
Overexpression of (P)RR in SHR and Renin-Induced HepG2 Cells Leads to Spontaneous Hypertension Combined with Metabolic Dysfunction-Associated Fatty Liver Disease
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
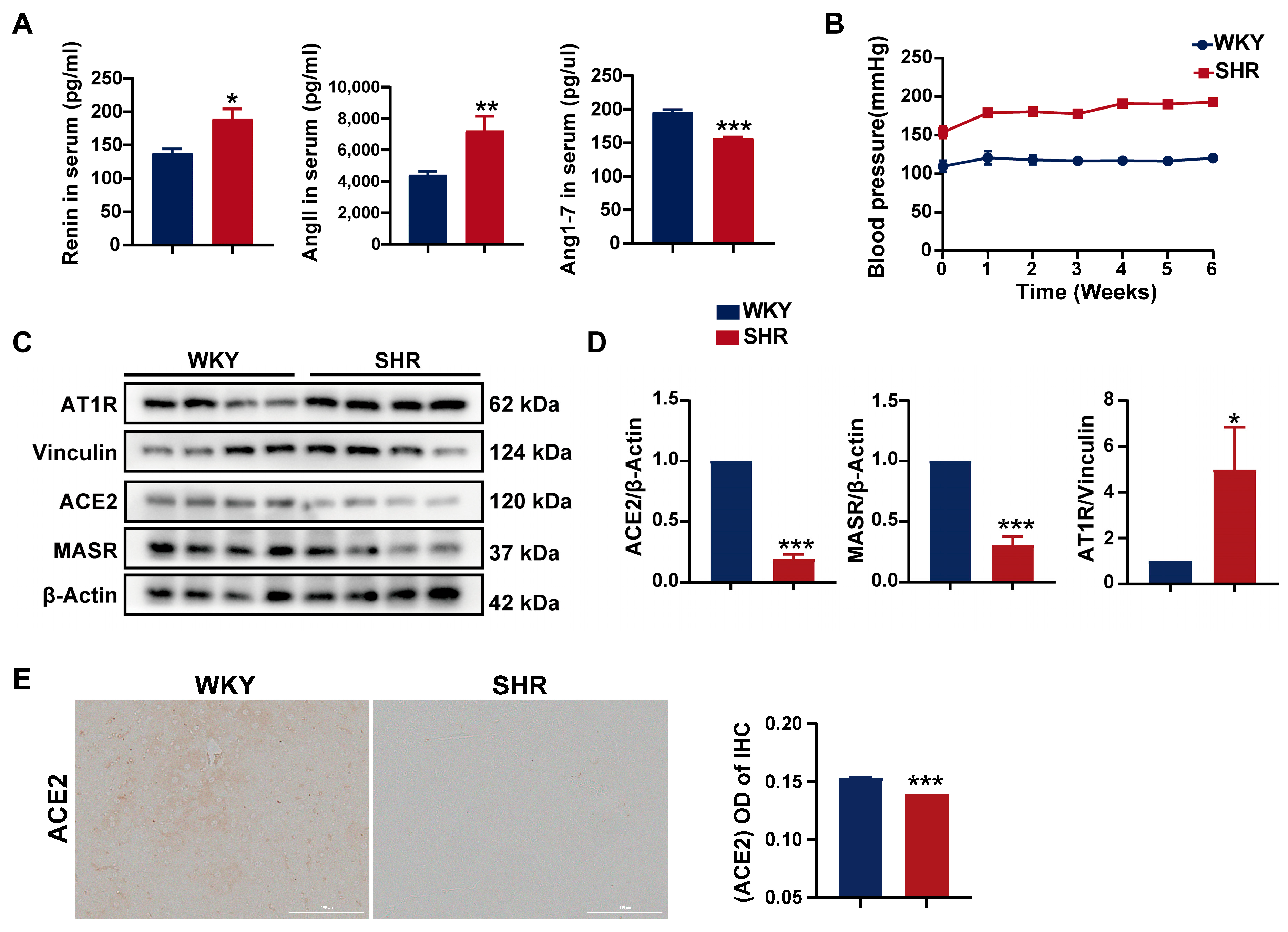
2.1. Systemic and Hepatic RAS Activation in SHRs
2.2. Significant Liver Injury and Lipid Accumulation in SHRs
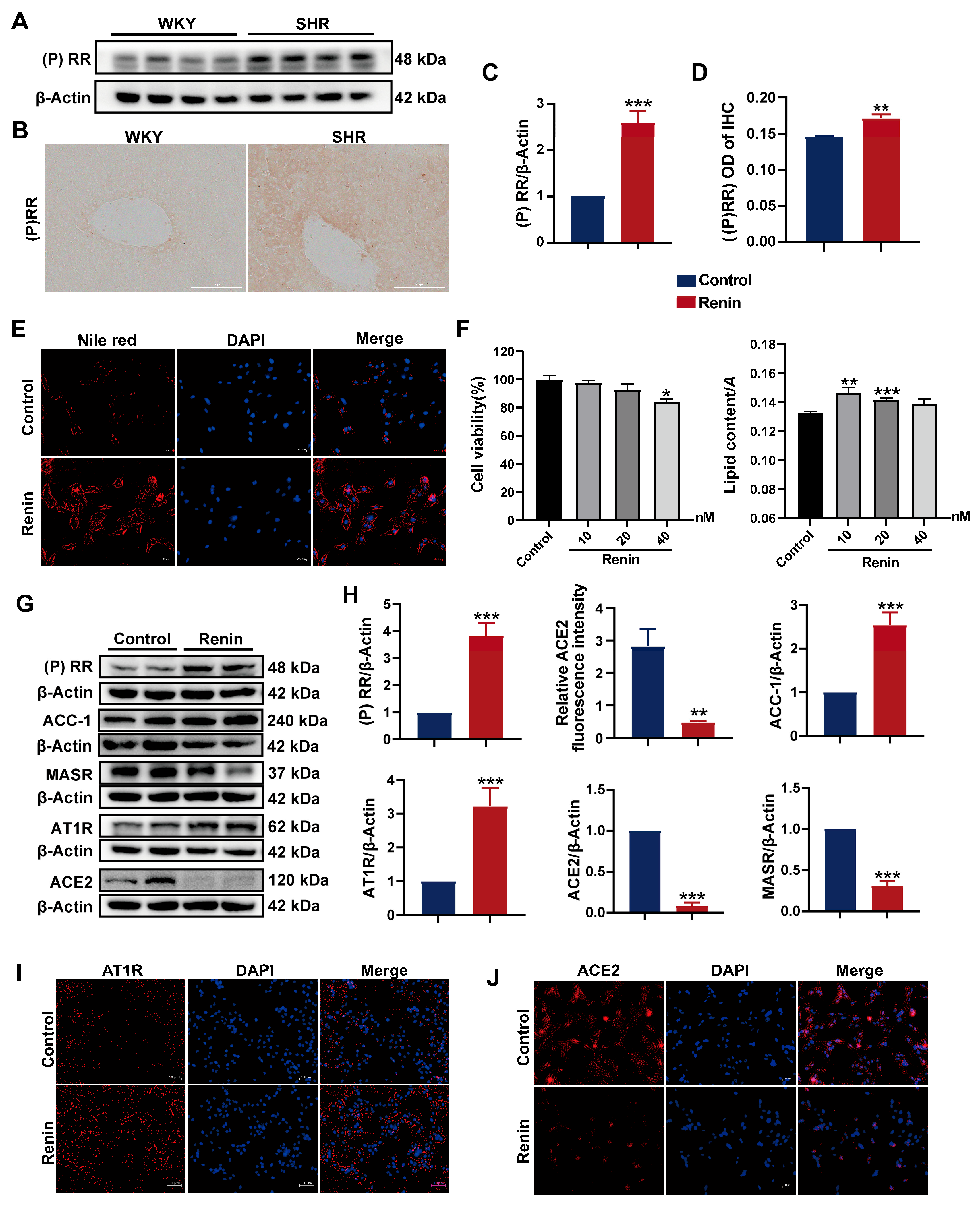
2.3. Activation of (P)RR Leads to Lipid Accumulation in Hepatocytes
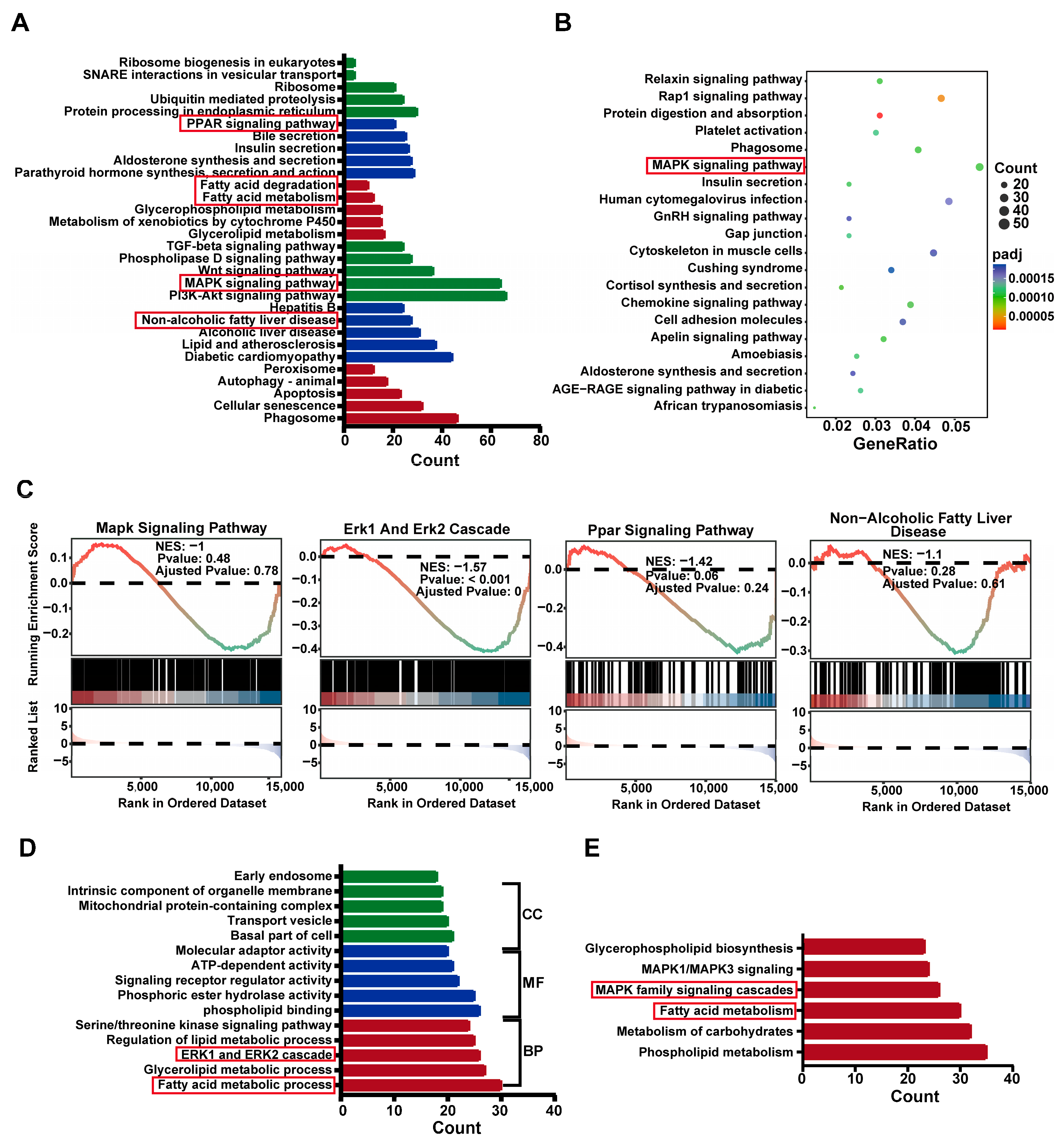
2.4. Differential Gene Expression Analysis in SHR Liver
2.5. Differential Gene Enrich in ERK Cascade and PPAR Pathways in SHR Liver
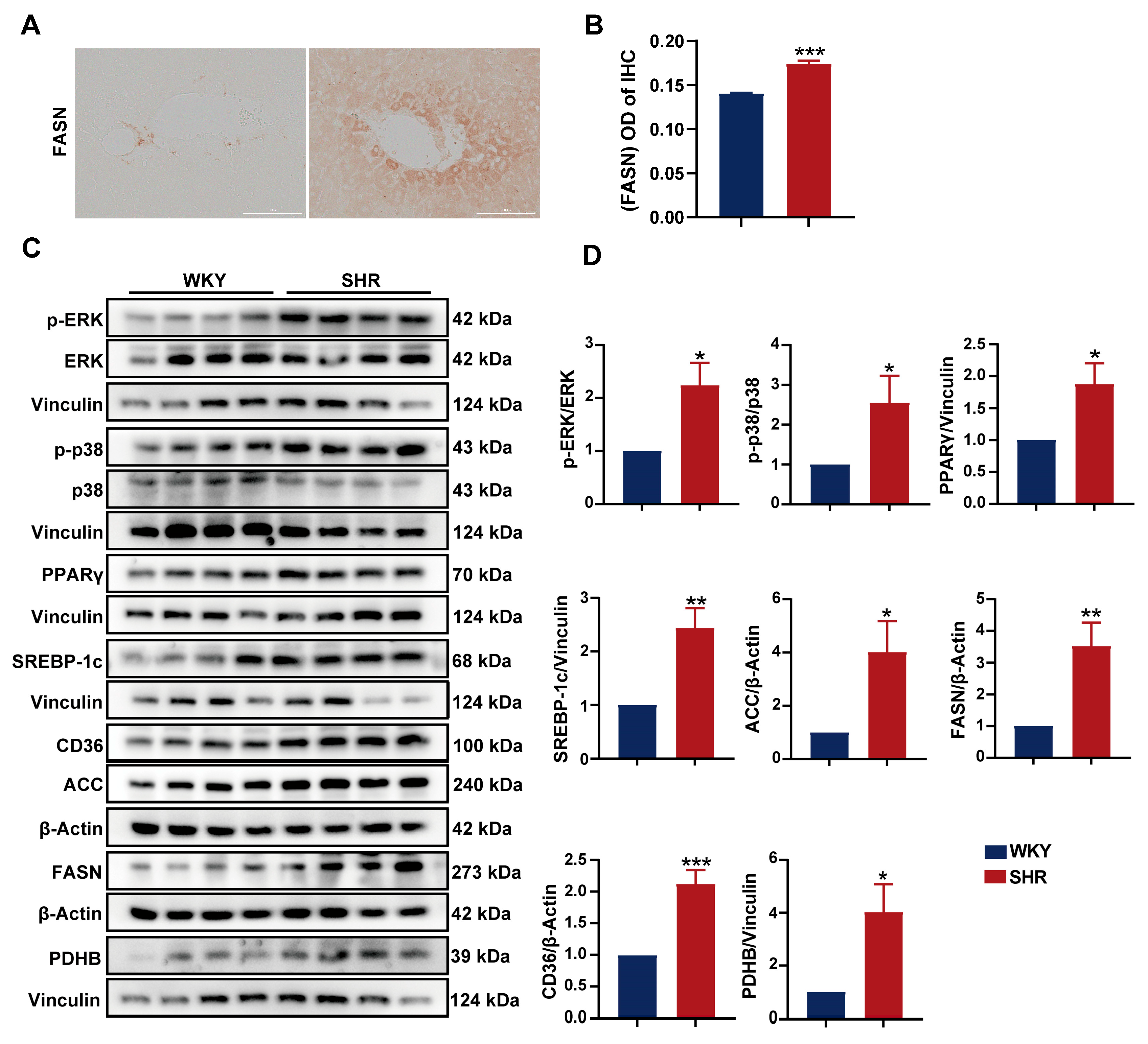
2.6. Activation of the ERK/PPARγ Pathway in SHR Liver
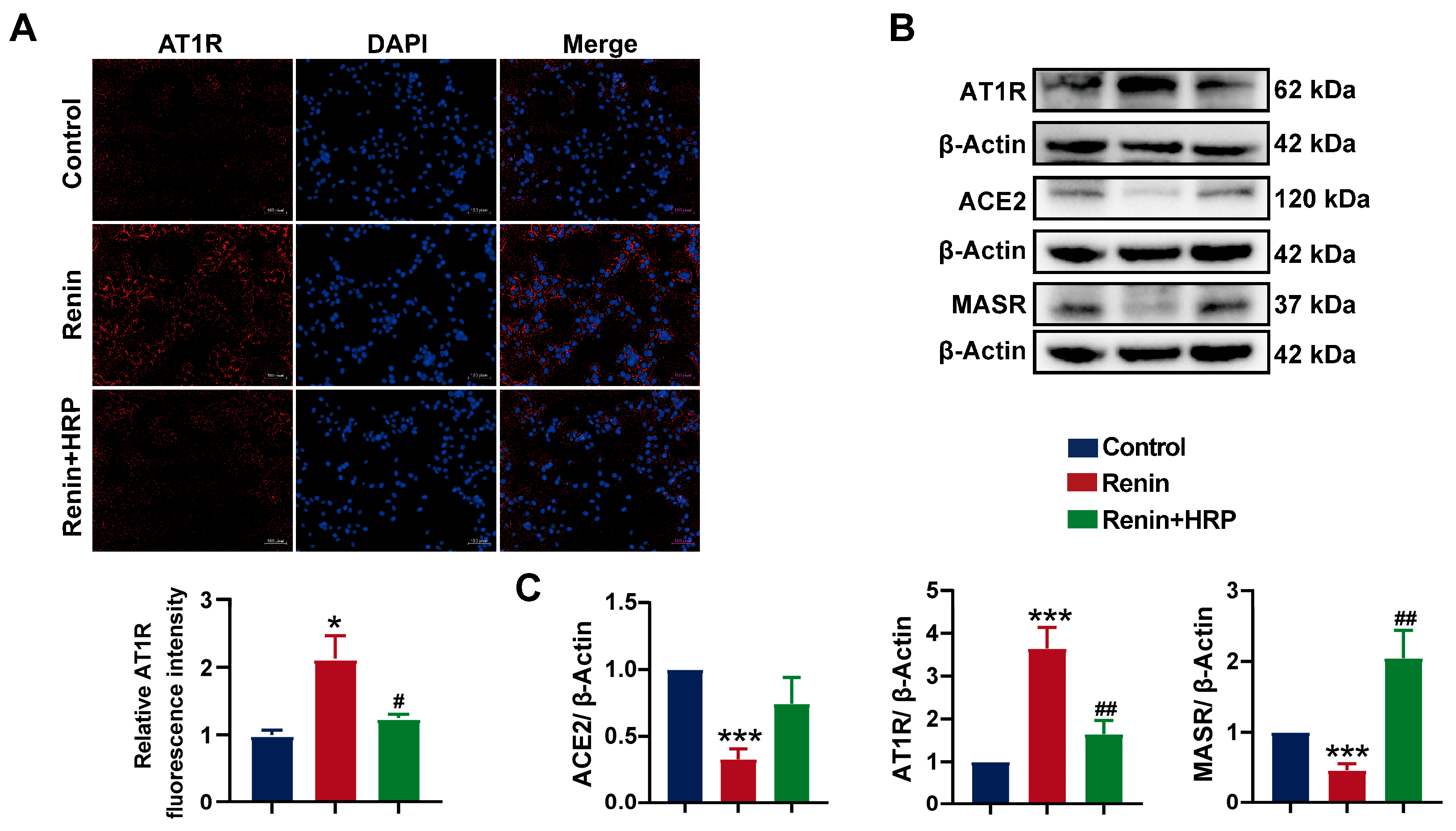
2.7. Involvement of the (P)RR/ERK/PPARγ Pathway in Hypertension Combined with MAFLD
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Measurement of Blood Pressure
4.3. Histology Examination
4.4. Biochemical Analysis
4.5. RNA Sequencing and Data Analysis
4.6. Cell Viability Assay and Renin-Induced HepG2 Cells
4.7. Nile Red Fluorescent Staining
4.8. Immunofluorescence and Nuclear Staining
4.9. Western Blot (WB)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Yuan, M.; He, J.; Hu, X.; Yao, L.; Chen, P.; Wang, Z.; Liu, P.; Xiong, Z.; Jiang, Y.; Li, L. Hypertension and NAFLD risk: Insights from the NHANES 2017–2018 and Mendelian randomization analyses. Chin. Med. J. 2024, 137, 457–464. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Ekstedt, M.; Wong, G.L.-H.; Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yin, X.; Liu, Z.; Wang, J. Non-Alcoholic Fatty Liver Disease (NAFLD) Pathogenesis and Natural Products for Prevention and Treatment. Int. J. Mol. Sci. 2022, 23, 15489. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef]
- Rong, L.; Zou, J.; Ran, W.; Qi, X.; Chen, Y.; Cui, H.; Guo, J. Advancements in the treatment of non-alcoholic fatty liver disease (NAFLD). Front. Endocrinol. 2023, 13, 1087260. [Google Scholar] [CrossRef]
- Zeng, X.F.; Varady, K.A.; Wang, X.D.; Targher, G.; Byrne, C.D.; Tayyem, R.; Latella, G.; Bergheim, I.; Valenzuela, R.; George, J.; et al. The role of dietary modification in the prevention and management of metabolic dysfunction-associated fatty liver disease: An international multidisciplinary expert consensus. Metab. Clin. Exp. 2024, 161, 156028. [Google Scholar] [CrossRef]
- Huh, J.H.; Lee, K.J.; Lim, J.S.; Lee, M.Y.; Park, H.J.; Kim, M.Y.; Kim, J.W.; Chung, C.H.; Shin, J.Y.; Kim, H.-S.; et al. High Dietary Sodium Intake Assessed by Estimated 24-h Urinary Sodium Excretion Is Associated with NAFLD and Hepatic Fibrosis. PLoS ONE 2015, 10, e0143222. [Google Scholar] [CrossRef]
- Latea, L.; Negrea, S.; Bolboaca, S. Primary non-alcoholic fatty liver disease in hypertensive patients. Australas. Med. J. 2013, 6, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Monti, T.; Sala, I.; Grassi, G.; Mancia, G.; Perseghin, G. Nonalcoholic Fatty Liver Disease and Advanced Fibrosis in US Adults Across Blood Pressure Categories. Hypertension 2020, 76, 562–568. [Google Scholar] [CrossRef]
- Song, Q.; Ling, Q.; Fan, L.; Deng, Y.; Gao, Q.; Yang, R.; Chen, S.; Wu, S.; Cai, J. Severity of non-alcoholic fatty liver disease is a risk factor for developing hypertension from prehypertension. Chin. Med. J. 2023, 136, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Lan, J.; Cen, J.; Han, Y.; Hu, H. Association Between Hypertension and New-Onset Non-Alcoholic Fatty Liver Disease in Chinese Non-Obese People: A Longitudinal Cohort Study. Diabetes Metab. Syndr. Obes. Targets Ther. 2023, 16, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Rassler, B. The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans. Pharmaceuticals 2010, 3, 940–960. [Google Scholar] [CrossRef]
- Yang, T. Soluble (Pro)Renin Receptor in Hypertension. Nephron 2023, 147, 234–243. [Google Scholar] [CrossRef]
- Peng, H.; Wu, Z.B.; Kong, S.S.; Li, L. Role of (pro)rennin receptor in cardiomyocytes of heart failure rat model. J. Huazhong Univ. Sci. Technol. 2013, 33, 640–643. [Google Scholar] [CrossRef]
- Mastoor, Z.; Diz-Chaves, Y.; González-Matías, L.C.; Mallo, F. Renin–Angiotensin System in Liver Metabolism: Gender Differences and Role of Incretins. Metabolites 2022, 12, 411. [Google Scholar] [CrossRef]
- Hennrikus, M.; Gonzalez, A.A.; Prieto, M.C. The prorenin receptor in the cardiovascular system and beyond. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H139–H145. [Google Scholar] [CrossRef]
- Fan, X.; Li, X.; Li, J.; Zhang, Y.; Wei, X.; Hu, H.; Zhang, B.; Du, H.; Zhao, M.; Zhu, R.; et al. Polystyrene nanoplastics induce glycolipid metabolism disorder via NF-κB and MAPK signaling pathway in mice. J. Environ. Sci. 2024, 137, 553–566. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef]
- Reddy Sankaran, K.; Ganjayi, M.S.; Oruganti, L.; Chippada, A.R.; Meriga, B. A bioactive fraction of Pterocarpus santalinus inhibits adipogenesis and inflammation in 3T3-L1 cells via modulation of PPAR-γ/SREBP-1c and TNF-α/IL-6. 3 Biotech 2021, 11, 233. [Google Scholar] [CrossRef]
- Svoboda, D.S.; Kawaja, M.D. Changes in hepatic protein expression in spontaneously hypertensive rats suggest early stages of non-alcoholic fatty liver disease. J. Proteom. 2012, 75, 1752–1763. [Google Scholar] [CrossRef]
- Ren, L.; Sun, Y.; Lu, H.; Ye, D.; Han, L.; Wang, N.; Daugherty, A.; Li, F.; Wang, M.; Su, F.; et al. (Pro)renin Receptor Inhibition Reprograms Hepatic Lipid Metabolism and Protects Mice From Diet-Induced Obesity and Hepatosteatosis. Circ. Res. 2018, 122, 730–741. [Google Scholar] [CrossRef]
- Nguyen, G.; Contrepas, A. The (pro)renin receptors. J. Mol. Med. 2008, 86, 643–646. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, Y.; Zhang, Y.; Bian, Y.; Zeng, Y.; Yao, X.; Wan, J.; Chen, X.; Li, J.; et al. S100A4 enhances protumor macrophage polarization by control of PPAR-γ-dependent induction of fatty acid oxidation. J. Immunother. Cancer 2021, 9, e002548. [Google Scholar] [CrossRef]
- Ferré, P.; Phan, F.; Foufelle, F. SREBP-1c and lipogenesis in the liver: An update1. Biochem. J. 2021, 478, 3723–3739. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E855–E862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, L.; Cao, Y.Y.; Gao, X.; Targher, G.; Byrne, C.D.; Sun, D.Q.; Zheng, M.H. MAFLD as part of systemic metabolic dysregulation. Hepatol. Int. 2024, 18, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Kahl, S.; Pesta, D.; Mastrototaro, L.; Dewidar, B.; Strassburger, K.; Sabah, E.; Esposito, I.; Weiß, J.; Sarabhai, T.; et al. Impaired Hepatic Mitochondrial Capacity in Nonalcoholic Steatohepatitis Associated with Type 2 Diabetes. Diabetes Care 2022, 45, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Hliwa, A.; Ramos-Molina, B.; Laski, D.; Mika, A.; Sledzinski, T. The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update. Int. J. Mol. Sci. 2021, 22, 6900. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, Y.; Fu, Y.; Guo, G.; Zhang, X. Liver fatty acid composition in mice with or without nonalcoholic fatty liver disease. Lipids Health Dis. 2011, 10, 234. [Google Scholar] [CrossRef]
- Dzau, V.J.; Hodgkinson, C.P. Precision Hypertension. Hypertension 2024, 81, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Badmus, O.O.; Kipp, Z.A.; Bates, E.A.; da Silva, A.A.; Taylor, L.C.; Martinez, G.J.; Lee, W.H.; Creeden, J.F.; Hinds, T.D., Jr.; Stec, D.E. Loss of hepatic PPARα in mice causes hypertension and cardiovascular disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2023, 325, R81–R95. [Google Scholar] [CrossRef]
- Sun, L.; Chi, B.; Xia, M.; Ma, Z.; Zhang, H.; Jiang, H.; Zhang, F.; Tian, Z. LC-MS-based lipidomic analysis of liver tissue sample from spontaneously hypertensive rats treated with extract hawthorn fruits. Front. Pharmacol. 2022, 13, 963280. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, D.; Loncar, G.; Radenovic, S.; Tahirovic, E.; Heidecke, H.; Schulze-Forster, K.; Muller, D.; Busjahn, A.; Buttner, P.; Veskovic, J.; et al. Soluble (pro)renin receptor in elderly chronic heart failure patients. Front. Biosci. 2020, 25, 1839–1853. [Google Scholar] [CrossRef]
- Du, X.Y.; Xiang, D.C.; Gao, P.; Peng, H.; Liu, Y.L. Inhibition of (Pro)renin Receptor-Mediated Oxidative Stress Alleviates Doxorubicin-Induced Heart Failure. Front. Oncol. 2022, 12, 874852. [Google Scholar] [CrossRef]
- Hu, J.; Tan, Y.; Chen, Y.; Mo, S.; Hekking, B.; Su, J.; Pu, M.; Lu, A.; Du, Y.; Symons, J.D.; et al. Role of (pro)renin receptor in cyclosporin A-induced nephropathy. Am. J. Physiol. -Ren. Physiol. 2022, 322, F437–F448. [Google Scholar] [CrossRef]
- Xie, S.; Song, S.; Liu, S.; Li, Q.; Zou, W.; Ke, J.; Wang, C. (Pro)renin receptor mediates tubular epithelial cell pyroptosis in diabetic kidney disease via DPP4-JNK pathway. J. Transl. Med. 2024, 22, 26. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Lee, K.C.; Lei, H.J.; Lan, K.H.; Huo, T.I.; Lin, Y.T.; Chan, C.C.; Schnabl, B.; Huang, Y.H.; Hou, M.C.; et al. (Pro)renin Receptor Knockdown Attenuates Liver Fibrosis Through Inactivation of ERK/TGF-β1/SMAD3 Pathway. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 813–838. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-C.; Wu, P.-S.; Lin, Y.-T.; Huang, Y.-H.; Hou, M.-C.; Lee, K.-C.; Lin, H.-C. (Pro)renin receptor inhibition attenuated liver steatosis, inflammation, and fibrosis in mice with steatohepatitis. FASEB J. 2022, 36, e22526. [Google Scholar] [CrossRef]
- Gayban, A.J.B.; Souza, L.A.C.; Cooper, S.G.; Regalado, E.; Kleemann, R.; Feng Earley, Y. (Pro)Renin Receptor Antagonism Attenuates High-Fat-Diet–Induced Hepatic Steatosis. Biomolecules 2023, 13, 142. [Google Scholar] [CrossRef]
- Yang, T. Potential of soluble (pro)renin receptor in kidney disease: Can it go beyond a biomarker? Am. J. Physiol. Ren. Physiol. 2022, 323, F507–F514. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Delarue, F.; Burcklé, C.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Prusty, D.; Park, B.H.; Davis, K.E.; Farmer, S.R. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2002, 277, 46226–46232. [Google Scholar] [CrossRef]
- Wang, F.; Luo, R.; Zou, C.J.; Xie, S.; Peng, K.; Zhao, L.; Yang, K.T.; Xu, C.; Yang, T. Soluble (pro)renin receptor treats metabolic syndrome in mice with diet-induced obesity via interaction with PPARγ. J. Clin. Investig. 2020, 5, 128061. [Google Scholar] [CrossRef]
- Wang, R.; Ji, C.L.; Feng, D.D.; Wu, Y.J.; Li, Y.; Olatunji, O.J.; Yu, L.J.; Zuo, J. Consumption of Saturated Fatty Acids-Rich Lard Benefits Recovery of Experimental Arthritis by Activating PPAR-γ. Mol. Nutr. Food Res. 2023, 67, e2200429. [Google Scholar] [CrossRef]
- Chen, H.; Tan, H.; Wan, J.; Zeng, Y.; Wang, J.; Wang, H.; Lu, X. PPAR-γ signaling in nonalcoholic fatty liver disease: Pathogenesis and therapeutic targets. Pharmacol. Ther. 2023, 245, 108391. [Google Scholar] [CrossRef]
- Xu, Q.; Jensen, D.D.; Peng, H.; Feng, Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol. Ther. 2016, 164, 126–134. [Google Scholar] [CrossRef]
- Yoshida, A.; Kanamori, H.; Naruse, G.; Minatoguchi, S.; Iwasa, M.; Yamada, Y.; Mikami, A.; Kawasaki, M.; Nishigaki, K.; Minatoguchi, S. (Pro)renin Receptor Blockade Ameliorates Heart Failure Caused by Chronic Kidney Disease. J. Card. Fail. 2019, 25, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Ellmers, L.J.; Rademaker, M.T.; Charles, C.J.; Yandle, T.G.; Richards, A.M. (Pro)renin Receptor Blockade Ameliorates Cardiac Injury and Remodeling and Improves Function After Myocardial Infarction. J. Card. Fail. 2016, 22, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Mishima, S.; Mitsui, T.; Tani, K.; Ooba, H.; Mitoma, T.; Ohira, A.; Maki, J.; Kirino, S.; Eto, E.; Hayata, K.; et al. Endothelin-1 production via placental (pro)renin receptor in a mouse model of preeclampsia. Placenta 2023, 138, 44–50. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Guo, X.; Zhang, L.; Lin, X.; Sun, H. Overexpression of (P)RR in SHR and Renin-Induced HepG2 Cells Leads to Spontaneous Hypertension Combined with Metabolic Dysfunction-Associated Fatty Liver Disease. Int. J. Mol. Sci. 2025, 26, 6541. https://doi.org/10.3390/ijms26136541
Gao C, Guo X, Zhang L, Lin X, Sun H. Overexpression of (P)RR in SHR and Renin-Induced HepG2 Cells Leads to Spontaneous Hypertension Combined with Metabolic Dysfunction-Associated Fatty Liver Disease. International Journal of Molecular Sciences. 2025; 26(13):6541. https://doi.org/10.3390/ijms26136541
Chicago/Turabian StyleGao, Chen, Xinyi Guo, Lingzhi Zhang, Xueman Lin, and Hua Sun. 2025. "Overexpression of (P)RR in SHR and Renin-Induced HepG2 Cells Leads to Spontaneous Hypertension Combined with Metabolic Dysfunction-Associated Fatty Liver Disease" International Journal of Molecular Sciences 26, no. 13: 6541. https://doi.org/10.3390/ijms26136541
APA StyleGao, C., Guo, X., Zhang, L., Lin, X., & Sun, H. (2025). Overexpression of (P)RR in SHR and Renin-Induced HepG2 Cells Leads to Spontaneous Hypertension Combined with Metabolic Dysfunction-Associated Fatty Liver Disease. International Journal of Molecular Sciences, 26(13), 6541. https://doi.org/10.3390/ijms26136541