degradation: Reverse Protein Flow of No Return. FASEB J. 1997, 11, 1227–1233. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, S.; Zhang, L.; Qin, X.; Zhu, Z.; Ma, T.; Lu, S.; Chen, J.; Shi, X.; Ren, H. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Is a Promising Signature to Predict Prognosis and Therapies for Hepatocellular Carcinoma (HCC). J. Clin. Med. 2023, 12, 1830. [Google Scholar] [CrossRef]
- Manganelli, V.; Longo, A.; Mattei, V.; Recalchi, S.; Riitano, G.; Caissutti, D.; Capozzi, A.; Sorice, M.; Misasi, R.; Garofalo, T. Role of ERLINs in the Control of Cell Fate through Lipid Rafts. Cells 2021, 10, 2408. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of Membrane/Lipid Rafts with the Cytoskeleton: Impact on Signaling and Function: Membrane/Lipid Rafts, Mediators of Cytoskeletal Arrangement and Cell Signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Gupta, N. Tether and Trap: Regulation of Membrane-Raft Dynamics by Actin-Binding Proteins. Nat. Rev. Immunol. 2007, 7, 889–896. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, W.; Farris, D.; Mishra, S. Merging Complexes: Properties of Membrane Raft Assembly during Lymphocyte Signaling. Trends Immunol. 2005, 26, 97–103. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum1: A Standalone Program for Generating PDBsum Analyses. Protein Sci. 2022, 31, e4473. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to Predict the Impact of Single Mutations on Protein Stability and Function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Jo, S. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Dickson, C.J.; Walker, R.C.; Gould, I.R. Lipid21: Complex Lipid Membrane Simulations with AMBER. J. Chem. Theory Comput. 2022, 18, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Perona, A.; Ros, M.P.; Mills, A.; Morreale, A.; Gago, F. Distinct Binding of Cetirizine Enantiomers to Human Serum Albumin and the Human Histamine Receptor H(1). J. Comput. Aided Mol. Des. 2020, 34, 1045–1062. [Google Scholar] [CrossRef]
- Liu, Q.R.; Lopez-Corcuera, B.; Mandiyan, S.; Nelson, H.; Nelson, N. Cloning and Expression of a Spinal Cord- and Brain-Specific Glycine Transporter with Novel Structural Features. J. Biol. Chem. 1993, 268, 22802–22808. [Google Scholar] [CrossRef] [PubMed]
- Carta, E.; Chung, S.K.; James, V.M.; Robinson, A.; Gill, J.L.; Remy, N.; Vanbellinghen, J.F.; Drew, C.J.; Cagdas, S.; Cameron, D.; et al. Mutations in the GlyT2 Gene (SLC6A5) Are a Second Major Cause of Startle Disease. J. Biol. Chem. 2012, 287, 28975–28985. [Google Scholar] [CrossRef]
- Benito-Muñoz, C.; Perona, A.; Felipe, R.; Pérez-Siles, G.; Núñez, E.; Aragón, C.; López-Corcuera, B. Structural Determinants of the Neuronal Glycine Transporter 2 for the Selective Inhibitors ALX1393 and ORG25543. ACS Chem. Neurosci. 2021, 12, 1860–1872. [Google Scholar] [CrossRef]
- Jiménez, E.; Fornés, A.; Felipe, R.; Núñez, E.; Aragón, C.; López-Corcuera, B. Calcium-Dependent Regulation of the Neuronal Glycine Transporter GlyT2 by M2 Muscarinic Acetylcholine Receptors. Neurochem. Res. 2022, 47, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Bolte, S.; Cordelieres, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Toledano-Zaragoza, A.; Enriquez-Zarralanga, V.; Naya-Forcano, S.; Briz, V.; Alfaro-Ruíz, R.; Parra-Martínez, M.; Mitroi, D.N.; Luján, R.; Esteban, J.A.; Ledesma, M.D. Enhanced mGluR(5) Intracellular Activity Causes Psychiatric Alterations in Niemann Pick Type C Disease. Cell Death Dis. 2024, 15, 771. [Google Scholar] [CrossRef]
- de la Rocha-Muñoz, A.; Núñez, E.; Vishwanath, A.A.; Gómez-López, S.; Dhanasobhon, D.; Rebola, N.; López-Corcuera, B.; de Juan-Sanz, J.; Aragón, C. The Presynaptic Glycine Transporter GlyT2 Is Regulated by the Hedgehog Pathway in Vitro and in Vivo. Commun. Biol. 2021, 4, 1197. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass. Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Orsburn, B.C. Proteome Discoverer-A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vázquez, J. Revisiting Peptide Identification by High-Accuracy Mass Spectrometry: Problems Associated with the Use of Narrow Mass Precursor Windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bandla, C.; Kundu, D.J.; Kamatchinathan, S.; Bai, J.; Hewapathirana, S.; John, N.S.; Prakash, A.; Walzer, M.; Wang, S.; et al. The PRIDE Database at 20 Years: 2025 Update. Nucleic Acids Res. 2025, 53, D543–D553. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Jorge, I.; Martinez-Val, A.; Barrero-Rodríguez, R.; Magni, R.; Núñez, E.; Laguillo, A.; Devesa, C.A.; López, J.A.; Camafeita, E.; et al. iSanXoT: A Standalone Application for the Integrative Analysis of Mass Spectrometry-Based Quantitative Proteomics Data. Comput. Struct. Biotechnol. J. 2024, 23, 452–459. [Google Scholar] [CrossRef]
- Navarro, P.; Trevisan-Herraz, M.; Bonzon-Kulichenko, E.; Núñez, E.; Martínez-Acedo, P.; Pérez-Hernández, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M.; et al. General Statistical Framework for Quantitative Proteomics by Stable Isotope Labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmiento-Jiménez, J.; Felipe, R.; Núñez, E.; Ferrando-Muñoz, A.; Benito-Muñoz, C.; Gago, F.; Vázquez, J.; Camafeita, E.; Clement, E.; Wilson, B.; et al. A New GlyT2 Variant Associated with Hyperekplexia. Int. J. Mol. Sci. 2025, 26, 6753. https://doi.org/10.3390/ijms26146753
Sarmiento-Jiménez J, Felipe R, Núñez E, Ferrando-Muñoz A, Benito-Muñoz C, Gago F, Vázquez J, Camafeita E, Clement E, Wilson B, et al. A New GlyT2 Variant Associated with Hyperekplexia. International Journal of Molecular Sciences. 2025; 26(14):6753. https://doi.org/10.3390/ijms26146753
Chicago/Turabian StyleSarmiento-Jiménez, Jorge, Raquel Felipe, Enrique Núñez, Alejandro Ferrando-Muñoz, Cristina Benito-Muñoz, Federico Gago, Jesús Vázquez, Emilio Camafeita, Emma Clement, Brian Wilson, and et al. 2025. "A New GlyT2 Variant Associated with Hyperekplexia" International Journal of Molecular Sciences 26, no. 14: 6753. https://doi.org/10.3390/ijms26146753
APA StyleSarmiento-Jiménez, J., Felipe, R., Núñez, E., Ferrando-Muñoz, A., Benito-Muñoz, C., Gago, F., Vázquez, J., Camafeita, E., Clement, E., Wilson, B., & López-Corcuera, B. (2025). A New GlyT2 Variant Associated with Hyperekplexia. International Journal of Molecular Sciences, 26(14), 6753. https://doi.org/10.3390/ijms26146753