
Genetic Variants in the Extracellular Matrix Gene TNXB Predicted to Alter Fibronectin III Domains in Arterial Aneurysmal and Dissection Diseases

 , , ,
, , ,
Abstract
1. Introduction
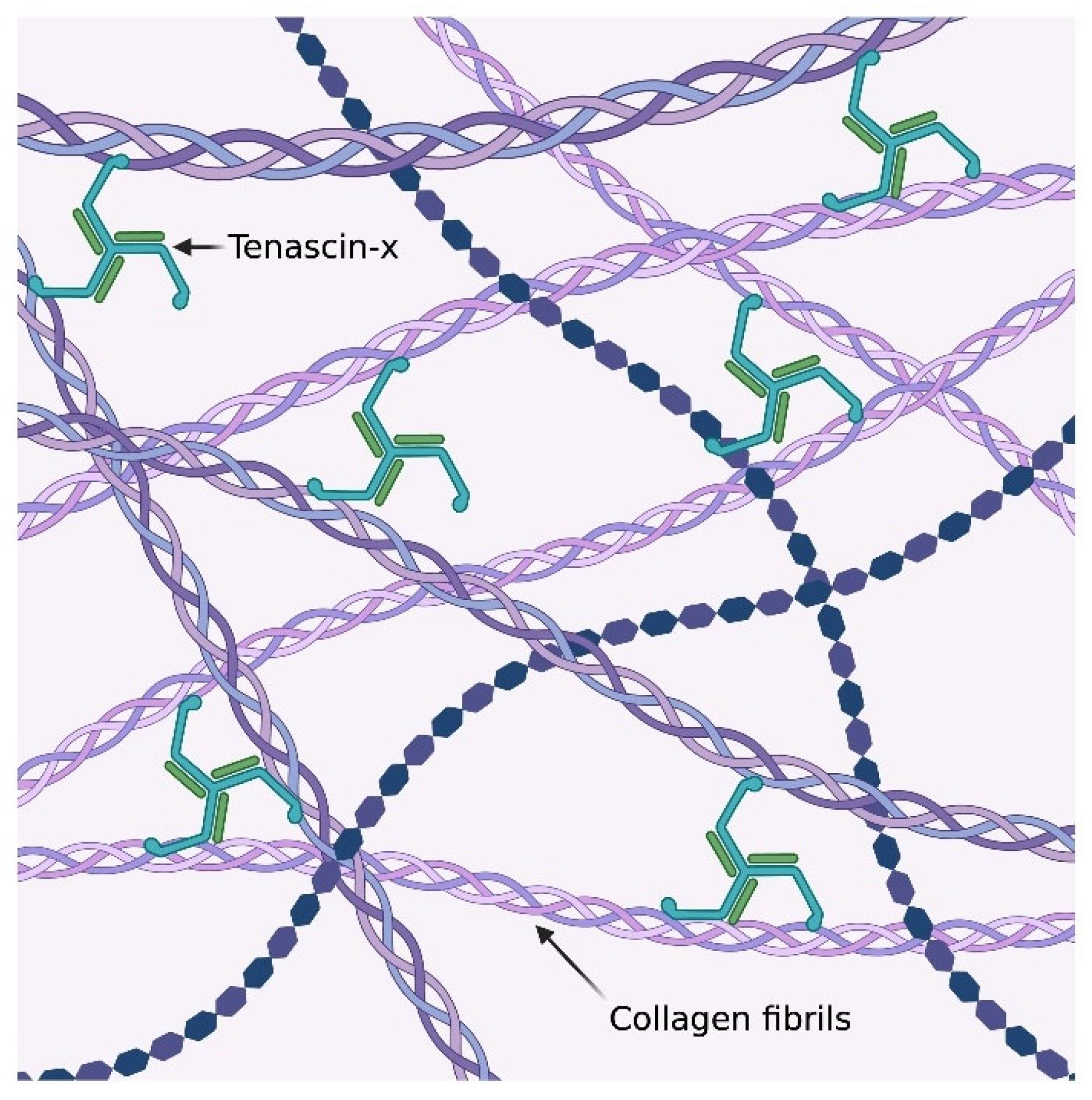
2. Results
2.1. Clinical Features
2.2. Genetic Results
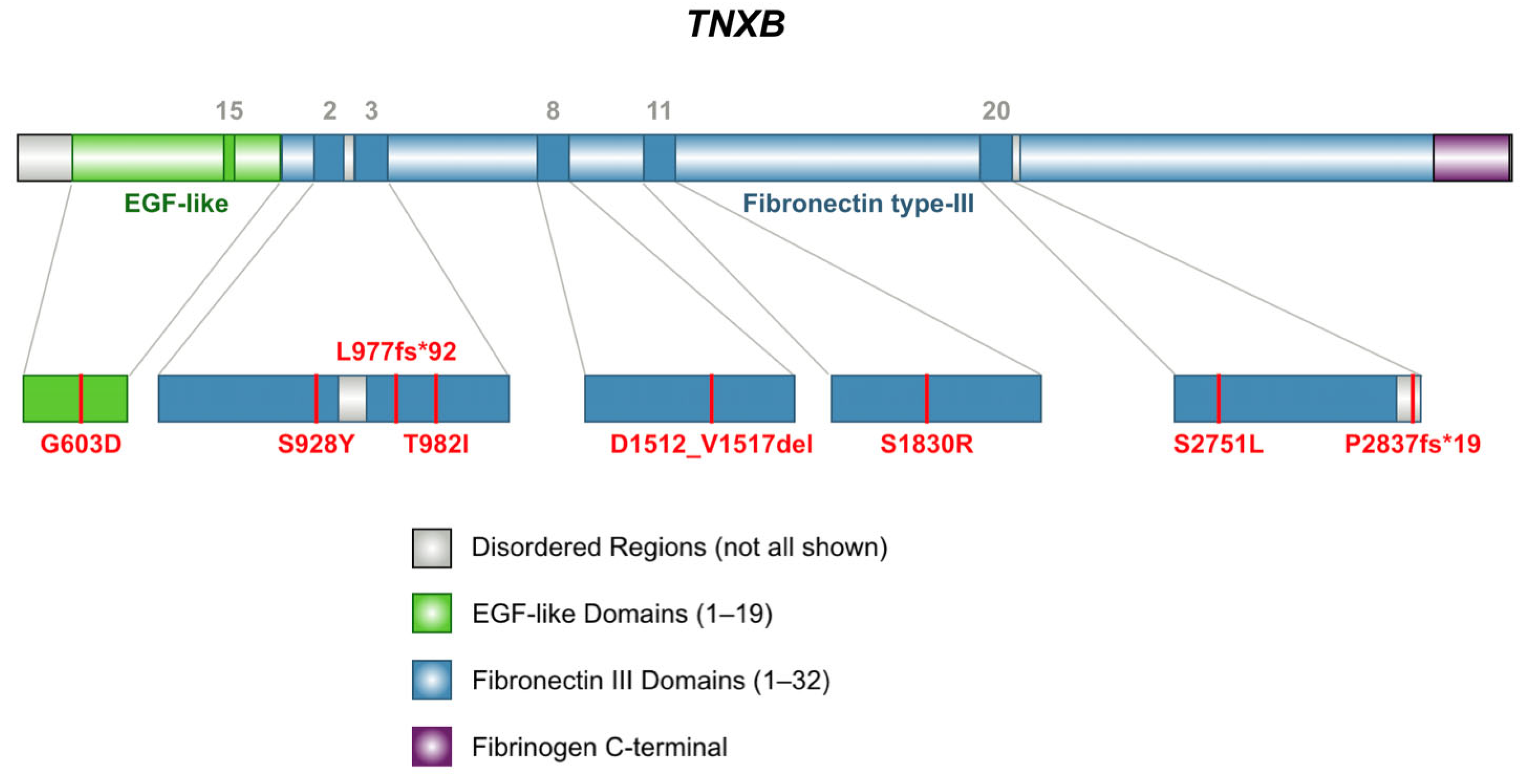
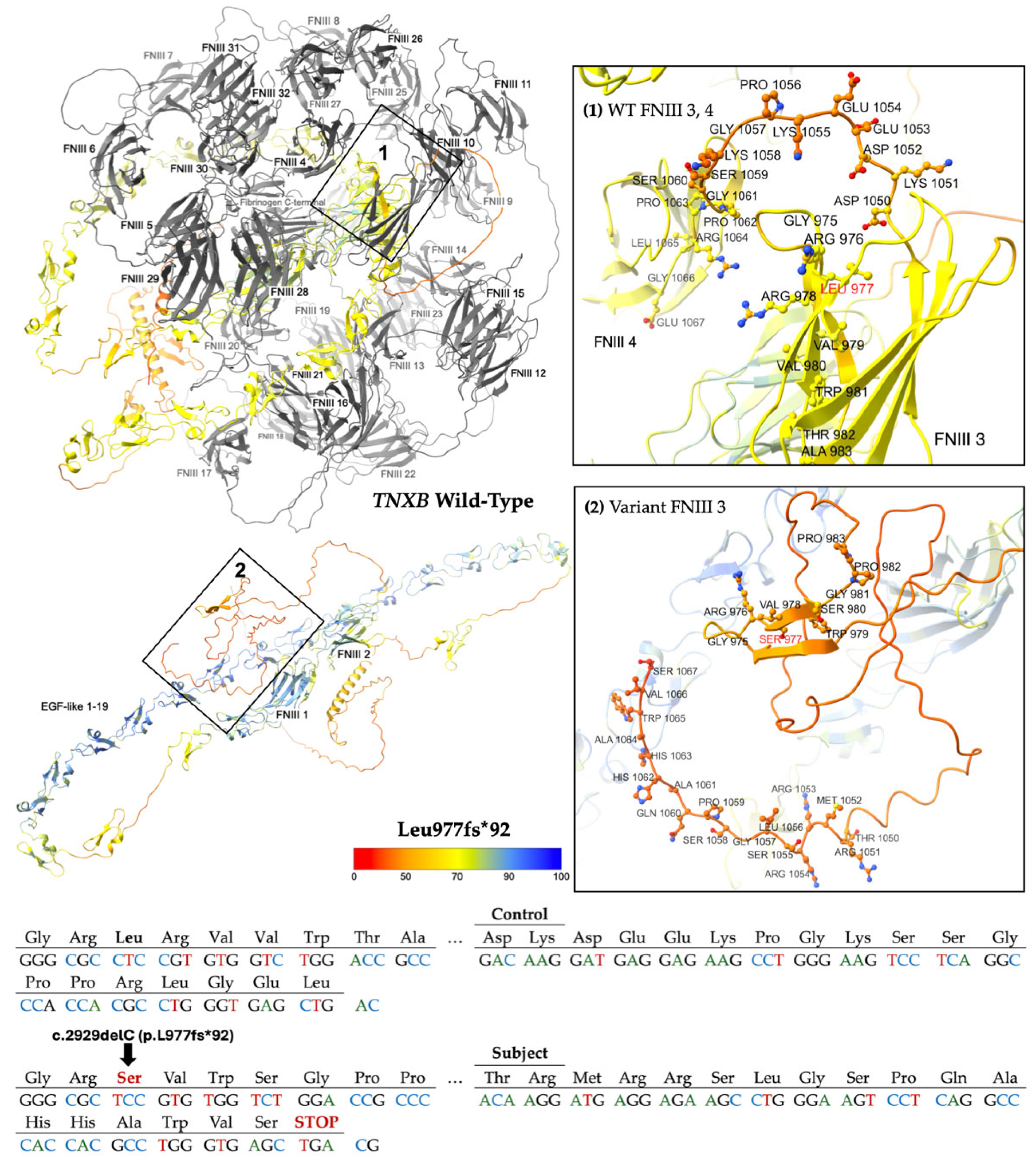
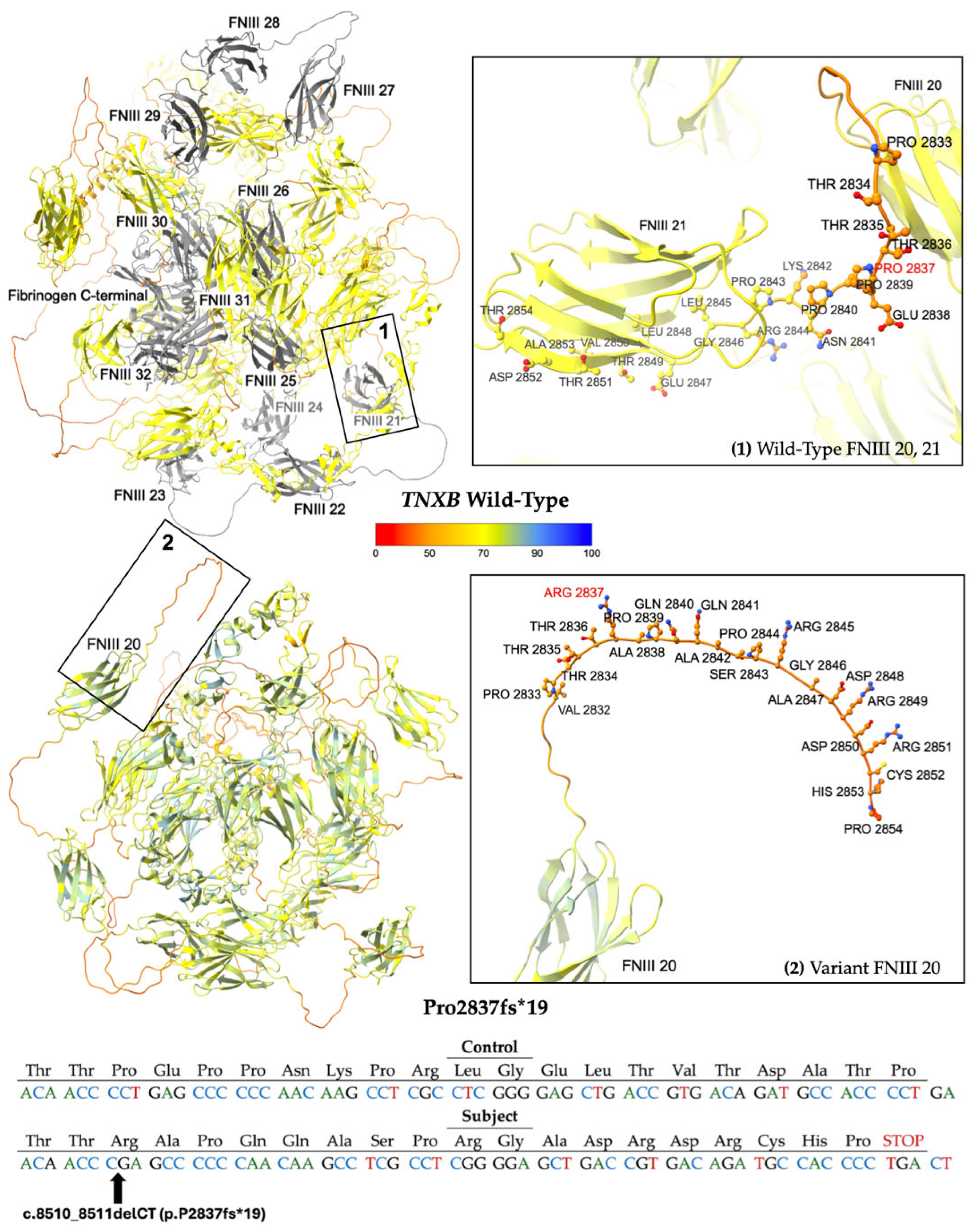
2.2.1. Frameshift Variants
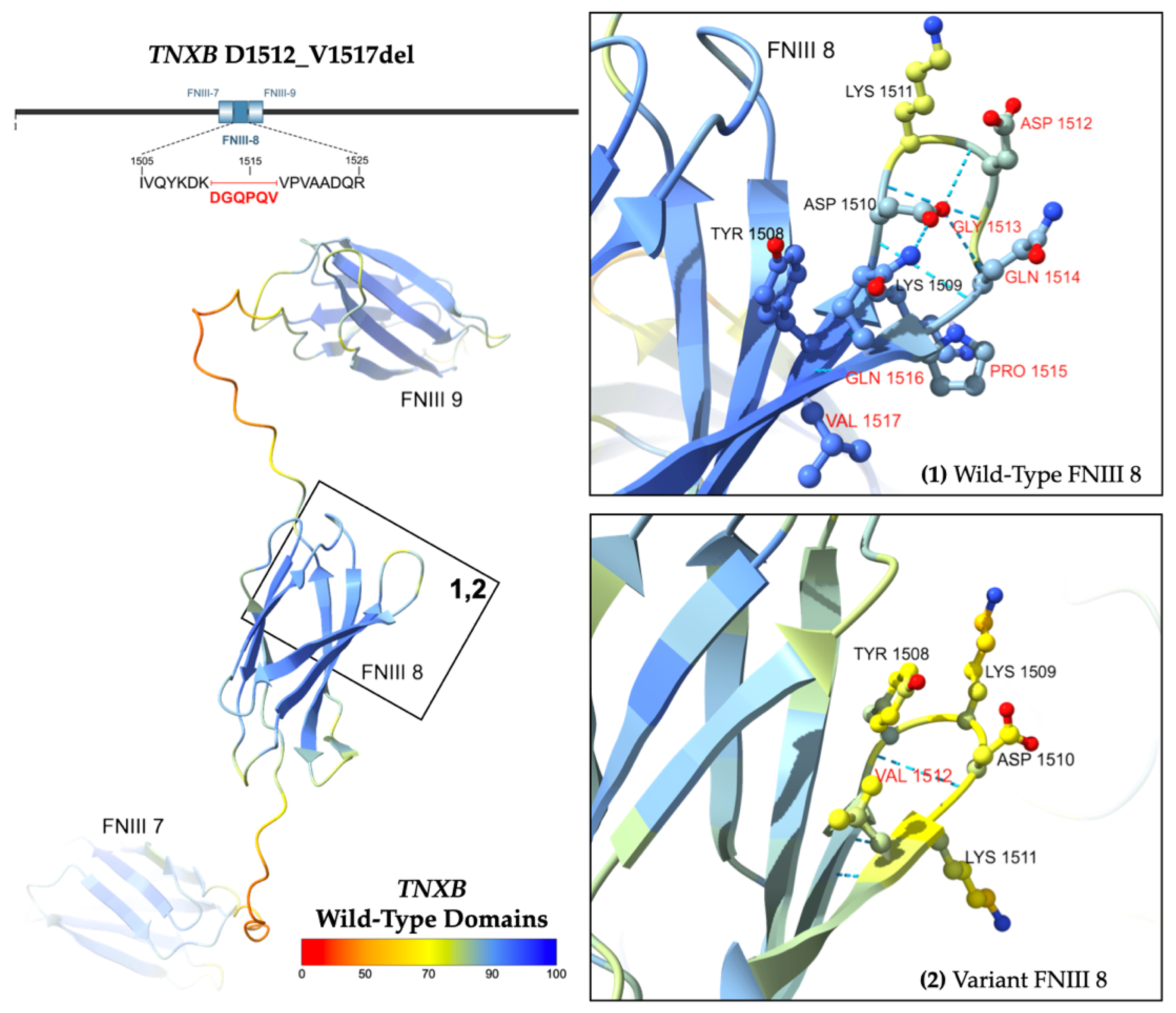
2.2.2. In-Frame Deletion Variant
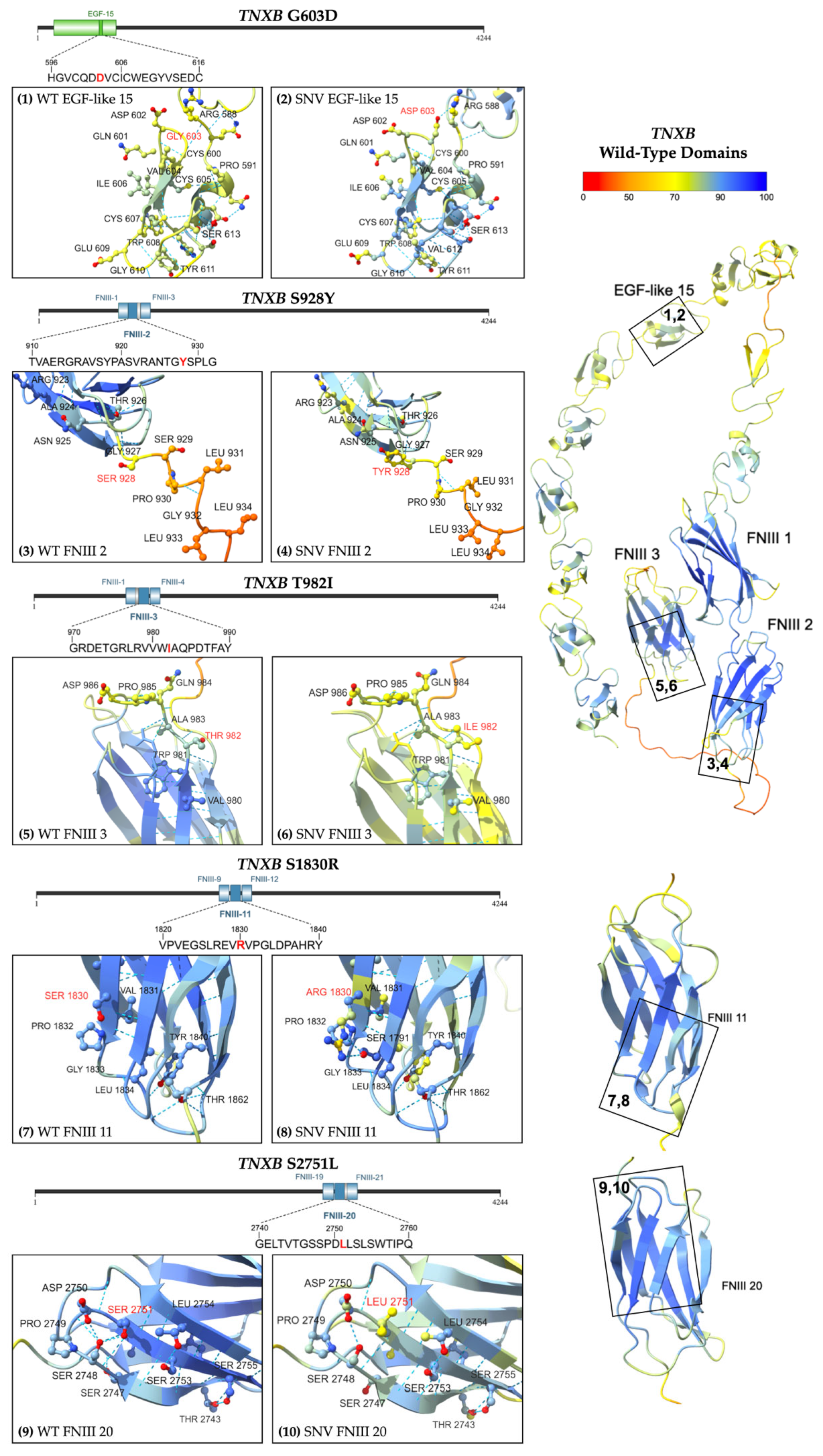
2.2.3. Missense Variants
3. Discussion
4. Materials and Methods
4.1. Next-Generation Sequencing Analysis
4.2. Variant Analysis and Computational Prediction
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bcharah, G.; Firth, C.E.; Abdou, M.M.; Ravi, S.N.; Ibrahim, R.; Pathangey, G.; Kumar, S.J.; Abdelnabi, M.; Wang, Y.; Osundiji, M.A.; et al. Gender- and Age-Based Differences in Nonsyndromic Arteriopathies in Younger Adults. Am. J. Cardiol. 2025, 239, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sakalihasan, N.; Michel, J.-B.; Katsargyris, A.; Kuivaniemi, H.; Defraigne, J.-O.; Nchimi, A.; Powell, J.T.; Yoshimura, K.; Hultgren, R. Abdominal aortic aneurysms. Nat. Rev. Dis. Primers 2018, 4, 34. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Preventza, O.; Black, J.H., III; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease. JACC 2022, 80, e223–e393. [Google Scholar] [CrossRef]
- Bossone, E.; Eagle, K.A. Epidemiology and management of aortic disease: Aortic aneurysms and acute aortic syndromes. Nat. Rev. Cardiol. 2021, 18, 331–348. [Google Scholar] [CrossRef]
- Hicks, K.L.; Byers, P.H.; Quiroga, E.; Pepin, M.G.; Shalhub, S. Testing patterns for genetically triggered aortic and arterial aneurysms and dissections at an academic center. J. Vasc. Surg. 2018, 68, 701–711. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef]
- Rega, S.; Farina, F.; Bouhuis, S.; de Donato, S.; Chiesa, M.; Poggio, P.; Cavallotti, L.; Bonalumi, G.; Giambuzzi, I.; Pompilio, G.; et al. Multi-omics in thoracic aortic aneurysm: The complex road to the simplification. Cell Biosci. 2023, 13, 131. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef]
- Valcourt, U.; Alcaraz, L.B.; Exposito, J.Y.; Lethias, C.; Bartholin, L. Tenascin-X: Beyond the architectural function. Cell Adhes. Migr. 2015, 9, 154–165. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.; Burrows, N.P.; Van Vlijmen-Willems, M.J.J.; Clark, S.M.; Schalkwijk, J. Tenascin-X deficiency and Ehlers–Danlos syndrome: A case report and review of the literature. Br. J. Dermatol. 2010, 163, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Matsumoto, K. Multiple Roles of Tenascins in Homeostasis and Pathophysiology of Aorta. Ann. Vasc. Dis. 2018, 11, 169–180. [Google Scholar] [CrossRef]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB Is Associated with Hypermobility Type of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef]
- Wortley, J. Created in BioRender. 2025. Available online: https://BioRender.com/03tl3gs (accessed on 27 May 2025).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Xie, Y.; Li, H.; Luo, X.; Gao, Q.; Zhang, L.; Teng, Y.; Zhao, Q.; Zuo, Z.; Ren, J. IBS 2.0: An upgraded illustrator for the visualization of biological sequences. Nucleic Acids Res. 2022, 50, W420–w426. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. VCV001797774.2-ClinVar-NCBI. 2025. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/1797774/ (accessed on 1 May 2025).
- National Center for Biotechnology Information. VCV001763690.2-ClinVar-NCBI. 2025. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/1763690/ (accessed on 15 May 2025).
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Tokhmafshan, F.; El Andalousi, J.; Murugapoopathy, V.; Fillion, M.L.; Campillo, S.; Capolicchio, J.P.; Jednak, R.; El Sherbiny, M.; Turpin, S.; Schalkwijk, J.; et al. Children with vesicoureteric reflux have joint hypermobility and occasional tenascin XB sequence variants. Can. Urol. Assoc. J. 2020, 14, E128–E136. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Sundaram, L.; Gao, H.; Padigepati, S.R.; McRae, J.F.; Li, Y.; Kosmicki, J.A.; Fritzilas, N.; Hakenberg, J.; Dutta, A.; Shon, J.; et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018, 50, 1161–1170. [Google Scholar] [CrossRef]
- Feng, B.J. PERCH: A Unified Framework for Disease Gene Prioritization. Hum. Mutat. 2017, 38, 243–251. [Google Scholar] [CrossRef]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024, 52, D1143–D1154. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Salmasi, M.Y.; Alwis, S.; Cyclewala, S.; Jarral, O.A.; Mohamed, H.; Mozalbat, D.; Nienaber, C.A.; Athanasiou, T.; Morris-Rosendahl, D.; Moore, J., Jr.; et al. The genetic basis of thoracic aortic disease: The future of aneurysm classification? Hell. J. Cardiol. 2023, 69, 41–50. [Google Scholar] [CrossRef]
- Miller, W.L. Tenascin-X—Discovery and Early Research. Front. Immunol. 2021, 11, 612497. [Google Scholar] [CrossRef]
- Lethias, C.; Carisey, A.; Comte, J.; Cluzel, C.; Exposito, J.Y. A model of tenascin-X integration within the collagenous network. FEBS Lett. 2006, 580, 6281–6285. [Google Scholar] [CrossRef]
- Liang, G.; Wang, S.; Shao, J.; Jin, Y.J.; Xu, L.; Yan, Y.; Günther, S.; Wang, L.; Offermanns, S. Tenascin-X Mediates Flow-Induced Suppression of EndMT and Atherosclerosis. Circ. Res. 2022, 130, 1647–1659. [Google Scholar] [CrossRef]
- Alcaraz, L.B.; Exposito, J.Y.; Chuvin, N.; Pommier, R.M.; Cluzel, C.; Martel, S.; Sentis, S.; Bartholin, L.; Lethias, C.; Valcourt, U. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J. Cell Biol. 2014, 205, 409–428. [Google Scholar] [CrossRef]
- Burch, G.H.; Bedolli, M.A.; McDonough, S.; Rosenthal, S.M.; Bristow, J. Embryonic expression of tenascin-X suggests a role in limb, muscle, and heart development. Dev. Dyn. 1995, 203, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Matsumoto, K.; Hara, M.; Sakakura, T.; Yoshida, T. The dynamic expression of tenascin-C and tenascin-X during early heart development in the mouse. Differentiation 2003, 71, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Aoki, H. The Roles of Tenascins in Cardiovascular, Inflammatory, and Heritable Connective Tissue Diseases. Front. Immunol. 2020, 11, 609752. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Merke, D.P. Tenascin-X, Congenital Adrenal Hyperplasia, and the CAH-X Syndrome. Horm. Res. Paediatr. 2018, 89, 352–361. [Google Scholar] [CrossRef]
- Neogi, A.; Towne, M.; Dykas, D.; Parsa, N.; Attar, A.; Fathzadeh, M.; Bale, A.; Mani, A. Abstract 554: The Association of Multiple Variants In the TNXB Gene With Vascular Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2022, 42, A554. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef]
- Mansoorshahi, S.; Yetman, A.T.; Bissell, M.M.; Kim, Y.Y.; Michelena, H.I.; De Backer, J.; Mosquera, L.M.; Hui, D.S.; Caffarelli, A.; Andreassi, M.G.; et al. Whole-exome sequencing uncovers the genetic complexity of bicuspid aortic valve in families with early-onset complications. Am. J. Hum. Genet. 2024, 111, 2219–2231. [Google Scholar] [CrossRef]
- Körfer, D.; Grond-Ginsbach, C.; Peters, A.S.; Burkart, S.; Hempel, M.; Schaaf, C.P.; Böckler, D.; Erhart, P. Genetic variants in patients with multiple arterial aneurysms. Langenbecks Arch. Surg. 2024, 409, 304. [Google Scholar] [CrossRef]
- Satoh, K.; Tsukamoto, M.; Shindoh, M.; Totsuka, Y.; Oda, T.; Matsumoto, K. Increased expression of tenascin-x in thoracic and abdominal aortic aneurysm tissues. Biol. Pharm. Bull. 2010, 33, 1898–1902. [Google Scholar] [CrossRef]
- Zweers, M.C.; Peeters, A.C.T.M.; Graafsma, S.; Kranendonk, S.; van der Vliet, J.A.; den Heijer, M.; Schalkwijk, J. Abdominal Aortic Aneurysm Is Associated With High Serum Levels of Tenascin-X and Decreased Aneurysmal Tissue Tenascin-X. Circulation 2006, 113, 1702–1707. [Google Scholar] [CrossRef]
- van Dijk, F.S.; Ghali, N.; Demirdas, S.; Baker, D. TNXB-Related Classical-Like Ehlers-Danlos Syndrome. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef]
- Merke, D.P.; Chen, W.; Morissette, R.; Xu, Z.; Van Ryzin, C.; Sachdev, V.; Hannoush, H.; Shanbhag, S.M.; Acevedo, A.T.; Nishitani, M.; et al. Tenascin-X Haploinsufficiency Associated with Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, E379–E387. [Google Scholar] [CrossRef]
- Cho, M.J.; Lee, M.R.; Park, J.G. Aortic aneurysms: Current pathogenesis and therapeutic targets. Exp. Mol. Med. 2023, 55, 2519–2530. [Google Scholar] [CrossRef]
- Minamitani, T.; Ikuta, T.; Saito, Y.; Takebe, G.; Sato, M.; Sawa, H.; Nishimura, T.; Nakamura, F.; Takahashi, K.; Ariga, H.; et al. Modulation of collagen fibrillogenesis by tenascin-X and type VI collagen. Exp. Cell Res. 2004, 298, 305–315. [Google Scholar] [CrossRef]
- Mao, J.R.; Taylor, G.; Dean, W.B.; Wagner, D.R.; Afzal, V.; Lotz, J.C.; Rubin, E.M.; Bristow, J. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 2002, 30, 421–425. [Google Scholar] [CrossRef]
- Margaron, Y.; Bostan, L.; Exposito, J.Y.; Malbouyres, M.; Trunfio-Sfarghiu, A.M.; Berthier, Y.; Lethias, C. Tenascin-X increases the stiffness of collagen gels without affecting fibrillogenesis. Biophys. Chem. 2010, 147, 87–91. [Google Scholar] [CrossRef]
- Liang, G.; Lv, X.F.; Huang, W.; Jin, Y.J.; Roquid, K.A.; Kawase, H.; Offermanns, S. Loss of Smooth Muscle Tenascin-X Inhibits Vascular Remodeling Through Increased TGF-β Signaling. Arter. Thromb. Vasc. Biol. 2024, 44, 1748–1763. [Google Scholar] [CrossRef]
- Clarke, J.; Hamill, S.J.; Johnson, C.M. Folding and stability of a fibronectin type III domain of human tenascin. J. Mol. Biol. 1997, 270, 771–778. [Google Scholar] [CrossRef]
- Boelman, M.B.; Hansen, T.V.O.; Smith, M.N.; Hammer-Hansen, S.; Christensen, A.H.; Diness, B.R. Aortic dissection in a young male with persistent ductus arteriosus and a novel variant in MYLK. Am. J. Med. Genet. A 2024, 194, e63458. [Google Scholar] [CrossRef]
- Granzier, H.L.; Labeit, S. Discovery of Titin and Its Role in Heart Function and Disease. Circ. Res. 2025, 136, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.; Nikoopour, R.; Alexandrovich, A.; Pfuhl, M.; Lopes, L.R.; Akhtar, M.M.; Syrris, P.; Elliott, P.; Carr-White, G.; Gautel, M. Structure determination and analysis of titin A-band fibronectin type III domains provides insights for disease-linked variants and protein oligomerisation. J. Struct. Biol. 2023, 215, 108009. [Google Scholar] [CrossRef]
- Ambry Genetics. TAADNext: Analyses of 35 Genes Associated with Thoracic Aortic Aneurysms and Dissections. Available online: https://www.ambrygen.com/providers/genetic-testing/12/cardiology/taadnext (accessed on 15 May 2025).
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [Google Scholar] [CrossRef]
- Garcia, F.A.d.O.; Andrade, E.S.d.; Palmero, E.I. Insights on variant analysis in silico tools for pathogenicity prediction. Front. Genet. 2022, 13, 1010327. [Google Scholar] [CrossRef]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 2024, 53, D1243–D1249. [Google Scholar] [CrossRef]
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]
- Nucleotide. Homo Sapiens Tenascin XB (TNXB), Transcript Variant XB, mRNA. 2025. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NM_019105.8 (accessed on 27 December 2024).
- Nucleotide. Homo sapiens tenascin XB (TNXB), Transcript Variant 3, mRNA. 2025. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NM_001365276.2 (accessed on 1 May 2025).
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef]
- Brandes, N.; Goldman, G.; Wang, C.H.; Ye, C.J.; Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 2023, 55, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Zhozhikov, L.; Vasilev, F.; Maksimova, N. Protein-Variant-Phenotype Study of NBAS Using AlphaFold in the Aspect of SOPH Syndrome. Proteins 2025, 93, 871–884. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Variant | Age Sex | PMHx | Clinical Features | CT Angiography | Transthoracic Echocardiogram |
|---|---|---|---|---|---|---|
| 1 | p.L977fs*92 | 48 M | HTN | Asymptomatic | 45 mm ascending aortic aneurysm | Normal structure, function. EF > 60% |
| 2 | p.2837fs*19 | 61 F | Stroke, HTN | Dizziness, tinnitus | 4.1 × 5.2 mm right paraclinoid internal carotid aneurysm with a 3.8 mm neck | EF > 60% |
| 3 | p.D1512_V1517Del | 57 F | HTN | Asymptomatic | 42 mm ascending aortic aneurysm | Normal structure, function. EF > 60% |
| 4 | p.G603D | 40 M | BAV s/p AVR for severe AI | Asymptomatic | 43 mm ascending aortic aneurysm | Mechanical AVR, EF > 60% |
| 5 | p.S928Y | 72 M | Splenic aneurysm and diffuse AVMs, HTN | GI bleeds | Splenic aneurysm, diffuse AVMs | Normal structure, function. |
| EF > 60% | ||||||
| 6 | p.T982I | 58 F | HTN | Asymptomatic | 44 mm ascending aortic aneurysm | Mild AI, |
| EF > 60% | ||||||
| 7 | p.S1830R | 90 M | HTN | Ascending aortic aneurysm repair and AVR | 51 mm ascending aortic aneurysm | Severe AI |
| EF> 60% | ||||||
| 8 | p.S1830R | 64 M | HTN | Asymptomatic | 43 mm fusiform ascending aortic aneurysm | Normal structure, function. EF > 60% |
| 9 | p.S2751L | 53 M | Ascending aortic dissection (>55 mm) with severe AI, HTN | Symptomatic dissection | Outside historical data | Mechanical AVR, EF > 60% |
| Tool | c.1808G>A, p.G603D | c.2783C>A, p.S928Y | c.2945C>T, p.T982I | c.5488A>C, p.S1830R | c.8252C>T p.S2751L |
|---|---|---|---|---|---|
| Align GVGD | 93.77 Grade: C65 | 143.11 Grade: C65 | 89.28 Grade: C65 | 109.21 Grade: C65 | 144.08 Grade: C65 |
| BayesDel (MaxAF) | 0.20017 deleterious | −0.455478 tolerated | −0.483146 tolerated | −0.517306 tolerated | −0.068 tolerated |
| CADD v1.7 | 26.4 1% most deleterious | 23.0 1% most deleterious | 24.2 1% most deleterious | 11.57 10% most deleterious | 25.5 1% most deleterious |
| Evolutionary Action (EA) | 43.08 neutral | 60.74 neutral | 54.27 neutral | 3.58 more neutral | 96.08 deleterious |
| FATHMM | 2.51 tolerated | 0.35 tolerated | 3.49 tolerated | 0.42 tolerated | −0.04 tolerated |
| GERP++ | 4.26 constrained | 4.03 constrained | 1.98 constrained | −2.02 unconstrained | 5.0 constrained |
| MutPred2 | 0.869 pathogenic | 0.269 benign | 0.431 benign | 0.218 benign | 0.51 pathogenic |
| PhyloP | 7.49995 conserved | 1.58957 conserved | 1.48181 conserved | −0.120591 fast-evolving | 4.417 conserved |
| PolyPhen-2 | 1.0 probably damaging | 0.852 possibly damaging | 0.999 probably damaging | 0.012 benign | 1.0 probably damaging |
| PrimateAI | 0.7044 uncertain | 0.6351 uncertain | 0.5129 tolerated | 0.3487 tolerated | 0.4389 tolerated |
| REVEL | 0.484 uncertain | 0.12 benign | 0.104 benign | 0.025 benign | 0.488 uncertain |
| SIFT | 0.00 deleterious | 0.40 tolerated | 0.16 tolerated | 0.44 tolerated | 0.01 deleterious |
| VEST4 | 0.566, p = 0.146 pathogenic | 0.363, p = 0.271 benign | 0.426, p = 0.225 benign | 0.175, p = 0.58 benign | 0.356, p = 0.279 benign |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norgan Radler, C.; Wang, T.; LeGate, J.; Crone, L.; Deo, P.; Wortley, J.; Moore, P.; Bryant, G.; Smitherman, K.; Sathyamoorthy, M. Genetic Variants in the Extracellular Matrix Gene TNXB Predicted to Alter Fibronectin III Domains in Arterial Aneurysmal and Dissection Diseases. Int. J. Mol. Sci. 2025, 26, 6535. https://doi.org/10.3390/ijms26136535
Norgan Radler C, Wang T, LeGate J, Crone L, Deo P, Wortley J, Moore P, Bryant G, Smitherman K, Sathyamoorthy M. Genetic Variants in the Extracellular Matrix Gene TNXB Predicted to Alter Fibronectin III Domains in Arterial Aneurysmal and Dissection Diseases. International Journal of Molecular Sciences. 2025; 26(13):6535. https://doi.org/10.3390/ijms26136535
Chicago/Turabian StyleNorgan Radler, Charlene, Tianci Wang, Jaden LeGate, Lily Crone, Parminder Deo, Jacob Wortley, Peyton Moore, Griffin Bryant, Katherine Smitherman, and Mohanakrishnan Sathyamoorthy. 2025. "Genetic Variants in the Extracellular Matrix Gene TNXB Predicted to Alter Fibronectin III Domains in Arterial Aneurysmal and Dissection Diseases" International Journal of Molecular Sciences 26, no. 13: 6535. https://doi.org/10.3390/ijms26136535
APA StyleNorgan Radler, C., Wang, T., LeGate, J., Crone, L., Deo, P., Wortley, J., Moore, P., Bryant, G., Smitherman, K., & Sathyamoorthy, M. (2025). Genetic Variants in the Extracellular Matrix Gene TNXB Predicted to Alter Fibronectin III Domains in Arterial Aneurysmal and Dissection Diseases. International Journal of Molecular Sciences, 26(13), 6535. https://doi.org/10.3390/ijms26136535