
Identification and Characterization of a Translational Mouse Model for Blood–Brain Barrier Leakage in Cerebral Small Vessel Disease

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
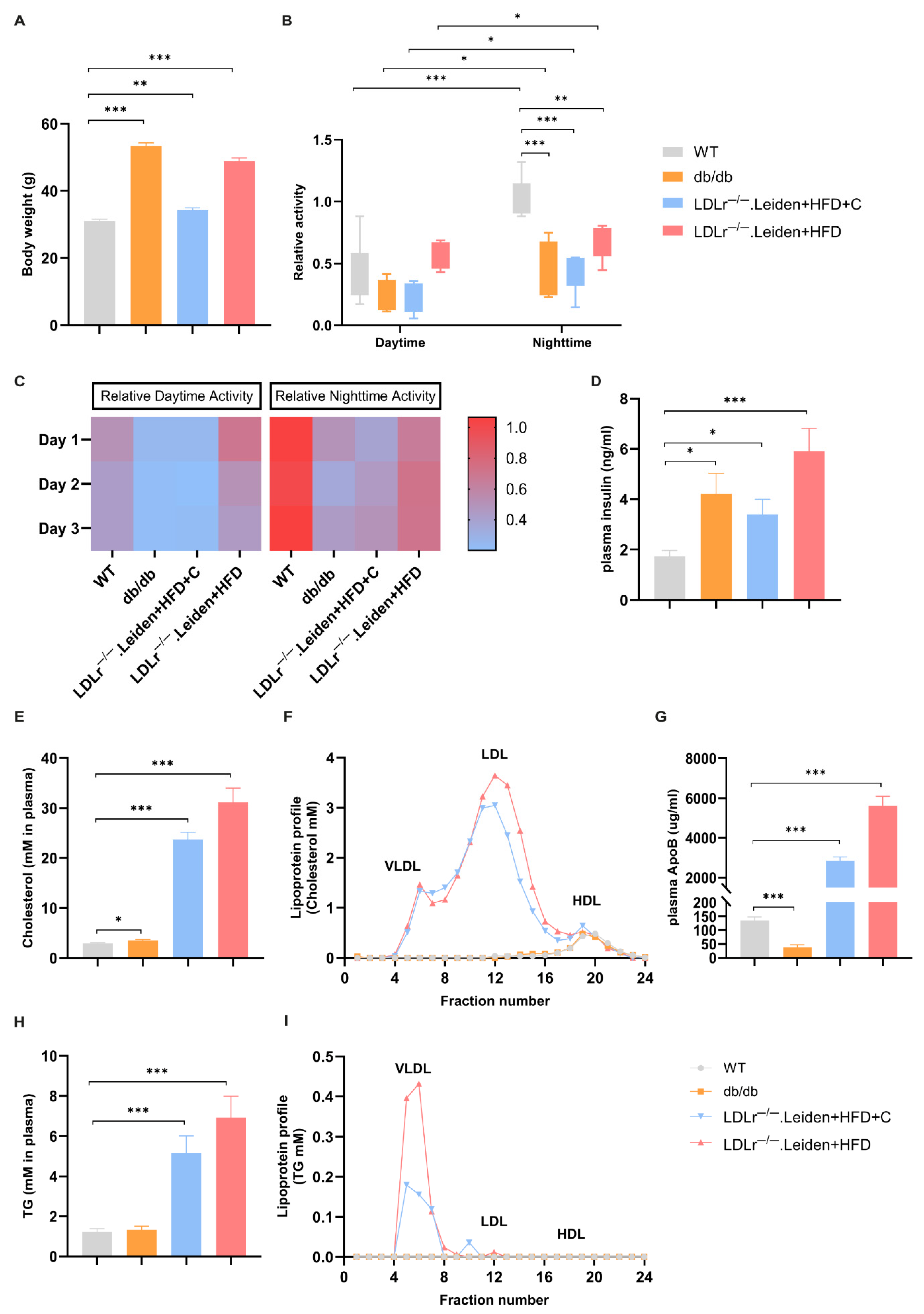
2.1. Body Weight, Plasma Lipid Changes and Home-Cage Activity
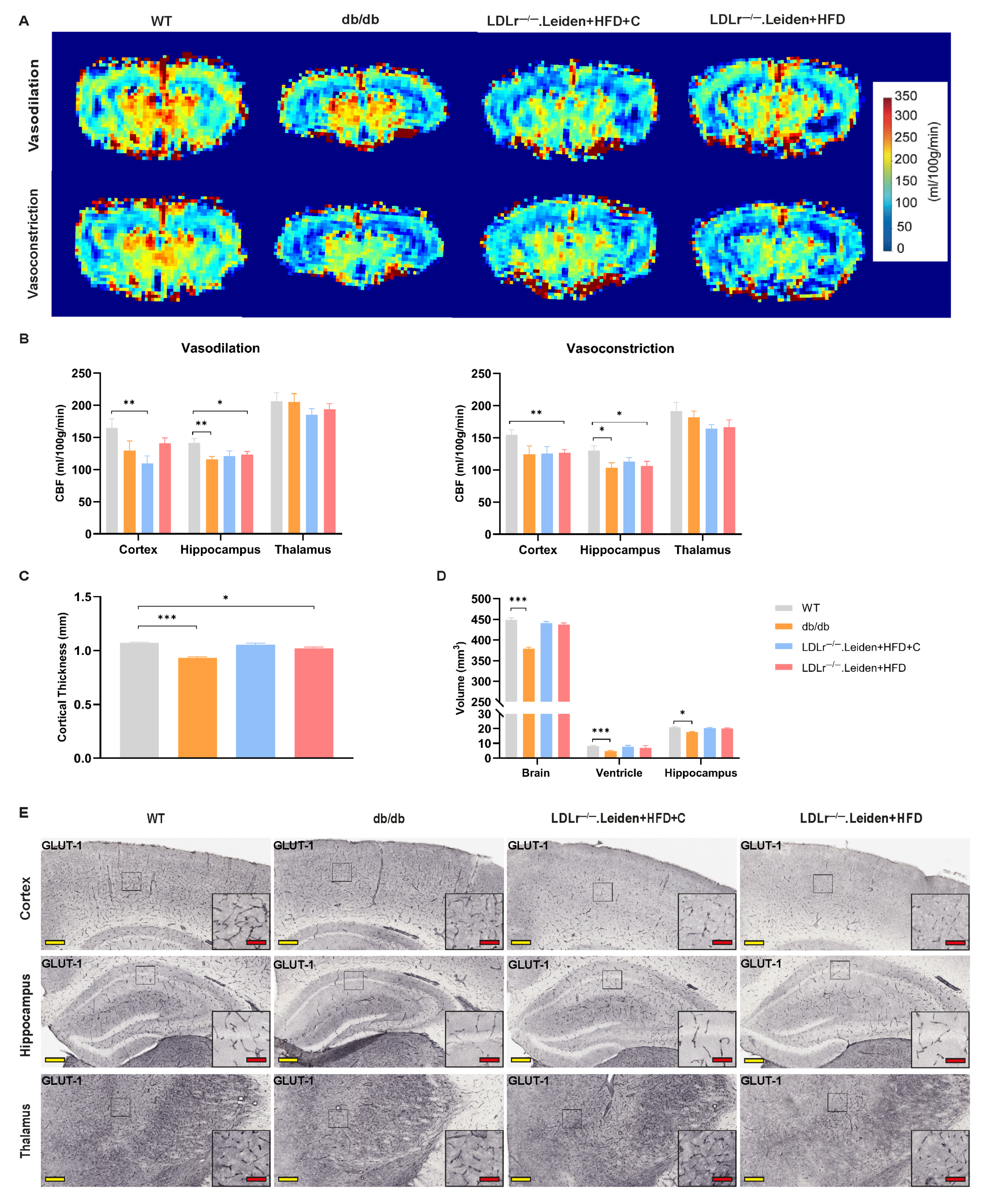
2.2. Changes in Cerebral Vascular Function and Brain Atrophy
2.2.1. CBF and Brain Atrophy
2.2.2. GLUT-1
2.2.3. Plasma Biomarkers of Endothelial and Vascular Inflammation
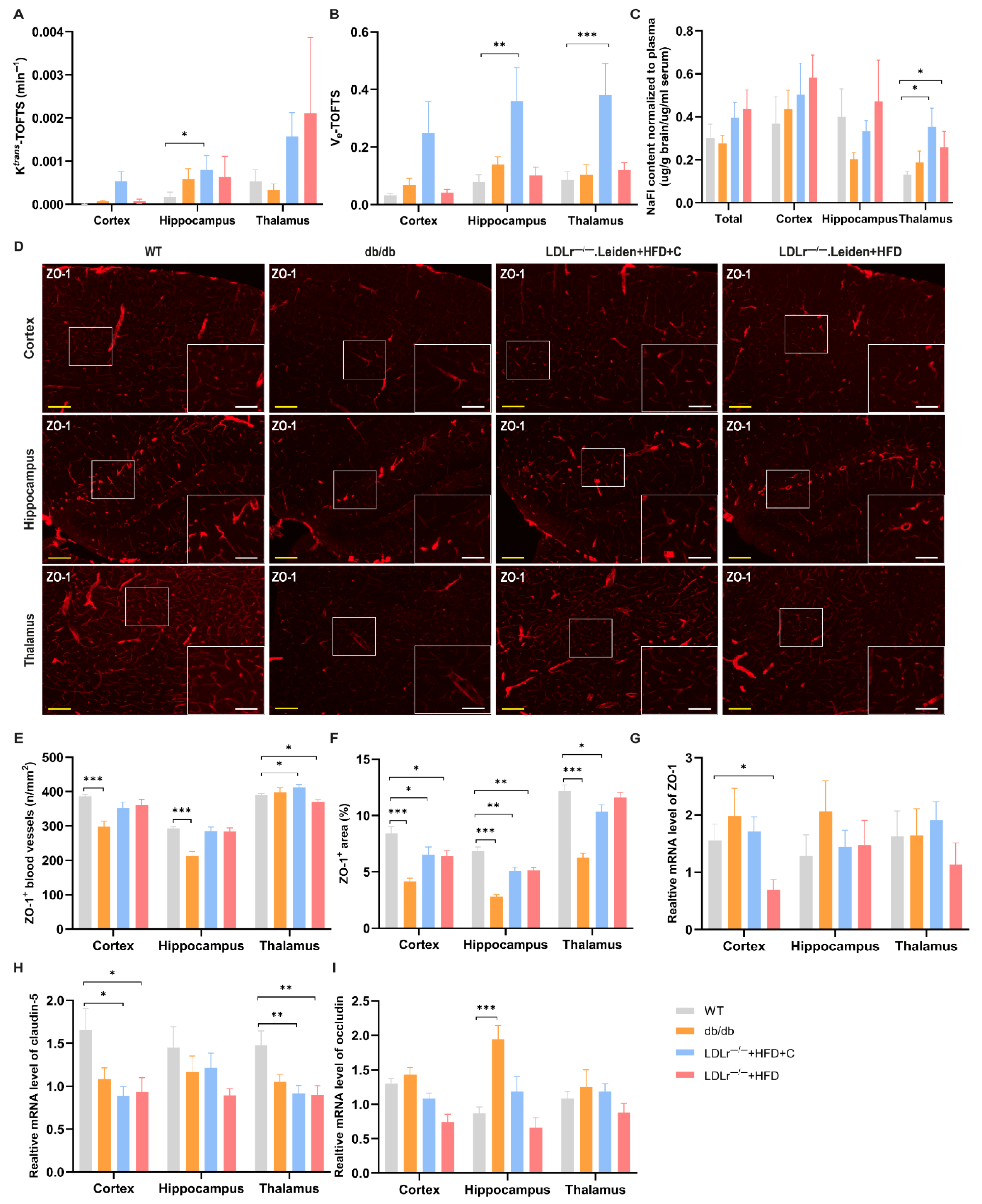
2.3. Changes in BBB Integrity
2.3.1. DCE-MRI
2.3.2. Sodium Fluorescein (NaFl)
2.3.3. TJs Integrity
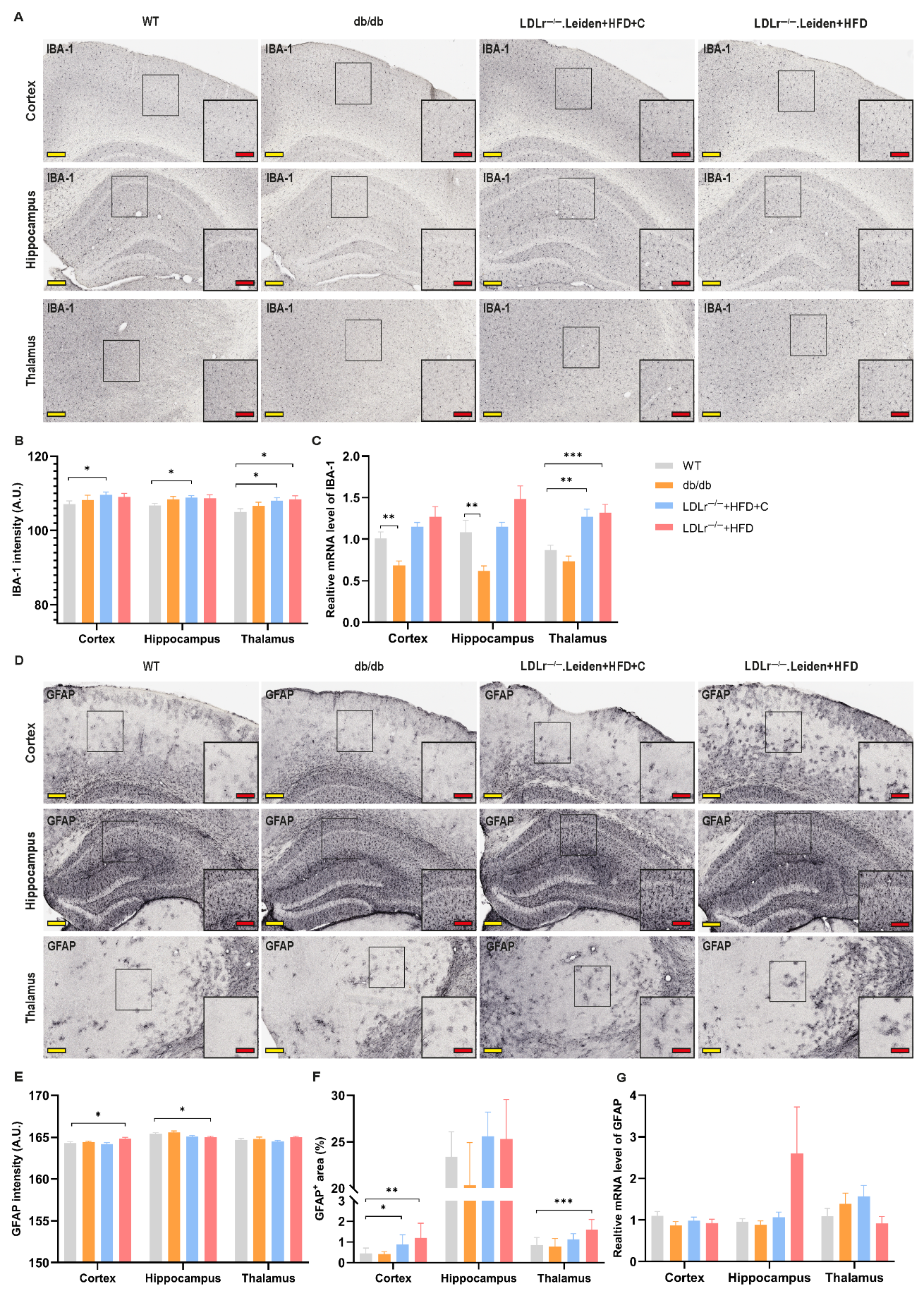
2.4. Neuroinflammation
2.4.1. Ionized Calcium-Binding Adapter Molecule 1 (IBA-1)
2.4.2. Glial Fibrillary Acidic Protein (GFAP)
3. Discussion
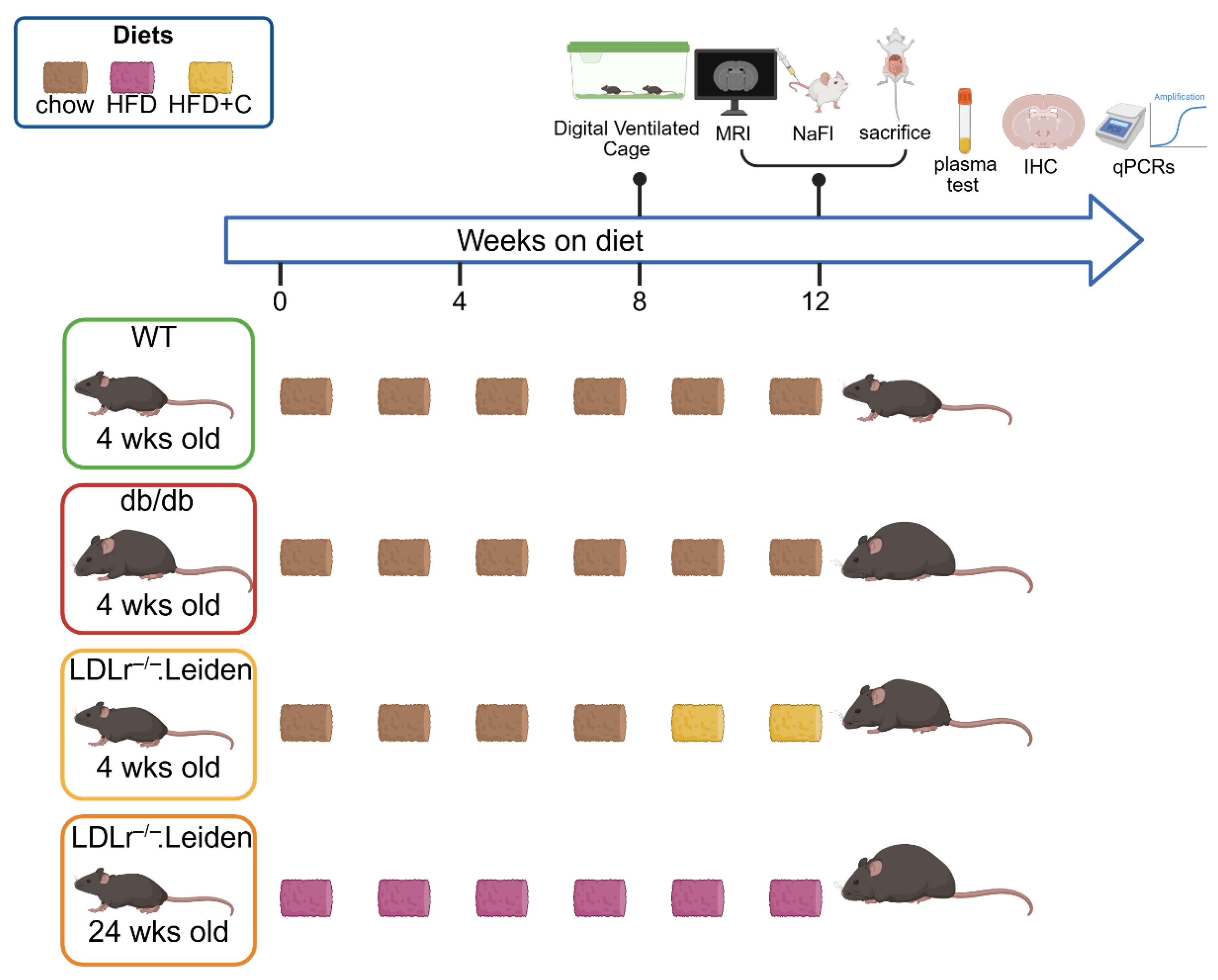
4. Materials and Methods
4.1. Animals
4.2. Home-Cage Activity: Digital Ventilated Cages (DVC)
4.3. Brain MRI
4.3.1. Dynamic Contrast-Enhanced Magnetic Resonance Imaging (DCE-MRI)
4.3.2. Cortical Thickness and Volumetry
4.3.3. Arterial Spin Labeling (ASL)
4.4. Perfusion and Tissue Preparation
4.5. Blood Chemistry
4.6. NaFl Measurement
4.7. Real-Time Quantitative PCR (RT-qPCR)
4.8. IHC
4.8.1. IBA-1, GFAP and GLUT-1 Staining
4.8.2. ZO-1 Staining
4.8.3. Quantification
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cSVD | cerebral small vessel disease |
| MRI | magnetic resonance imaging |
| WMH | white matter hyperintensities |
| CBF | cerebral blood flow |
| BBB | blood–brain barrier |
| ECs | endothelial cells |
| TJs | tight junctions |
| ICAM-1 | intercellular adhesion molecule 1 |
| VCAM-1 | vascular cell adhesion molecule 1 |
| HFD | high-fat diet |
| HFD+C | high-fat diet with 1% cholesterol |
| ASL | arterial spin labeling |
| DVC | digital ventilated cages |
| NaFl | sodium fluorescein |
| PBS | phosphate-buffered saline |
| EDTA | ethylenediaminetetraacetic acid |
| ELISA | enzyme-linked immunosorbent assay |
| TG | triglycerides |
| PAI-1 | plasminogen activator inhibitor-1 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HPRT | hypoxanthine guanine phosphoribosyl transferase |
| IHC | immunohistochemistry |
| IBA-1 | ionized calcium-binding adapter molecule 1 |
| GFAP | glial fibrillary acidic protein |
| GLUT-1 | glucose transporter-1 |
| ZO-1 | zonula occludens 1 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EES | extravascular extracellular space |
References
- Evans, L.E.; Taylor, J.L.; Smith, C.J.; Pritchard, H.A.T.; Greenstein, A.S.; Allan, S.M. Cardiovascular comorbidities, inflammation, and cerebral small vessel disease. Cardiovasc. Res. 2021, 117, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S.; de Leeuw, F.E. Cerebral small vessel disease: Recent advances and future directions. Int. J. Stroke. 2023, 18, 4–14. [Google Scholar] [CrossRef]
- Xu, T.; Wang, Y.; Yuan, J.; Chen, Y.; Luo, H. Small Vessel Disease Burden and Outcomes of Mechanical Thrombectomy in Ischemic Stroke: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 602037. [Google Scholar] [CrossRef]
- Inoue, Y.; Shue, F.; Bu, G.; Kanekiyo, T. Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 46. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef]
- Quick, S.; Moss, J.; Rajani, R.M.; Williams, A. A Vessel for Change: Endothelial Dysfunction in Cerebral Small Vessel Disease. Trends Neurosci. 2021, 44, 289–305. [Google Scholar] [CrossRef]
- Shin, Y.; Choi, S.H.; Kim, E.; Bylykbashi, E.; Kim, J.A.; Chung, S.; Kim, D.Y.; Kamm, R.D.; Tanzi, R.E. Blood-Brain Barrier Dysfunction in a 3D In Vitro Model of Alzheimer’s Disease. Adv. Sci. 2019, 6, 1900962. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Z.; Guan, J.; Lu, W.; Zhan, C. Unraveling GLUT-mediated transcytosis pathway of glycosylated nanodisks. Asian J. Pharm. Sci. 2021, 16, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef]
- Bai, T.; Yu, S.; Feng, J. Advances in the Role of Endothelial Cells in Cerebral Small Vessel Disease. Front. Neurol. 2022, 13, 861714. [Google Scholar] [CrossRef] [PubMed]
- Cifu, A.; Janes, F.; Mio, C.; Domenis, R.; Pessa, M.E.; Garbo, R.; Curcio, F.; Valente, M.; Fabris, M. Brain Endothelial Cells Activate Neuroinflammatory Pathways in Response to Early Cerebral Small Vessel Disease (CSVD) Patients’ Plasma. Biomedicines 2023, 11, 3055. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, D.; Lin, J.; Thomas, A.M.; Miao, J.; Chen, D.; Li, S.; Chu, C. Cerebral small vessel disease: Pathological mechanisms and potential therapeutic targets. Front. Aging Neurosci. 2022, 14, 961661. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Liao, F.F.; Lin, G.; Chen, X.; Chen, L.; Zheng, W.; Raghow, R.; Zhou, F.M.; Shih, A.Y.; Tan, X.L. Endothelial Nitric Oxide Synthase-Deficient Mice: A Model of Spontaneous Cerebral Small-Vessel Disease. Am. J. Pathol. 2021, 191, 1932–1945. [Google Scholar] [CrossRef]
- Li, L.; He, G.; Shi, M.; Zhu, J.; Cheng, Y.; Chen, Y.; Chen, J.; Xue, Q. Edaravone dexborneol ameliorates cognitive impairment by regulating the NF-kappaB pathway through AHR and promoting microglial polarization towards the M2 phenotype in mice with bilateral carotid artery stenosis (BCAS). Eur. J. Pharmacol. 2023, 957, 176036. [Google Scholar] [CrossRef]
- Song, X.; Zhu, Z.; Qian, X.; Liu, X.; Chen, S.; Tang, H. Multi-Omics Characterization of Type 2 Diabetes Mellitus-Induced Cognitive Impairment in the db/db Mouse Model. Molecules 2022, 27, 1904. [Google Scholar] [CrossRef]
- Guilbaud, A.; Howsam, M.; Niquet-Leridon, C.; Delguste, F.; Boulanger, E.; Tessier, F.J. The LepR(db/db) mice model for studying glycation in the context of diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3103. [Google Scholar] [CrossRef]
- Cannistraro, R.J.; Badi, M.; Eidelman, B.H.; Dickson, D.W.; Middlebrooks, E.H.; Meschia, J.F. CNS small vessel disease: A clinical review. Neurology 2019, 92, 1146–1156. [Google Scholar] [CrossRef]
- van den Hoek, A.M.; Verschuren, L.; Worms, N.; van Nieuwkoop, A.; de Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Egelandsdal, B.; Harvei, S.; Rocha, S.D.C.; Pieterman, E.J.; Kleemann, R.; Carlsen, H. Differential effects of plant and animal fats on obesity-induced dyslipidemia and atherosclerosis in Ldlr-/-.Leiden mice. FASEB J. 2023, 37, e23096. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Chen, Y.; Joo, J.; Chu, J.M.; Chang, R.C.; Wong, G.T. Downregulation of the glucose transporter GLUT 1 in the cerebral microvasculature contributes to postoperative neurocognitive disorders in aged mice. J. Neuroinflammation 2023, 20, 237. [Google Scholar] [CrossRef]
- Mosteiro, A.; Pedrosa, L.; Amaro, S.; Menendez-Giron, S.; Reyes, L.; de Riva, N.; Misis, M.; Blasco, J.; Vert, C.; Dominguez, C.J.; et al. Understanding the Importance of Blood-Brain Barrier Alterations in Brain Arteriovenous Malformations and Implications for Treatment: A Dynamic Contrast-Enhanced-MRI-Based Prospective Study. Neurosurgery 2025, 96, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Sole-Guardia, G.; Kiliaan, A.J. Blood-brain barrier pathology in cerebral small vessel disease. Neural. Regen. Res. 2024, 19, 1233–1240. [Google Scholar] [CrossRef]
- Olga, L.; Bobeldijk-Pastorova, I.; Bas, R.C.; Seidel, F.; Snowden, S.G.; Furse, S.; Ong, K.K.; Kleemann, R.; Koulman, A. Lipid profiling analyses from mouse models and human infants. STAR Protoc. 2022, 3, 101679. [Google Scholar] [CrossRef]
- Gart, E.; van Duyvenvoorde, W.; Snabel, J.M.; de Ruiter, C.; Attema, J.; Caspers, M.P.M.; Lek, S.; van Heuven, B.J.; Speksnijder, A.; Giera, M.; et al. Translational characterization of the temporal dynamics of metabolic dysfunctions in liver, adipose tissue and the gut during diet-induced NASH development in Ldlr-/-.Leiden mice. Heliyon 2023, 9, e13985. [Google Scholar] [CrossRef]
- Seidel, F.; Morrison, M.C.; Arnoldussen, I.; Verweij, V.; Attema, J.; de Ruiter, C.; van Duyvenvoorde, W.; Snabel, J.; Geenen, B.; Franco, A.; et al. Obesity accelerates age-related memory deficits and alters white matter tract integrity in Ldlr-/-.Leiden mice. Brain Behav. Immun. Health 2025, 45, 100991. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Lu, W.; Yu, C.; Wang, L.; Wang, F.; Qiu, J. Perfusion heterogeneity of cerebral small vessel disease revealed via arterial spin labeling MRI and machine learning. Neuroimage Clin. 2022, 36, 103165. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.; Ying, C.; Chen, Y.; Ford, A.L.; An, H.; Lee, J.M. Oxygen Metabolic Stress and White Matter Injury in Patients with Cerebral Small Vessel Disease. Stroke 2022, 53, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Sabayan, B.; Westendorp, R.G.; Grond, J.; Stott, D.J.; Sattar, N.; van Osch, M.J.; van Buchem, M.A.; de Craen, A.J. Markers of endothelial dysfunction and cerebral blood flow in older adults. Neurobiol. Aging 2014, 35, 373–377. [Google Scholar] [CrossRef]
- Claassen, J.; Thijssen, D.H.J.; Panerai, R.B.; Faraci, F.M. Regulation of cerebral blood flow in humans: Physiology and clinical implications of autoregulation. Physiol. Rev. 2021, 101, 1487–1559. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, H.; Doubal, F.N.; Duering, M. Advanced MRI in cerebral small vessel disease. Int. J. Stroke 2023, 18, 28–35. [Google Scholar] [CrossRef]
- Ghaznawi, R.; Zwartbol, M.H.; Zuithoff, N.P.; Bresser, J.; Hendrikse, J.; Geerlings, M.I.; UCC-SMART Study Group. Reduced parenchymal cerebral blood flow is associated with greater progression of brain atrophy: The SMART-MR study. J. Cereb. Blood Flow Metab. 2021, 41, 1229–1239. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, X.; Su, Y.; Yin, G.; Wang, S.; Yu, B.; Li, H.; Qi, J.; Chen, H.; Zeng, W.; et al. Activation of Wnt/beta-catenin pathway mitigates blood-brain barrier dysfunction in Alzheimer’s disease. Brain 2022, 145, 4474–4488. [Google Scholar] [CrossRef]
- Beran, M.; Jansen, W.J.; Oomens, J.E.; Moonen, J.E.F.; Slagboom, P.E.; Huisman, M.; Kok, A.A.L.; Nooyens, A.C.J.; Verschuren, W.M.M.; Stehouwer, C.D.A.; et al. Biomarkers of endothelial dysfunction and cognition: A two-step IPD meta-analysis. Alzheimers Dement. 2024, 20, 8402–8411. [Google Scholar] [CrossRef]
- Markova, I.; Huttl, M.; Gayova, N.; Miklankova, D.; Cerna, K.; Kavanova, M.; Skaroupkova, P.; Cacanyiova, S.; Malinska, H. Visceral Adipose Tissue Inflammation and Vascular Complications in a Rat Model with Severe Dyslipidemia: Sex Differences and PAI-1 Tissue Involvement. Biomolecules 2024, 15, 19. [Google Scholar] [CrossRef]
- Low, A.; Mak, E.; Rowe, J.B.; Markus, H.S.; O’Brien, J.T. Inflammation and cerebral small vessel disease: A systematic review. Ageing Res. Rev. 2019, 53, 100916. [Google Scholar] [CrossRef]
- Sharma, V.; Singh, T.G.; Mannan, A. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem Res 2022, 47, 2173–2186. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.K.; Xian, Y.X.; Zhang, L.; Lai, H.; Hou, X.G.; Xu, Y.X.; Yu, T.; Xu, F.Y.; Song, J.; Fu, C.L.; et al. Influence of blood glucose on the expression of glucose trans-porter proteins 1 and 3 in the brain of diabetic rats. Chin. Med. J. 2007, 120, 1704–1709. [Google Scholar] [CrossRef]
- Koranyi, L.; Bourey, R.E.; James, D.; Mueckler, M.; Fiedorek, F.T., Jr.; Permutt, M.A. Glucose transporter gene expression in rat brain: Pretranslational changes associated with chronic insulin-induced hypoglycemia, fasting, and diabetes. Mol. Cell. Neurosci. 1991, 2, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Nation, D.A.; Kisler, K.; Toga, A.W.; Zlokovic, B.V. Imaging subtle leaks in the blood-brain barrier in the aging human brain: Potential pitfalls, challenges, and possible solutions. Geroscience 2022, 44, 1339–1351. [Google Scholar] [CrossRef]
- de Lussanet, Q.G.; Langereis, S.; Beets-Tan, R.G.; van Genderen, M.H.; Griffioen, A.W.; van Engelshoven, J.M.; Backes, W.H. Dynamic contrast-enhanced MR imaging kinetic parameters and molecular weight of dendritic contrast agents in tumor angiogenesis in mice. Radiology 2005, 235, 65–72. [Google Scholar] [CrossRef]
- Alshammari, M.A.; Alshehri, A.O.; Alqahtani, F.; Khan, M.R.; Bakhrebah, M.A.; Alasmari, F.; Alshammari, T.K.; Alsharari, S.D. Increased Permeability of the Blood-Brain Barrier in a Diabetic Mouse Model (Leprdb/db Mice). Int. J. Mol. Sci. 2024, 25, 7768. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Hao, S.; Dey, A.; Yu, X.; Baban, B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 2016, 36, 2108–2121. [Google Scholar] [CrossRef]
- Wang, H.K.; Su, Y.T.; Ho, Y.C.; Lee, Y.K.; Chu, T.H.; Chen, K.T.; Wu, C.C. HDAC1 is Involved in Neuroinflammation and Blood-Brain Barrier Damage in Stroke Pathogenesis. J. Inflamm. Res. 2023, 16, 4103–4116. [Google Scholar] [CrossRef]
- Hou, Y.; Fan, F.; Xie, N.; Zhang, Y.; Wang, X.; Meng, X. Rhodiola crenulata alleviates hypobaric hypoxia-induced brain injury by maintaining BBB integrity and balancing energy metabolism dysfunction. Phytomedicine 2024, 128, 155529. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Lee, H.G.; Lee, J.H.; Flausino, L.E.; Quintana, F.J. Neuroinflammation: An astrocyte perspective. Sci. Transl. Med. 2023, 15, eadi7828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, C.; Shi, R.; Zhou, S.; Shan, H.; Deng, L.; Chen, T.; Guo, Y.; Zhang, Z.; Yang, G.Y.; et al. Blocking C3d(+)/GFAP(+) A1 Astrocyte Conversion with Semaglutide Attenuates Blood-Brain Barrier Disruption in Mice after Ischemic Stroke. Aging Dis. 2022, 13, 943–959. [Google Scholar] [CrossRef]
- Li, X.; Cai, Y.; Zhang, Z.; Zhou, J. Glial and Vascular Cell Regulation of the Blood-Brain Barrier in Diabetes. Diabetes Metab. J. 2022, 46, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Moca, E.N.; Lecca, D.; Hope, K.T.; Etienne, F.; Schaler, A.W.; Espinoza, K.; Chappell, M.S.; Gray, D.T.; Tweedie, D.; Sidhu, S.; et al. Microglia Drive Pockets of Neuroinflammation in Middle Age. J. Neurosci. 2022, 42, 3896–3918. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR-/-.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Gart, E.; van Duyvenvoorde, W.; Caspers, M.P.M.; van Trigt, N.; Snabel, J.; Menke, A.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Intervention with isoleucine or valine corrects hyperinsulinemia and reduces intrahepatic diacylglycerols, liver steatosis, and inflammation in Ldlr-/-.Leiden mice with manifest obesity-associated NASH. FASEB J. 2022, 36, e22435. [Google Scholar] [CrossRef]
- Kuhnast, S.; van der Hoorn, J.W.; Pieterman, E.J.; van den Hoek, A.M.; Sasiela, W.J.; Gusarova, V.; Peyman, A.; Schafer, H.L.; Schwahn, U.; Jukema, J.W.; et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J. Lipid Res. 2014, 55, 2103–2112. [Google Scholar] [CrossRef]
- Gomes, F.C.; Ferreira, M.Y.; Larcipretti, A.L.L.; Freitas, B.C.B.; Andreao, F.F.; Turpin, J.; Bertani, R.; Singha, S.; Polverini, A.D.; Ferreira, C.; et al. Sodium fluorescein and 5-aminolevulinic acid fluorescence- guided biopsy in brain lesions: A systematic review and meta-analysis. J. Neurooncol. 2024, 170, 11–29. [Google Scholar] [CrossRef]
- Jaspers, K.; Aerts, H.J.; Leiner, T.; Oostendorp, M.; van Riel, N.A.; Post, M.J.; Backes, W.H. Reliability of pharmacokinetic parameters: Small vs. medium-sized contrast agents. Magn. Reson. Med. 2009, 62, 779–787. [Google Scholar] [CrossRef]
- Liang, W.; Xu, W.; Zhu, J.; Zhu, Y.; Gu, Q.; Li, Y.; Guo, C.; Huang, Y.; Yu, J.; Wang, W.; et al. Ginkgo biloba extract improves brain uptake of ginsenosides by increasing blood-brain barrier permeability via activating A1 adenosine receptor signaling pathway. J. Ethnopharmacol. 2020, 246, 112243. [Google Scholar] [CrossRef]
- Lohkamp, K.J.; van den Hoek, A.M.; Sole-Guardia, G.; Lisovets, M.; Alves Hoffmann, T.; Velanaki, K.; Geenen, B.; Verweij, V.; Morrison, M.C.; Kleemann, R.; et al. The Preventive Effect of Exercise and Oral Branched-Chain Amino Acid Supplementation on Obesity-Induced Brain Changes in Ldlr-/-.Leiden Mice. Nutrients 2023, 15, 1716. [Google Scholar] [CrossRef] [PubMed]
- Iannello, F. Non-intrusive high throughput automated data collection from the home cage. Heliyon 2019, 5, e01454. [Google Scholar] [CrossRef]
- Voikar, V.; Gaburro, S. Three Pillars of Automated Home-Cage Phenotyping of Mice: Novel Findings, Refinement, and Reproducibility Based on Literature and Experience. Front. Behav. Neurosci. 2020, 14, 575434. [Google Scholar] [CrossRef] [PubMed]
- Golini, E.; Rigamonti, M.; Iannello, F.; De Rosa, C.; Scavizzi, F.; Raspa, M.; Mandillo, S. A Non-invasive Digital Biomarker for the Detection of Rest Disturbances in the SOD1G93A Mouse Model of ALS. Front. Neurosci. 2020, 14, 896. [Google Scholar] [CrossRef]
- Arnoldussen, I.A.C.; Morrison, M.C.; Wiesmann, M.; van Diepen, J.A.; Worms, N.; Voskuilen, M.; Verweij, V.; Geenen, B.; Gualdo, N.P.; van der Logt, L.; et al. Milk fat globule membrane attenuates high fat diet-induced neuropathological changes in obese Ldlr-/-.Leiden mice. Int. J. Obes. 2022, 46, 342–349. [Google Scholar] [CrossRef]
- Tengeler, A.C.; Gart, E.; Wiesmann, M.; Arnoldussen, I.A.C.; van Duyvenvoorde, W.; Hoogstad, M.; Dederen, P.J.; Verweij, V.; Geenen, B.; Kozicz, T.; et al. Propionic acid and not caproic acid, attenuates nonalcoholic steatohepatitis and improves (cerebro) vascular functions in obese Ldlr(-/-).Leiden mice. FASEB J. 2020, 34, 9575–9593. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T. Gadobutrol. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Barnes, S.R.; Ng, T.S.; Santa-Maria, N.; Montagne, A.; Zlokovic, B.V.; Jacobs, R.E. ROCKETSHIP: A flexible and modular software tool for the planning, processing and analysis of dynamic MRI studies. BMC Med. Imaging 2015, 15, 19. [Google Scholar] [CrossRef]
- Backhaus, P.; Buther, F.; Wachsmuth, L.; Frohwein, L.; Buchholz, R.; Karst, U.; Schafers, K.; Hermann, S.; Schafers, M.; Faber, C. Toward precise arterial input functions derived from DCE-MRI through a novel extracorporeal circulation approach in mice. Magn. Reson. Med. 2020, 84, 1404–1415. [Google Scholar] [CrossRef]
- Yen, T.T.; Stienmetz, J.; Simpson, P.J. Blood volume of obese (ob-ob) and diabetic (db-db) mice. Proc. Soc. Exp. Biol. Med. 1970, 133, 307–308. [Google Scholar] [CrossRef]
- Tofts, P.S.; Brix, G.; Buckley, D.L.; Evelhoch, J.L.; Henderson, E.; Knopp, M.V.; Larsson, H.B.; Lee, T.Y.; Mayr, N.A.; Parker, G.J.; et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: Standardized quantities and symbols. J. Magn. Reson. Imaging 1999, 10, 223–232. [Google Scholar] [CrossRef]
- Fries, P.; Muller, A.; Seidel, R.; Robert, P.; Denda, G.; Menger, M.D.; Schneider, G.; Buecker, A. P03277-A New Approach to Achieve High-Contrast Enhancement: Initial Results of an Experimental Extracellular Gadolinium-Based Magnetic Resonance Contrast Agent. Investig. Radiol. 2015, 50, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Goerner, F.L.; Snyder, C.; Morelli, J.N.; Hao, D.; Hu, D.; Li, X.; Runge, V.M. T1 relaxivities of gadolinium-based magnetic resonance contrast agents in human whole blood at 1.5, 3, and 7 T. Investig. Radiol. 2015, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Wiesmann, M.; Zerbi, V.; Jansen, D.; Lutjohann, D.; Veltien, A.; Heerschap, A.; Kiliaan, A.J. Hypertension, cerebrovascular impairment, and cognitive decline in aged AbetaPP/PS1 mice. Theranostics 2017, 7, 1277–1289. [Google Scholar] [CrossRef]
- Ruzek, D.; Salat, J.; Singh, S.K.; Kopecky, J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS ONE 2011, 6, e20472. [Google Scholar] [CrossRef] [PubMed]
- Calahorra, J.; Shenk, J.; Wielenga, V.H.; Verweij, V.; Geenen, B.; Dederen, P.J.; Peinado, M.A.; Siles, E.; Wiesmann, M.; Kiliaan, A.J. Hydroxytyrosol, the Major Phenolic Compound of Olive Oil, as an Acute Therapeutic Strategy after Ischemic Stroke. Nutrients 2019, 11, 2430. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward | Reverse |
|---|---|---|
| Occludin | CCACCCCCATCTGACTATGC | TTCAGGCACCAGAGGTGTTG |
| Claudin-5 | GTTAAGGCACGGGTAGCACT | TACTTCTGTGACACCGGCAC |
| ZO-1 | CTCCGTTGCCCTCACAGTAC | ACTGAGTTGCCTTCACCCTG |
| GLUT-1 | GATCCCAGCAGCAAGAAGGT | TAGCCGAACTGCAGTGATCC |
| IBA-1 | GGATTTGCAGGGAGGAAAAG | TGGGATCATCGAGGAATTG |
| GFAP | TCGGCCAGTTACCAGGAGG | ATGGTGATGCGGTTTTCTTCG |
| HPRT | TGATTAGCGATGATGAACCAGGT | AGCAAGTCTTTCAGTCCTGTCC |
| GAPDH | GTCGGTGTGAACGGATTTGG | ACAATCTCCACTTTGCCACTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, R.; Solé-Guardia, G.; Verweij, V.; Snabel, J.M.; Geenen, B.; Tuladhar, A.M.; Kleemann, R.; Kiliaan, A.J.; Wiesmann, M. Identification and Characterization of a Translational Mouse Model for Blood–Brain Barrier Leakage in Cerebral Small Vessel Disease. Int. J. Mol. Sci. 2025, 26, 6706. https://doi.org/10.3390/ijms26146706
Jia R, Solé-Guardia G, Verweij V, Snabel JM, Geenen B, Tuladhar AM, Kleemann R, Kiliaan AJ, Wiesmann M. Identification and Characterization of a Translational Mouse Model for Blood–Brain Barrier Leakage in Cerebral Small Vessel Disease. International Journal of Molecular Sciences. 2025; 26(14):6706. https://doi.org/10.3390/ijms26146706
Chicago/Turabian StyleJia, Ruxue, Gemma Solé-Guardia, Vivienne Verweij, Jessica M. Snabel, Bram Geenen, Anil Man Tuladhar, Robert Kleemann, Amanda J. Kiliaan, and Maximilian Wiesmann. 2025. "Identification and Characterization of a Translational Mouse Model for Blood–Brain Barrier Leakage in Cerebral Small Vessel Disease" International Journal of Molecular Sciences 26, no. 14: 6706. https://doi.org/10.3390/ijms26146706
APA StyleJia, R., Solé-Guardia, G., Verweij, V., Snabel, J. M., Geenen, B., Tuladhar, A. M., Kleemann, R., Kiliaan, A. J., & Wiesmann, M. (2025). Identification and Characterization of a Translational Mouse Model for Blood–Brain Barrier Leakage in Cerebral Small Vessel Disease. International Journal of Molecular Sciences, 26(14), 6706. https://doi.org/10.3390/ijms26146706