
LZTR1: c.1260+1del Variant as a Significant Predictor of Early-Age Breast Cancer Development: Case Report Combined with In Silico Analysis

 , ,
, ,
Abstract
1. Introduction
1.1. Genetics of Breast Cancer
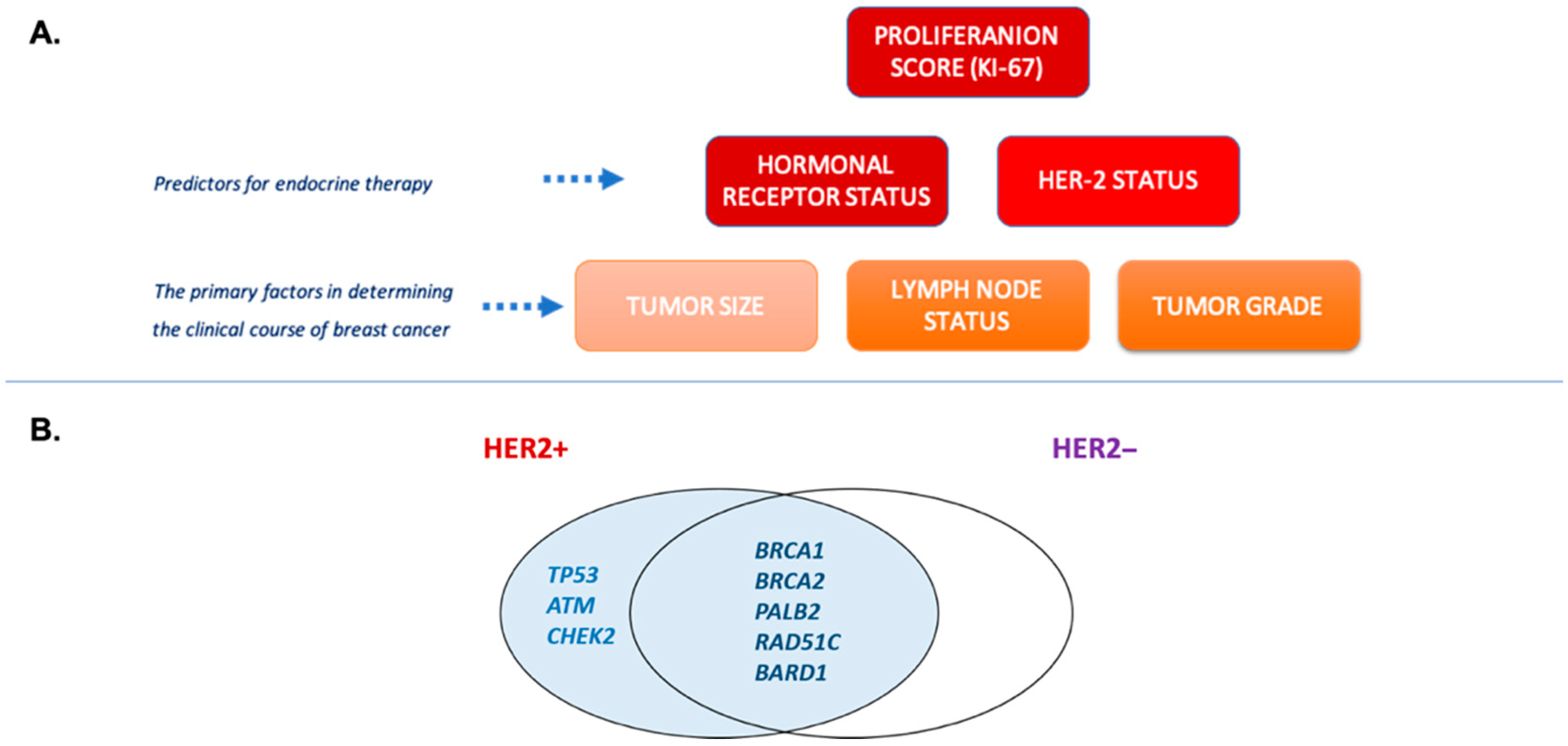
1.2. Biological Role of LZTR1 Protein
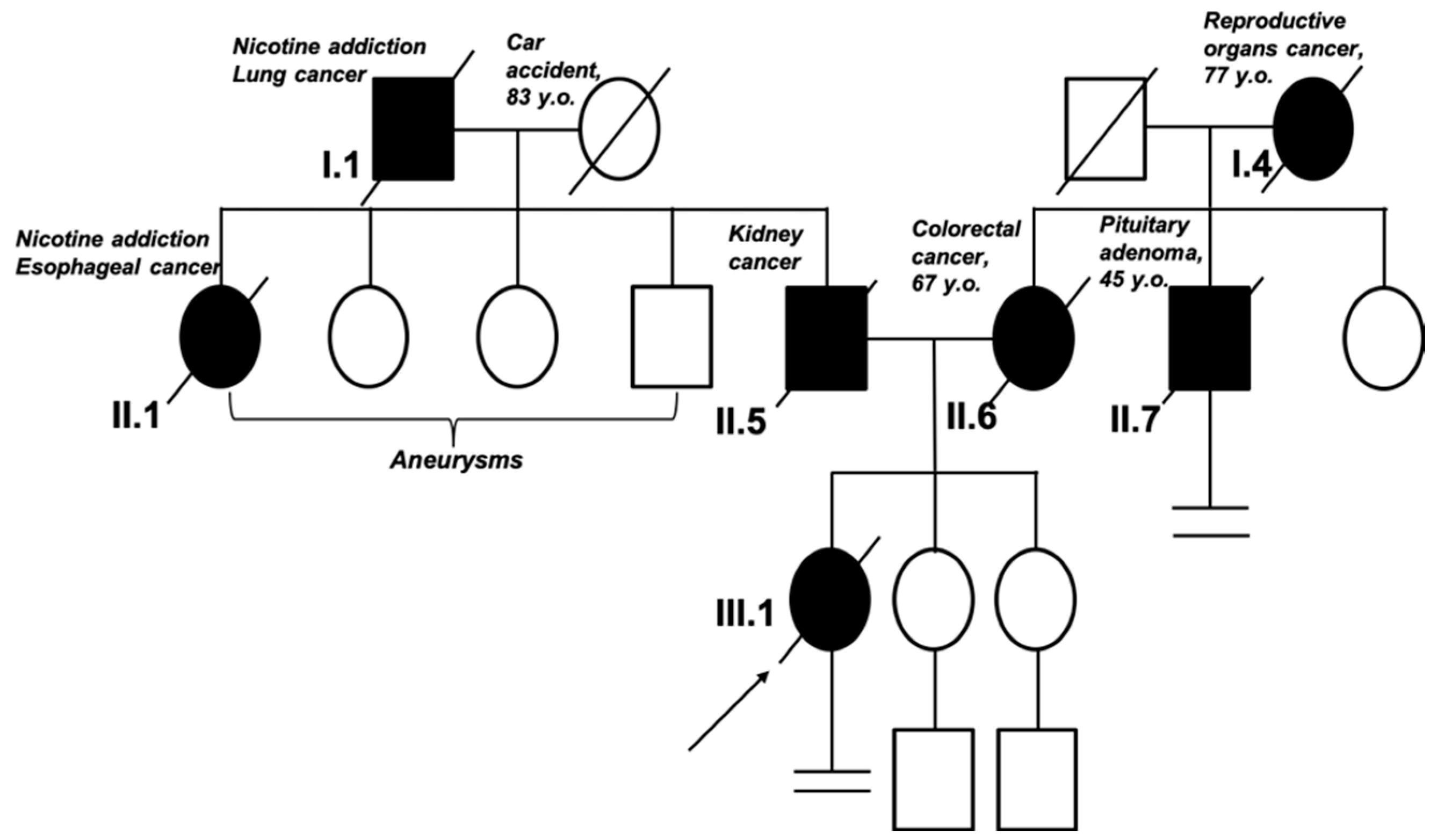
1.3. Clinical Case Description: Clinical Examination
2. Results
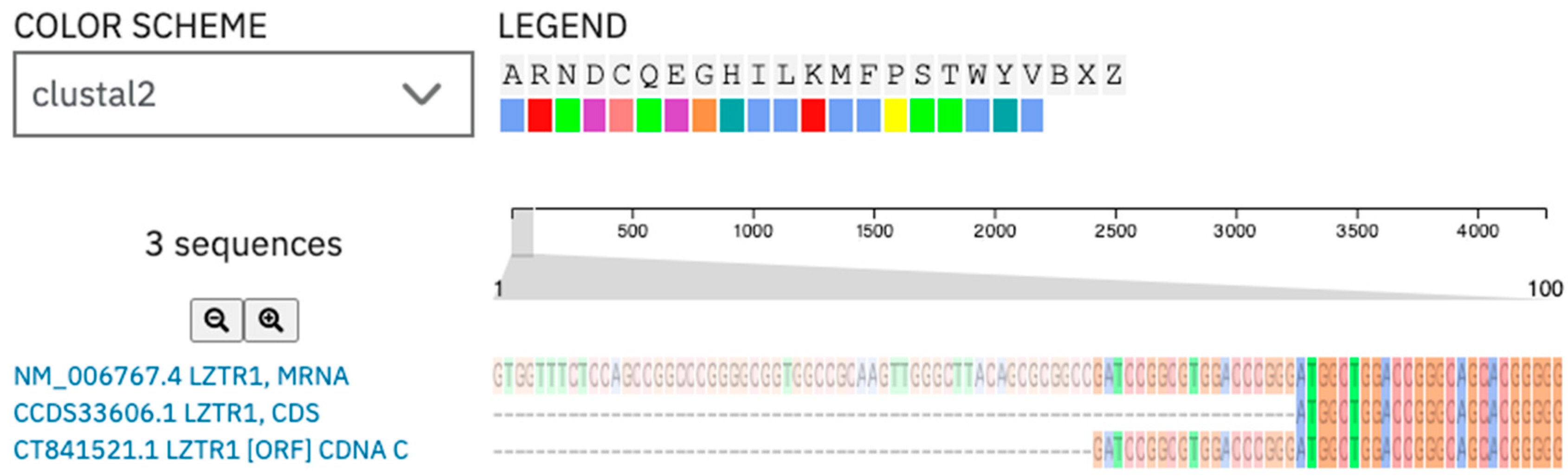
2.1. Molecular Diagnostic
2.2. Bioinformatical Analysis of the Gene Variant
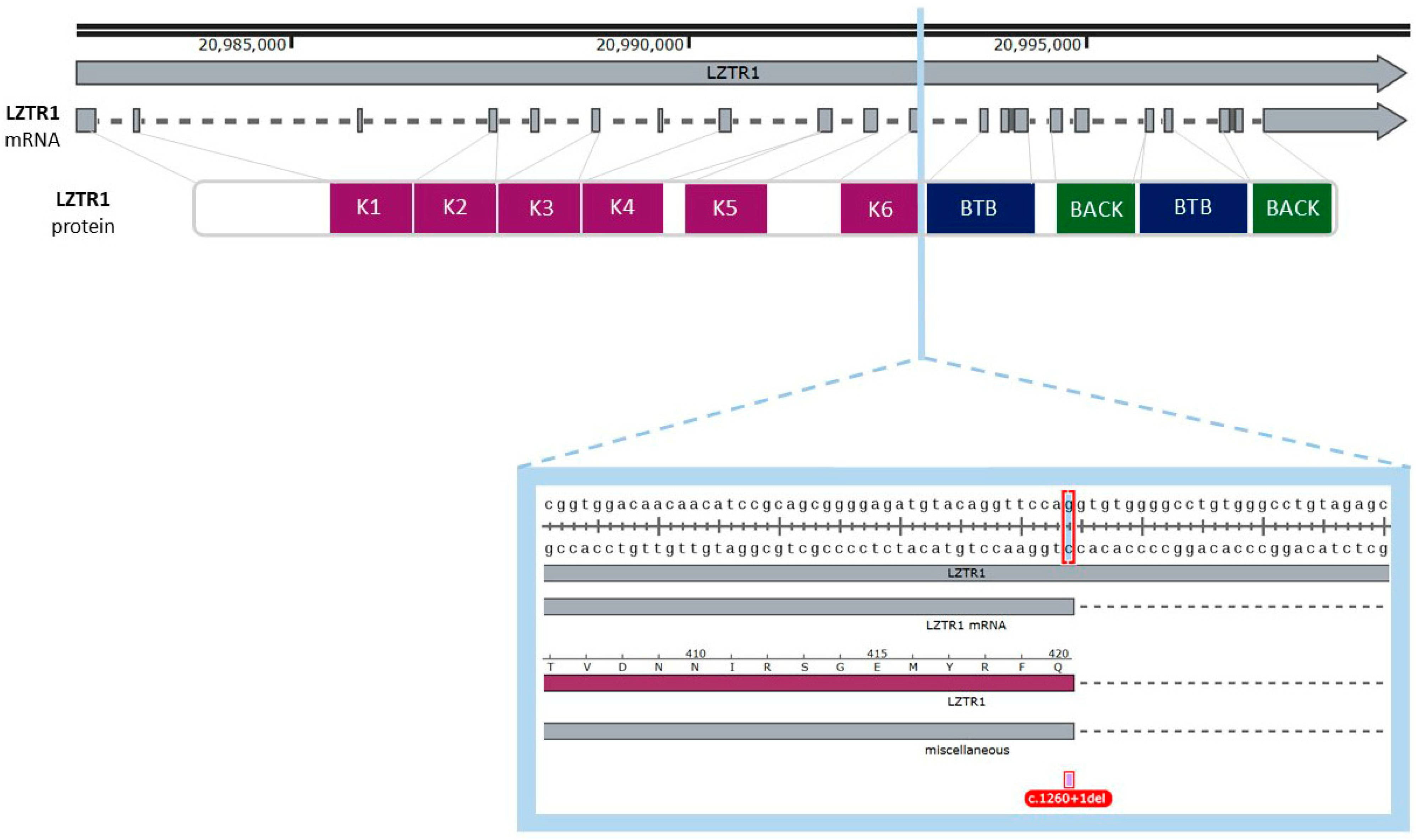
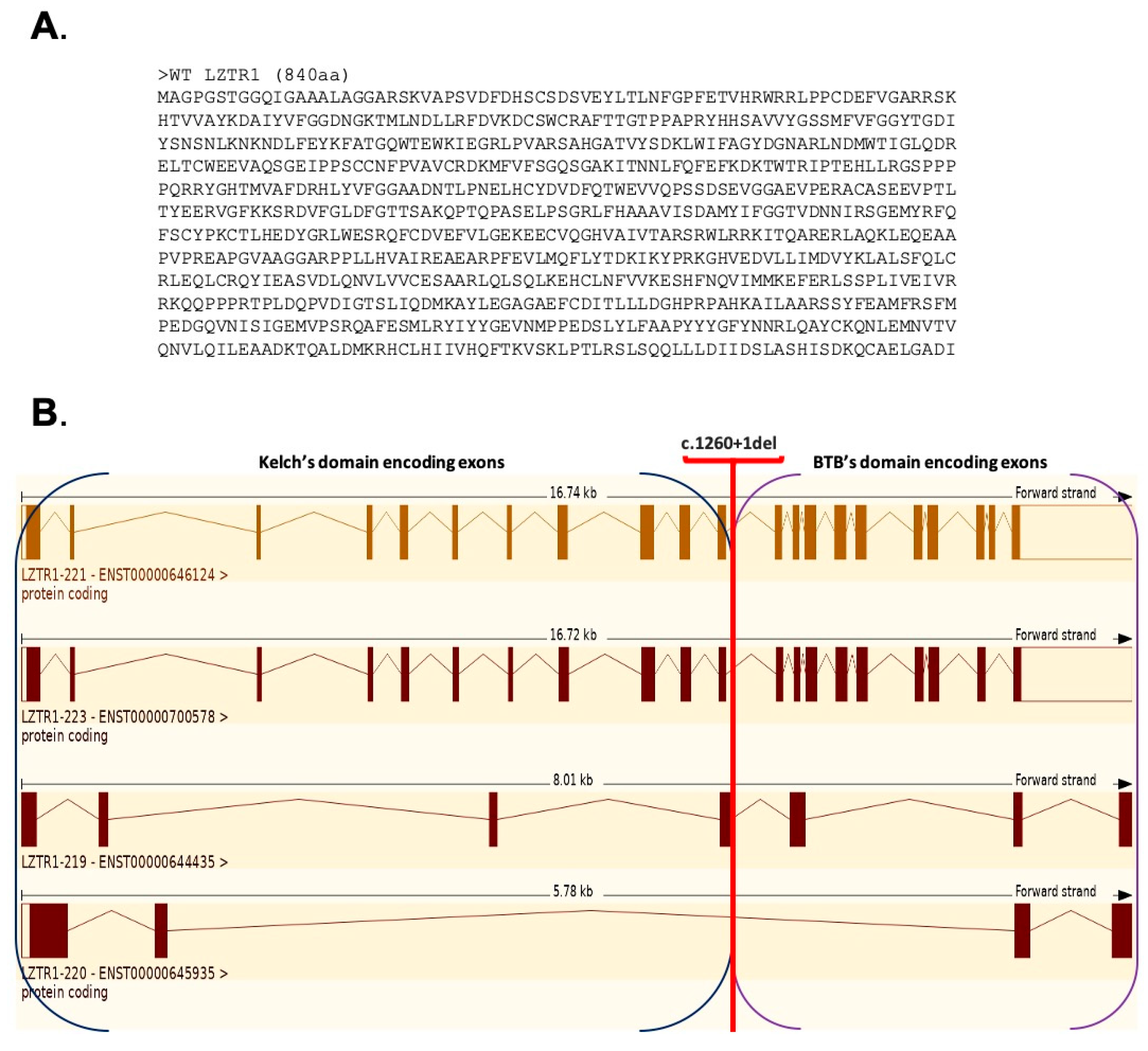
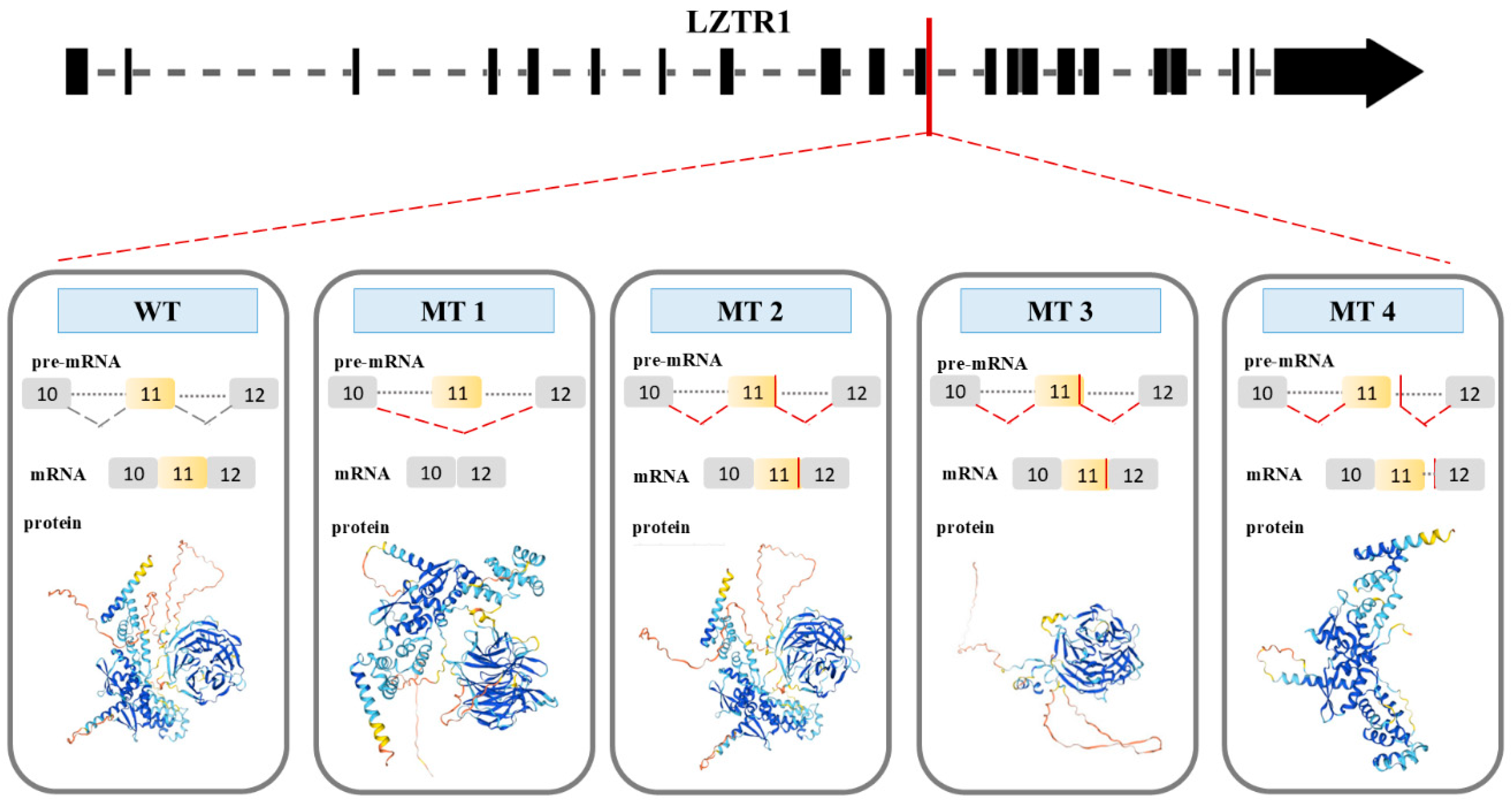
2.2.1. Analysis of the Gene and Transcript Structures Combined with LZTR1: c.1260+1del Pathogenicity Assessment
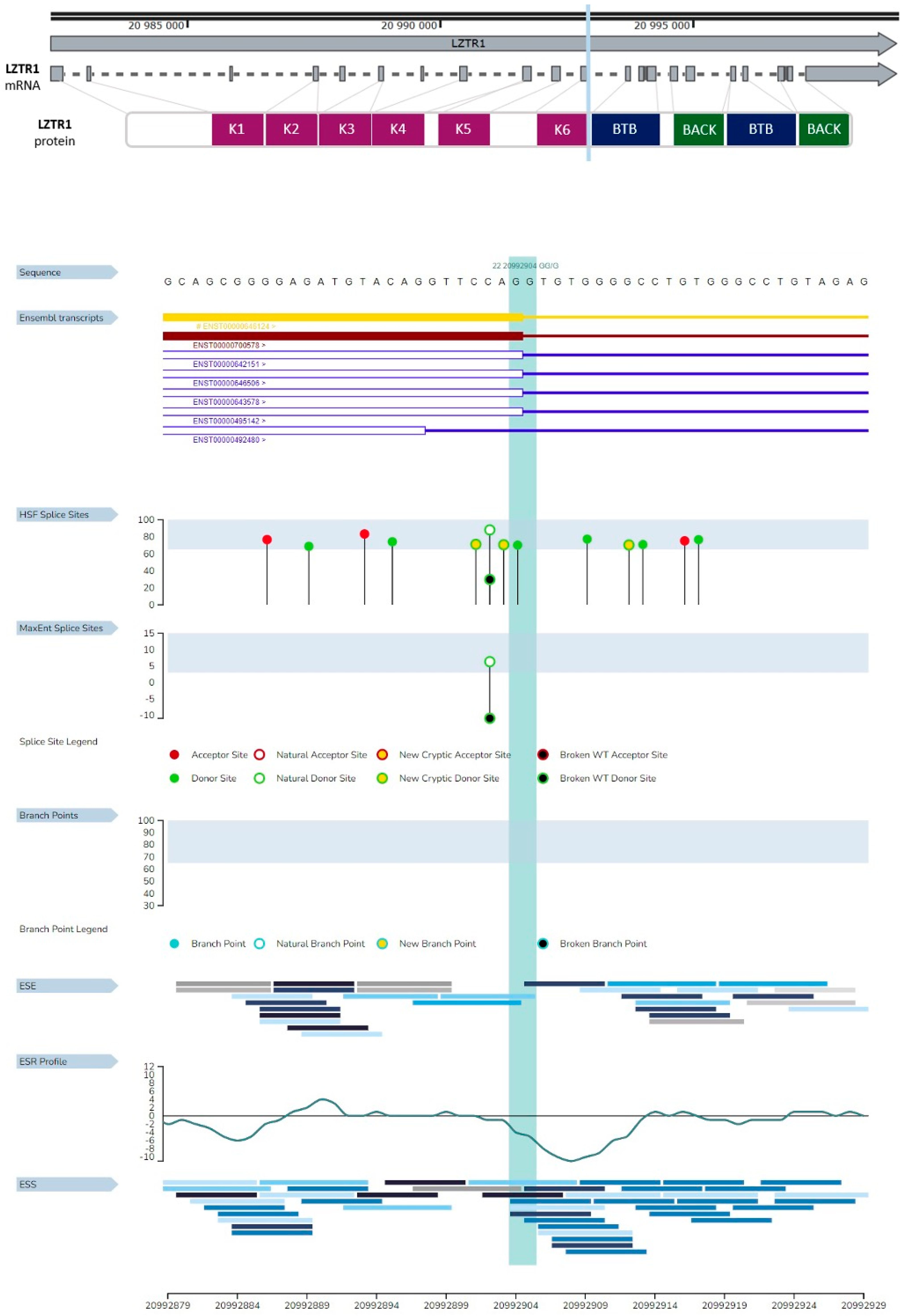
2.2.2. Analysis of Genetic Variant Influence on Splicing
2.2.3. Gene Variant Transcript Prediction, Protein Structure Design, and Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | MT1 | MT2 | MT3 | MT4 | |
|---|---|---|---|---|---|
| Family | Leucine-zipper-like transcriptional regulator | Leucine-zipper-like transcriptional regulator | Leucine-zipper-like transcriptional regulator | Leucine-zipper-like transcriptional regulator | Leucine-zipper-like transcriptional regulator |
| Domains | 4 Kelch motifs 6 Kelch type beta propellers 4 kelc_smart BTB1_POZ_LZTR1 BACK1_LZTR1 BTB2_POZ_LZTR1 BACK2_LZTR1 | 2 Kelch motifs 4 Kelch type beta propellers 4 kelc_smart BTB1_POZ_LZTR1 BACK1_LZTR1 BTB2_POZ_LZTR1 BACK2_LZTR1 | 3 Kelch motifs 6 Kelch type beta propellers 4 kelc_smart BTB1_POZ_LZTR1 BACK1_LZTR1 BTB2_POZ_LZTR1 BACK2_LZTR1 | 3 Kelch motifs 6 Kelch type beta propellers 4 kelc_smart | BTB1_POZ_LZTR1 BACK1_LZTR1 BTB2_POZ_LZTR1 BACK2_LZTR1 |
| Link for the analysis results | [35] | [36] | [37] | [38] | [39] |
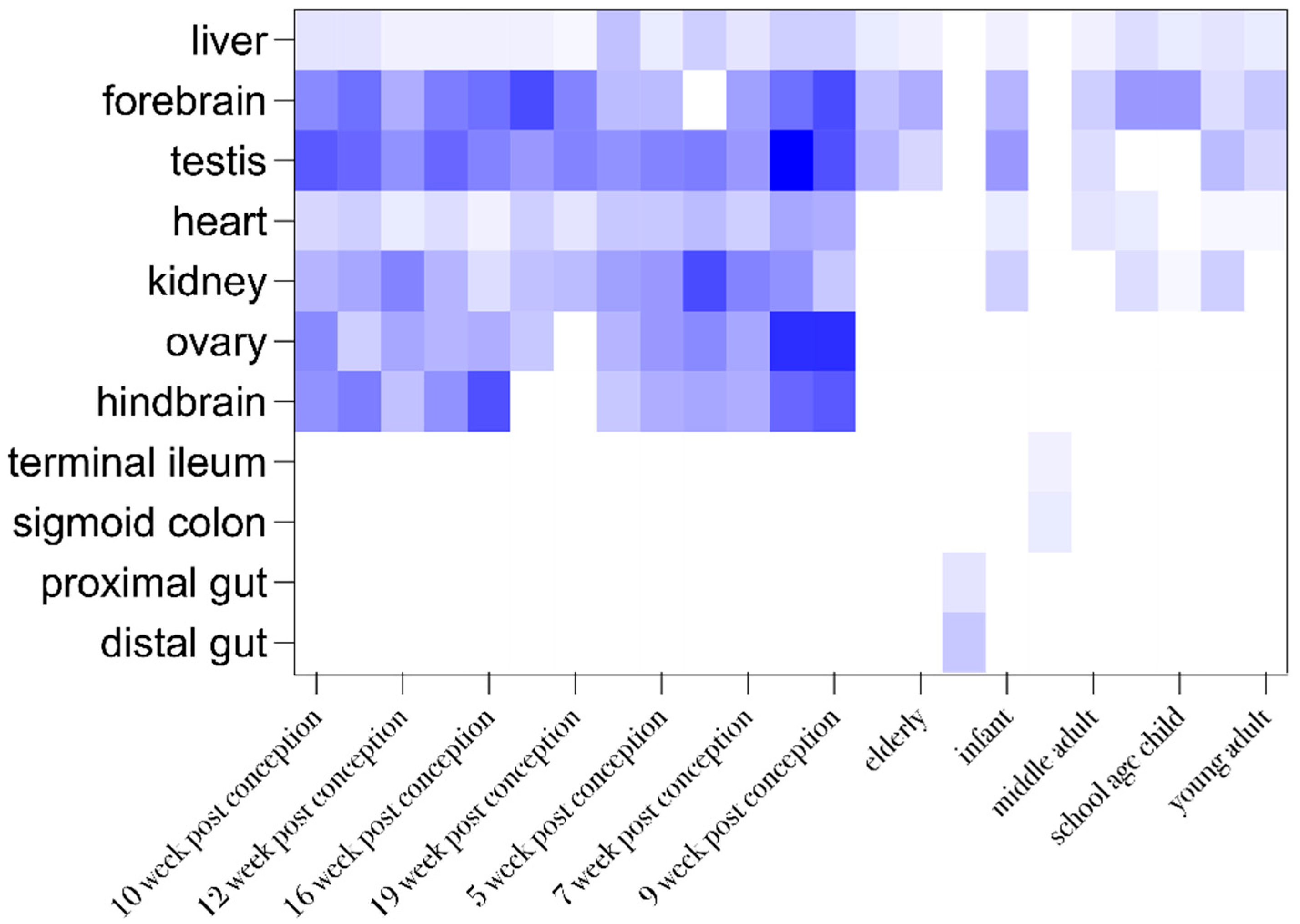
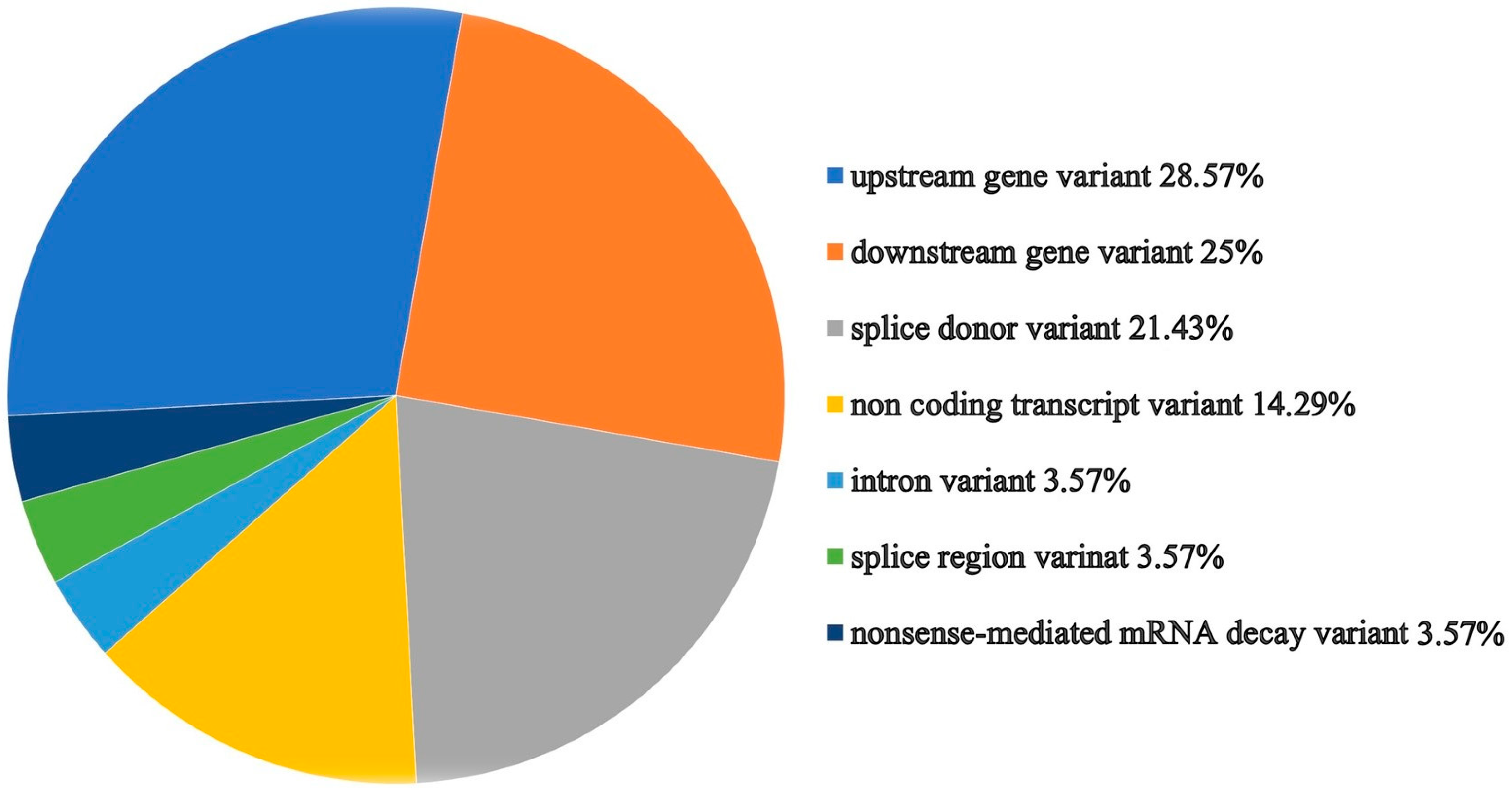
2.3. Analysis of Gene-Related Phenotypes
2.4. Analysis of Protein–Protein Interactions
2.5. Protein Homology Analysis
2.6. Model Organism’s Phenotype Analysis
3. Discussion
4. Materials and Methods
4.1. Molecular Diagnostics
4.2. Bioinformatical Analysis Was Performed with the Use of Available Datasets and Bioinformatical Tools
Analysis of Gene and Transcripts Structures Combined with LZTR1: c.1260+1del Pathogenicity Assessment
4.3. Analysis of Genetic Variant Influence on Splicing
4.4. Gene Variant Transcript Prediction, Protein Structure Design, and Evaluation
4.5. Analysis of Gene-Related Phenotypes
4.6. Analysis of Protein–Protein Interactions
4.7. Protein Homology Analysis
4.8. Model Organism’s Phenotype Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASCO | American Society of Clinical Oncology |
| ESMO | European Society of Medical Oncology |
| BRCA1/2 | breast cancer gene |
| LZTR1 | leucine zipper-like transcriptional regulator 1 |
| NCCN | National Comprehensive Cancer Network |
| HER-2 | human epidermal growth factor receptor 2 |
| ER | estrogen receptor |
| PR | progesterone receptor |
| TP53 | tumor protein 53 gene |
| PTEN | tensin homology gene |
| CDH1 | cadherin1 gene |
| STK11 | serine/threonine kinase 11 gene |
| ATM | ataxia-telangiectasia mutated gene |
| BARD1 | BRCA1-associated RING domain 1gene |
| CHECK2 | checkpoint kinase 2 |
| PALB2 | partner and localizer of BRCA2 gene |
| RAD51 gene | adiation sensitive protein 51 |
| Ki-67 | protein marker of cell proliferation |
| NOS | no otherwise specified according to the WHO 2018 classification |
| CDK4/6 | cyclin-dependent kinase 4 and 6. |
| AIP | aryl hydrocarbon receptor-interacting gene. |
| ALK | anaplastic lymphoma kinase gene |
| ATM | ataxia telangiectasia mutated |
| AXIN2 | axis inhibition protein 2 |
| BAP1 | BRCA1-associated protein 1 |
| BARD1 | BRCA1-associated RING domain 1 |
| BLM | Bloom syndrome RecQ-like helicase |
| BMPR1A | bone morphogenetic protein receptor type 1A |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| BRIP1 | BRCA1 interacting protein C-terminal helicase 1 |
| CDC73 | cell division cycle 73 |
| CDH1 | cadherin 1 |
| CDK4 | cyclin-dependent kinase 4 |
| CDKN1B | cyclin-dependent kinase inhibitor 1B |
| CDKN2A | cyclin-dependent kinase inhibitor 2A |
| CEBPA | CCAAT enhancer binding protein alpha |
| CHEK2 | checkpoint kinase 2 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DDB2 | damage specific DNA binding protein 2 |
| DICER1 | dicer 1, ribonuclease III |
| EPCAM | epithelial cell adhesion molecule |
| FANCC | Fanconi anemia complementation group C |
| FH | fumarate hydratase |
| FLCN | folliculin |
| GALNT12 | polypeptide N-acetylgalactosaminyltransferase 12 |
| GREM1 | gremlin 1, DAN family BMP antagonist |
| HNF1A | hepatocyte nuclear factor 1 alpha |
| HOXB13 | homeobox B13 |
| HRAS | Harvey rat sarcoma viral oncogene homolog |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LZTR1 | leucine zipper-like transcription regulator 1 |
| MAX | MYC-associated factor X |
| MEN1 | multiple endocrine neoplasia type 1 |
| MET | MET proto-oncogene, receptor tyrosine kinase |
| MITF | Microphthalmia-associated transcription factor |
| MLH1 | MutL homolog 1 |
| MLH3 | MutL homolog 3 |
| MRE11 | MRE11 homolog, double-strand break repair nuclease |
| MSH2 | MutS homolog 2 |
| MSH3 | MutS homolog 3 |
| MSH6 | MutS homolog 6 |
| MUTYH | MutY DNA glycosylase |
| NBN | nibrin |
| NF1 | neurofibromin 1 |
| NF2 | neurofibromin 2 |
| NOD2 | nucleotide-binding oligomerization domain containing 2 |
| NTHL1 | endonuclease III-like 1 |
| PALB2 | partner and localizer of BRCA2 |
| PMS1 | PMS1 homolog 1, mismatch repair system component |
| PMS2 | PMS2 homolog 2, mismatch repair system component |
| POLD1 | DNA polymerase delta 1 |
| POLE | DNA polymerase epsilon |
| POT1 | protection of telomeres 1 |
| PRF1 | perforin 1 |
| PRKAR1A | protein kinase CAMP-dependent regulatory subunit alpha |
| PRSS1 | protease, serine 1 |
| PTCH1 | patched 1 |
| PTEN | phosphatase and tensin homolog |
| RAD50 | RAD50 double-strand break repair protein |
| RAD51C | RAD51 paralog C |
| RAD51D | RAD51 paralog D |
| RB1 | retinoblastoma 1 |
| RET | RET proto-oncogene |
| SDHA | succinate dehydrogenase complex flavoprotein subunit A |
| SDHAF2 | succinate dehydrogenase complex assembly factor 2 |
| SDHB | succinate dehydrogenase complex iron sulfur subunit B |
| SDHC | succinate dehydrogenase complex subunit C |
| SDHD | succinate dehydrogenase complex subunit D |
| SMAD4 | SMAD family member 4 |
| SMARCB1 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 |
| STK11 | serine/threonine kinase 11 |
| TMEM127 | transmembrane protein 127 |
| TP53 | tumor protein P53 |
| TSC1 | tuberous sclerosis complex 1 |
| TSC2 | tuberous sclerosis complex 2 |
| VHL | Von Hippel–Lindau tumor suppressor |
| WT1 | Wilms tumor 1 |
| XRCC2 | X-ray repair cross complementing 2 |
| NGS | next-generation sequencing |
| dsSNP | Database of Single Nucleotide Polymorphisms |
| GRCh38 | Genome Reference Consortium Human Build 38 |
| GnomAD | Genome Aggregation Database |
| BTB | broad-complex, tramtrack, and bric-a-brac |
| BACK | BTB and C-terminal Kelch |
| Kelch | Kelch repeat domain |
| E3 | E3 ubiquitin ligase |
| CUL3 | cullin 3 |
| ORF | open reading frame |
| WT | wild type |
| MT1/2/3/4 | mutation variant1/2/3/4 |
| ccds | consensus coding sequence |
| NCBI | National Center for Biotechnology Information |
| MIT | Massachusetts Institute of Technology |
| RAT | Rattus norvegicus |
| MOUSE | Mus musculus |
| DANRE | Danio rerio |
| CHIC | Gallus gallus |
| XENTR | Xenopus tropicalis |
| ZFIN database | Zebrafish Model Organism Database |
| RAS | rat sarcoma viral oncogene homolog |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase (mitogen-activated protein kinase/extracellular signal-regulated kinase) |
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological Characteristics of and Risk Factors for Breast Cancer in the World. Breast Cancer Dove Med. Press 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.H.; Evans, D.G.; Agarwal, G.; Buccimazza, I.; Kwong, A.; Morant, R.; Prakash, I.; Song, C.Y.; Taib, N.A.; Tausch, C.; et al. Global Disparities in Breast Cancer Genetics Testing, Counselling and Management. World J. Surg. 2019, 43, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Premature Mortality Trends in 183 Countries by Cancer Type, Sex, WHO Region, and World Bank Income Level in 2000–2019: A Retrospective, Cross-Sectional, Population-Based Study—The Lancet Oncology. Available online: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(24)00274-2/fulltext (accessed on 23 March 2025).
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Abramson, V.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Bailey, J.; Burstein, H.J.; et al. Breast Cancer, Version 3.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2024, 22, 331–357. [Google Scholar] [CrossRef]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer—Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef]
- Huber-Keener, K.J. Cancer Genetics and Breast Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2022, 82, 3–11. [Google Scholar] [CrossRef]
- Mavaddat, N.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Keeman, R.; Bolla, M.K.; Dennis, J.; Wang, Q.; Ahearn, T.U.; et al. Pathology of Tumors Associated With Pathogenic Germline Variants in 9 Breast Cancer Susceptibility Genes. JAMA Oncol. 2022, 8, e216744. [Google Scholar] [CrossRef] [PubMed]
- Impact of High-to-Moderate Penetrance Genes on Genetic Testing: Looking over Breast Cancer. Available online: https://www.mdpi.com/2073-4425/14/8/1530 (accessed on 25 April 2025).
- Cybulski, C.; Kluźniak, W.; Huzarski, T.; Wokołorczyk, D.; Kashyap, A.; Rusak, B.; Stempa, K.; Gronwald, J.; Szymiczek, A.; Bagherzadeh, M.; et al. The Spectrum of Mutations Predisposing to Familial Breast Cancer in Poland. Int. J. Cancer 2019, 145, 3311–3320. [Google Scholar] [CrossRef]
- Reid, S.; Spalluto, L.B.; Lang, K.; Weidner, A.; Pal, T. An Overview of Genetic Services Delivery for Hereditary Breast Cancer. Breast Cancer Res. Treat. 2022, 191, 491–500. [Google Scholar] [CrossRef]
- Sessa, C.; Balmaña, J.; Bober, S.L.; Cardoso, M.J.; Colombo, N.; Curigliano, G.; Domchek, S.M.; Evans, D.G.; Fischerova, D.; Harbeck, N.; et al. Risk Reduction and Screening of Cancer in Hereditary Breast-Ovarian Cancer Syndromes: ESMO Clinical Practice Guideline. Ann. Oncol. 2023, 34, 33–47. [Google Scholar] [CrossRef]
- Rowlands, C.F.; Allen, S.; Balmaña, J.; Domchek, S.M.; Evans, D.G.; Hanson, H.; Hoogerbrugge, N.; James, P.A.; Nathanson, K.L.; Robson, M.; et al. Population-Based Germline Breast Cancer Gene Association Studies and Meta-Analysis to Inform Wider Mainstream Testing. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2024, 35, 892–901. [Google Scholar] [CrossRef]
- Gene2Phenotype. Available online: https://www.ebi.ac.uk/gene2phenotype/lgd/G2P03479 (accessed on 30 June 2025).
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical Features of Schwannomatosis: A Retrospective Analysis of 87 Patients. Oncologist 2012, 17, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Blondeaux, E.; Arecco, L.; Punie, K.; Graffeo, R.; Toss, A.; De Angelis, C.; Trevisan, L.; Buzzatti, G.; Linn, S.C.; Dubsky, P.; et al. Germline TP53 Pathogenic Variants and Breast Cancer: A Narrative Review. Cancer Treat. Rev. 2023, 114, 102522. [Google Scholar] [CrossRef] [PubMed]
- cBioPortal for Cancer Genomics. Available online: http://www.cbioportal.org/study/summary?id=brca_tcga (accessed on 30 June 2025).
- Nacak, T.G.; Leptien, K.; Fellner, D.; Augustin, H.G.; Kroll, J. The BTB-Kelch Protein LZTR-1 Is a Novel Golgi Protein That Is Degraded upon Induction of Apoptosis. J. Biol. Chem. 2006, 281, 5065–5071. [Google Scholar] [CrossRef]
- Farncombe, K.M.; Thain, E.; Barnett-Tapia, C.; Sadeghian, H.; Kim, R.H. LZTR1 Molecular Genetic Overlap with Clinical Implications for Noonan Syndrome and Schwannomatosis. BMC Med. Genom. 2022, 15, 160. [Google Scholar] [CrossRef]
- Piech, S.; Brüschweiler, S.; Westphalen, J.; Siess, K.M.; García Murias, J.; Konrat, R.; Bigenzahn, J.W.; Superti-Furga, G. Identification and Characterization of Novel Small-Molecule Enhancers of the CUL3LZTR1 E3 Ligase KRAS Complex. ACS Chem. Biol. 2024, 19, 1942–1952. [Google Scholar] [CrossRef]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 Drive Human Disease by Dysregulating RAS Ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Downs, G.S.; Jauhal, J.J.; Nandra, S.K.; Privé, G.G. Sequence and Structural Analysis of BTB Domain Proteins. Genome Biol. 2005, 6, R82. [Google Scholar] [CrossRef]
- Shi, X.; Xiang, S.; Cao, J.; Zhu, H.; Yang, B.; He, Q.; Ying, M. Kelch-like Proteins: Physiological Functions and Relationships with Diseases. Pharmacol. Res. 2019, 148, 104404. [Google Scholar] [CrossRef]
- Adams, J.; Kelso, R.; Cooley, L. The Kelch Repeat Superfamily of Proteins: Propellers of Cell Function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar] [CrossRef]
- Pilcher, C.; Buco, P.A.V.; Truong, J.Q.; Ramsland, P.A.; Smeets, M.F.; Walkley, C.R.; Holien, J.K. Characteristics of the Kelch Domain Containing (KLHDC) Subfamily and Relationships with Diseases. FEBS Lett. 2025, 599, 1094–1112. [Google Scholar] [CrossRef]
- Search Results < Expression Atlas < EMBL-EBI. Available online: https://www.ebi.ac.uk/gxa/genes/lztr1?bs=%7B%22homo%20sapiens%22%3A%5B%22CELL_TYPE%22%5D%7D#baseline (accessed on 13 June 2025).
- Franklin. Available online: https://franklin.genoox.com/clinical-db/home (accessed on 16 April 2025).
- Homo Sapiens Chromosome 22, GRCh38.P14 Primary Assembly 2024. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.40/ (accessed on 13 June 2025).
- Hansen, T.J.; Hodges, E. ATAC-STARR-Seq Reveals Transcription Factor-Bound Activators and Silencers within Chromatin-Accessible Regions of the Human Genome. Genome Res. 2022, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The Consensus Coding Sequence (CCDS) Project: Identifying a Common Protein-Coding Gene Set for the Human and Mouse Genomes. Genome Res. 2009, 19, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Furuno, M.; Kasukawa, T.; Saito, R.; Adachi, J.; Suzuki, H.; Baldarelli, R.; Hayashizaki, Y.; Okazaki, Y. CDS Annotation in Full-Length cDNA Sequence. Genome Res. 2003, 13, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Variant Effect Predictor—Homo_Sapiens—Ensembl Genome Browser 113. Available online: https://www.ensembl.org/Tools/VEP (accessed on 16 April 2025).
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/uniprotkb/Q8N653/entry (accessed on 8 May 2025).
- Phillips, C.L.; So, C.; Gillis, M.F.; Harrison, J.; Hsu, C.-H.; Armao, D.; Snider, N.T. The Kelch 3 Motif on Gigaxonin Mediates the Interaction with NUDCD3 and Regulates Vimentin Filament Morphology. bioRxiv 2025. [Google Scholar] [CrossRef]
- InterProScan Search Result (Undefined)—Protein—InterPro. Available online: https://www.ebi.ac.uk/interpro/result/InterProScan/iprscan5-R20250430-122859-0354-19954911-p1m/internal-1746012531319-86-1/ (accessed on 8 May 2025).
- InterProScan Search Result (Undefined)—Protein—InterPro. Available online: https://www.ebi.ac.uk/interpro/result/InterProScan/iprscan5-R20250417-072109-0229-54863226-p1m/internal-1744870863913-44-1/ (accessed on 8 May 2025).
- InterProScan Search Result (Undefined)—Protein—InterPro. Available online: https://www.ebi.ac.uk/interpro/result/InterProScan/iprscan5-R20250417-082513-0970-83110158-p1m/internal-1744874704669-57-1/ (accessed on 8 May 2025).
- InterProScan Search Result (Undefined)—Protein—InterPro. Available online: https://www.ebi.ac.uk/interpro/result/InterProScan/iprscan5-R20250417-082547-0366-54327965-p1m/internal-1744874739853-59-1/ (accessed on 8 May 2025).
- InterProScan Search Result (Undefined)—Protein—InterPro. Available online: https://www.ebi.ac.uk/interpro/result/InterProScan/iprscan5-R20250417-082623-0539-35128666-p1m/internal-1744874770164-61-1/ (accessed on 8 May 2025).
- ZFIN The Zebrafish Information Network. Available online: https://zfin.org/ (accessed on 17 April 2025).
- [Alliance Genome Source]. Available online: https://www.alliancegenome.org/ (accessed on 17 April 2025).
- Cuevas-Navarro, A.; Van, R.; Cheng, A.; Urisman, A.; Castel, P.; McCormick, F. The RAS GTPase RIT1 Compromises Mitotic Fidelity through Spindle Assembly Checkpoint Suppression. Curr. Biol. CB 2021, 31, 3915–3924.e9. [Google Scholar] [CrossRef]
- Abe, T.; Morisaki, K.; Niihori, T.; Terao, M.; Takada, S.; Aoki, Y. Dysregulation of RAS Proteostasis by Autosomal-Dominant LZTR1 Mutation Induces Noonan Syndrome-like Phenotypes in Mice. JCI Insight 2024, 9, e182382. [Google Scholar] [CrossRef]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 Is a Regulator of RAS Ubiquitination and Signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef]
- Sewduth, R.N.; Ivanisevic, T.; Zhao, P.; Sablina, A.A. Novel Therapeutic Approaches for KRAS-Mutated Lung Cancer Involving LZTR1 Genetic Alteration. Med. Sci. Forum 2023, 20, 6. [Google Scholar] [CrossRef]
- Garutti, M.; Foffano, L.; Mazzeo, R.; Michelotti, A.; Da Ros, L.; Viel, A.; Miolo, G.; Zambelli, A.; Puglisi, F. Hereditary Cancer Syndromes: A Comprehensive Review with a Visual Tool. Genes 2023, 14, 1025. [Google Scholar] [CrossRef]
- Barbero, A.I.S.; Valenzuela, I.; Fernández-Alvarez, P.; Vazquez, É.; Cueto-Gonzalez, A.M.; Lasa-Aranzasti, A.; Trujillano, L.; Masotto, B.; Arumí, E.G.; Tizzano, E.F. New Insights Into the Spectrum of RASopathies: Clinical and Genetic Data in a Cohort of 121 Spanish Patients. Am. J. Med. Genet. A. 2025, 197, e63905. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Hasanain, M.; Oh, Y.T.; D’Angelo, F.; Sommer, D.; Frangaj, B.; Tran, S.; Bielle, F.; Pollo, B.; Paterra, R.; et al. LZTR1 Mutation Mediates Oncogenesis through Stabilization of EGFR and AXL. Cancer Discov. 2023, 13, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Fricke, J.; Mambetsariev, I.; Velasquez, G.; Nadaf-Rahrov, R.; Dingal, S.T.; Kim, P.; Babikian, R.; Amini, A.; Afkhami, M.; et al. Novel LZTR1 Germline Mutation as a Mechanism of Resistance to Osimertinib in EGFR-Mutated Lung Adenocarcinoma: A Case Report. Transl. Lung Cancer Res. 2025, 14, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kanno, S.-I.; Niihori, T.; Terao, M.; Takada, S.; Aoki, Y. LZTR1 Deficiency Exerts High Metastatic Potential by Enhancing Sensitivity to EMT Induction and Controlling KLHL12-Mediated Collagen Secretion. Cell Death Dis. 2023, 14, 556. [Google Scholar] [CrossRef]
- Pae, J.; Cinalli, R.M.; Marzio, A.; Pagano, M.; Lehmann, R. GCL and CUL3 Control the Switch between Cell Lineages by Mediating Localized Degradation of an RTK. Dev. Cell 2017, 42, 130–142.e7. [Google Scholar] [CrossRef]
- Zipper, L.; Wagener, R.; Fischer, U.; Hoffmann, A.; Yasin, L.; Brandes, D.; Soura, S.; Anwar, A.; Walter, C.; Varghese, J.; et al. Hyperdiploid Acute Lymphoblastic Leukemia in Children with LZTR1 Germline Variants. HemaSphere 2024, 8, e26. [Google Scholar] [CrossRef]
- Chaves Rabelo, N.; Gomes, M.E.; de Oliveira Moraes, I.; Cantagalli Pfisterer, J.; Loss de Morais, G.; Antunes, D.; Caffarena, E.R.; Llerena, J., Jr.; Gonzalez, S. RASopathy Cohort of Patients Enrolled in a Brazilian Reference Center for Rare Diseases: A Novel Familial LZTR1 Variant and Recurrent Mutations. Appl. Clin. Genet. 2022, 15, 153–170. [Google Scholar] [CrossRef]
- Aoki, Y.; Niihori, T.; Inoue, S.; Matsubara, Y. Recent Advances in RASopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef]
- Wilcox, N.; Dumont, M.; González-Neira, A.; Carvalho, S.; Joly Beauparlant, C.; Crotti, M.; Luccarini, C.; Soucy, P.; Dubois, S.; Nuñez-Torres, R.; et al. Exome Sequencing Identifies Breast Cancer Susceptibility Genes and Defines the Contribution of Coding Variants to Breast Cancer Risk. Nat. Genet. 2023, 55, 1435–1439. [Google Scholar] [CrossRef]
- New GENSCAN Web Server at MIT. Available online: http://hollywood.mit.edu/GENSCAN.html (accessed on 2 April 2025).
- ORFfinder Home—NCBI. Available online: https://www.ncbi.nlm.nih.gov/orffinder/ (accessed on 2 April 2025).
- Genomnis_HSF_Pro. Available online: https://hsf.genomnis.com/welcome (accessed on 2 April 2025).
- MutationTaster. Available online: https://www.mutationtaster.org/ (accessed on 2 April 2025).
- Results from MutationTester for c.1260+1del in LZTR1. Available online: https://www.mutationtaster.org/MT69/MutationTaster69.cgi?gene=lztr1&sequence_type=cDNA&end_insdel=1261&transcript_stable_id_text=ENST00000215739&start_insdel=1259 (accessed on 2 April 2025).
- BDGP: Splice Site Prediction by Neural Network. Available online: https://www.fruitfly.org/seq_tools/splice.html (accessed on 16 April 2025).
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Expasy—Translate Tool. Available online: https://web.expasy.org/translate/ (accessed on 2 April 2025).
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling Protein Tertiary and Quaternary Structure Using Evolutionary Information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- SAVESv6.1—Structure Validation Server. Available online: https://saves.mbi.ucla.edu/ (accessed on 16 April 2025).
- InterPro. Available online: https://www.ebi.ac.uk/interpro/ (accessed on 17 April 2025).
- Expasy—PROSITE. Available online: https://prosite.expasy.org/ (accessed on 8 May 2025).
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- STRING: Functional Protein Association Networks. Available online: https://string-db.org/ (accessed on 12 June 2025).
- COBALT:Multiple Alignment Tool. Available online: https://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi (accessed on 16 April 2025).

| HSF Gen Omnis | Mutation Taster |
|---|---|
| GTTCCAGTTCTC | GTGTTTGG |
| GGTTCCATTTCT | TTTGGGGG |
| TGTGGGGCCTGT | TGGGGTGC |
| Database | Reported Phenotypes Related with LZTR1 |
|---|---|
| OMIM | Schwannomatosis 2, Noonan syndrome 2, Noonan syndrome 10 |
| ClinVar | Noonan syndrome 10, Noonan syndrome 2, DiGeorge syndrome, epilepsy, intellectual disability, hydronephrosis, neurodevelopmental delay, global developmental delay, inborn genetic diseases, cat eye syndrome, aganglionic megacolon, rasopathy, inherited immunodeficiency diseases, congenital diaphragmatic hernia, oppositional defiant disorder, 22q11.2 central duplication syndrome, 22q11.2 central deletion syndrome, chromosome 22q11.2 microduplication syndrome, velocardiofacial syndrome, neurodevelopmental disorder, bladder exstrophy, schizophrenia, autistic disorder, cognitive impairment, ear malformation, VATER association, autism spectrum disorder, premature ovarian failure |
| Orphanet | Familial isolated café-au-lait macules, full schwannomatosis, giant cell glioblastoma, glioblastoma, gliosarcoma, Noonan syndrome |
| Tool | Latest Version/Platform Info | Thresholds/Notes |
|---|---|---|
| gnomAD | v4.1 (2024) and v2.1.1 (legacy) | LOEUF threshold: <0.6 (v4.1), <0.35 (v2.1.1); GroupMax FAF used for BA1/BS1 variant filtering |
| SnapGene Viewer | v8.1.0 (2025) | No specific thresholds; version compatibility: Windows 10+ (64-bit), macOS 10.14+ |
| Variant Effect Predictor (VEP) | v112 and v113.3 (Ensembl) | No fixed thresholds; plugin-dependent; supports GRCh37 and GRCh38 |
| ORF Finder | Web version (NCBI) and standalone Linux version | Default min ORF length: 75–300 nt; start codon: ATG or alternatives; nested ORFs optional |
| GENSCAN | Web server (MIT); original version from 1997 | No numeric thresholds; organism-specific models; supports sequences up to 1 Mbp |
| HSF GenOmnis | https://hsf.genomnis.com/, accessed on 10 October 2024 | Splice site impact: MaxEntScan + HSF matrix; ESE/ESS prediction: sensitivity ~0.83, specificity ~0.81 |
| MutationTaster | MutationTaster2021 (GRCh37) | No fixed thresholds; integrates gnomAD, ExAC, and splice prediction; scores are probabilistic |
| SpliceAI | v1.3.1 (Illumina); also used via Broad’s SpliceAI Lookup | Recommended delta score threshold: ≥0.5 (high confidence), ≥0.2 (high recall); max distance: 50 bp |
| BDGP Splice Site Prediction | NNSPLICE v0.9 (1997) | Score threshold: typically ≥0.4–0.6 for reliable splice site prediction |
| SWISS-MODEL | Web-based (2025); version 101.0 of InterPro integration | No thresholds; uses QMEAN and LDDT for model quality; AlphaFold templates integrated |
| Prosite | Release 2025_03 (June 2025) | No thresholds; pattern/profile-based domain detection; integrated with InterPro |
| Smart | Integrated in InterPro (2025) | No thresholds; domain detection via HMMs |
| InterPro | Version 101.0 (2025) | No thresholds; integrates 13 databases including Pfam, SMART, PROSITE, etc. |
| STRING | Version 12.0 (2025) | Confidence score thresholds: low (0.15), medium (0.4), high (0.7), highest (0.9) |
| GenMANIA | v3.4.0Web-based; Cytoscape plugin available | No thresholds; uses label propagation and network weighting for gene function prediction |
| PipeAlign2 | Web-based (LBGI); updated version of PipeAlign | No thresholds; used for MACS alignment and subfamily clustering |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieleba, I.; Smoleń, P.; Czukiewska, E.; Szcześniak, D.; Filip, A.A. LZTR1: c.1260+1del Variant as a Significant Predictor of Early-Age Breast Cancer Development: Case Report Combined with In Silico Analysis. Int. J. Mol. Sci. 2025, 26, 6704. https://doi.org/10.3390/ijms26146704
Wieleba I, Smoleń P, Czukiewska E, Szcześniak D, Filip AA. LZTR1: c.1260+1del Variant as a Significant Predictor of Early-Age Breast Cancer Development: Case Report Combined with In Silico Analysis. International Journal of Molecular Sciences. 2025; 26(14):6704. https://doi.org/10.3390/ijms26146704
Chicago/Turabian StyleWieleba, Irena, Paulina Smoleń, Ewa Czukiewska, Dominika Szcześniak, and Agata A. Filip. 2025. "LZTR1: c.1260+1del Variant as a Significant Predictor of Early-Age Breast Cancer Development: Case Report Combined with In Silico Analysis" International Journal of Molecular Sciences 26, no. 14: 6704. https://doi.org/10.3390/ijms26146704
APA StyleWieleba, I., Smoleń, P., Czukiewska, E., Szcześniak, D., & Filip, A. A. (2025). LZTR1: c.1260+1del Variant as a Significant Predictor of Early-Age Breast Cancer Development: Case Report Combined with In Silico Analysis. International Journal of Molecular Sciences, 26(14), 6704. https://doi.org/10.3390/ijms26146704