
Molecular Hybrids of Thiazolidinone: Bridging Redox Modulation and Cancer Therapy

 , , ,
, , ,
Abstract

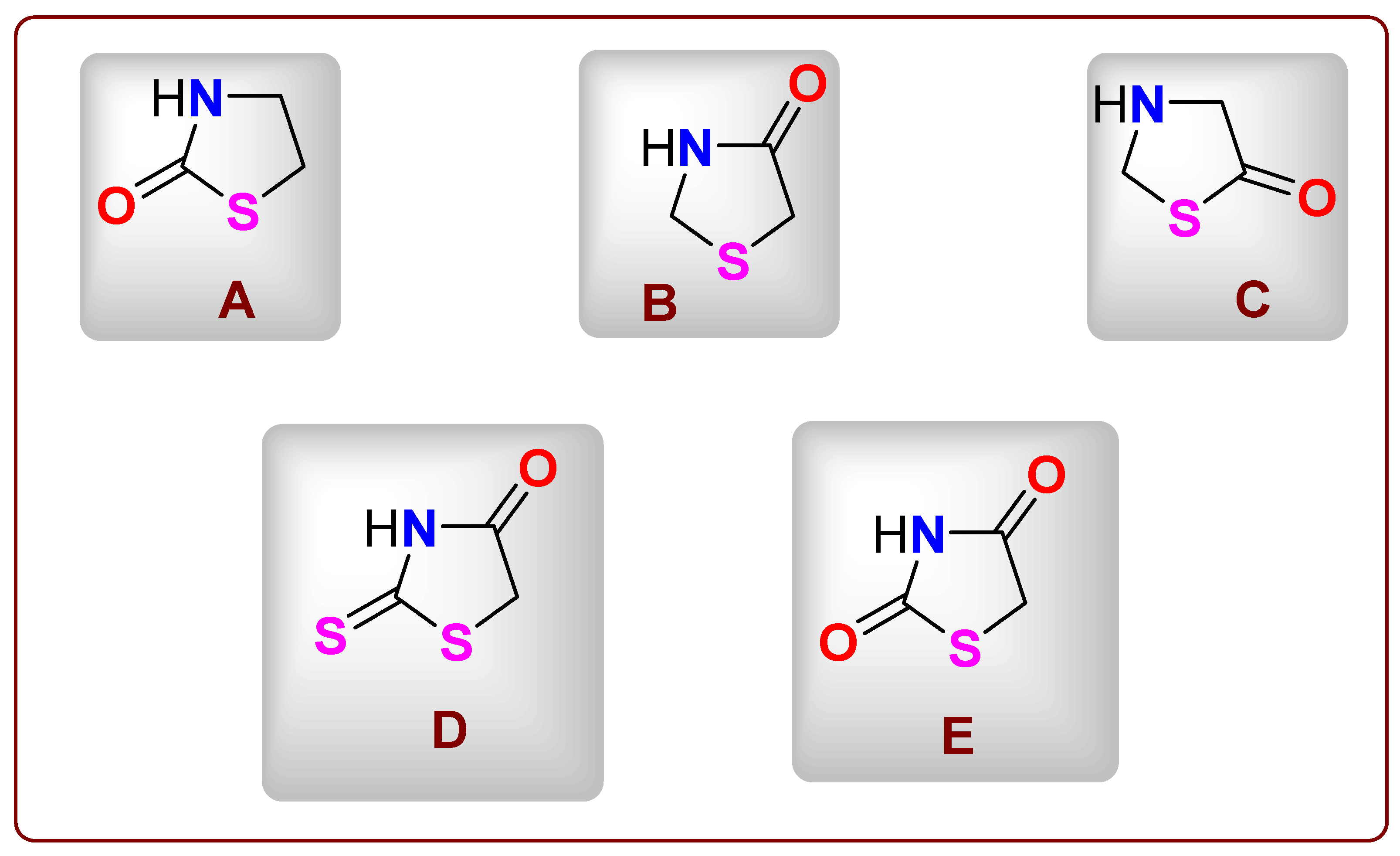
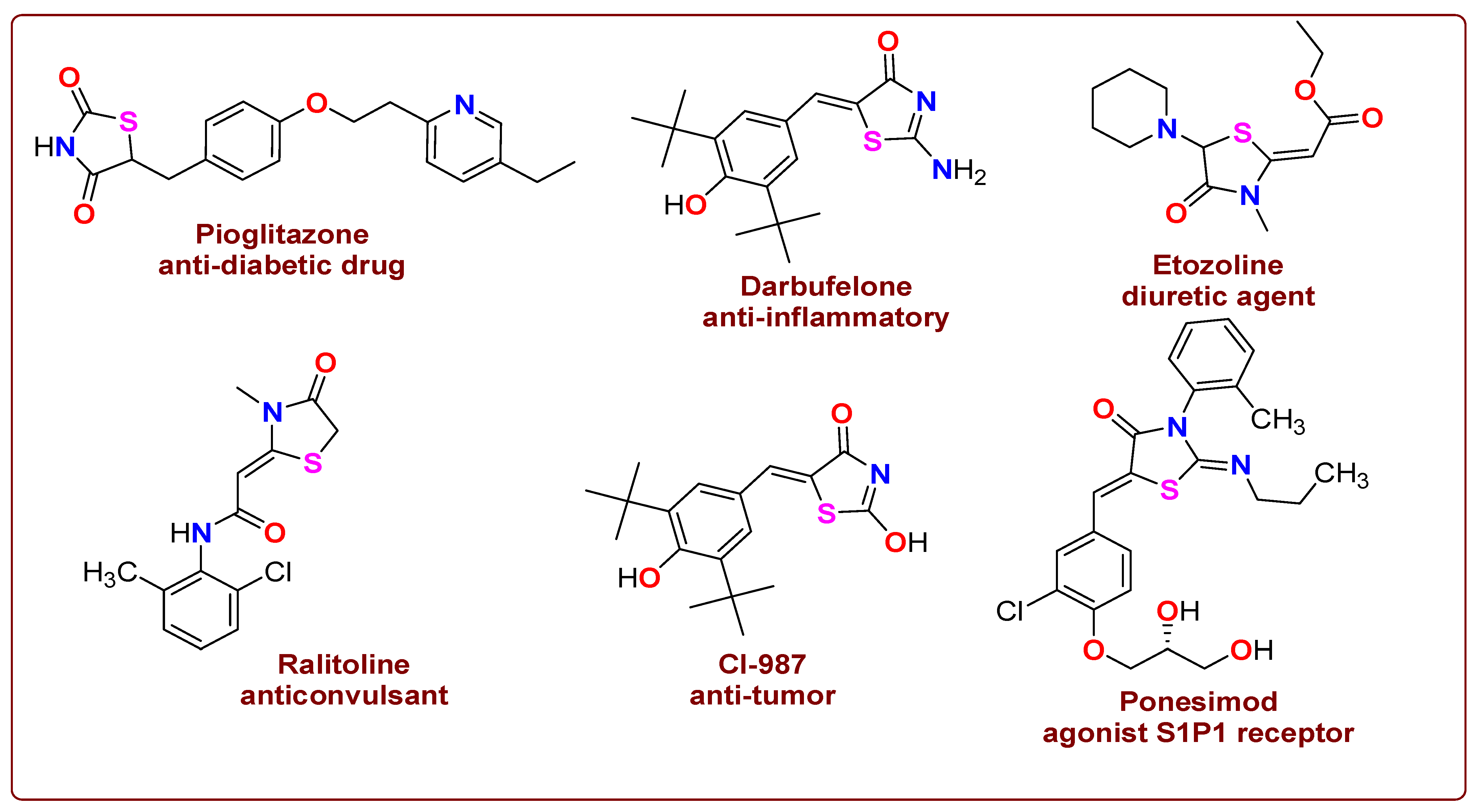
1. Introduction
2. Results and Discussion
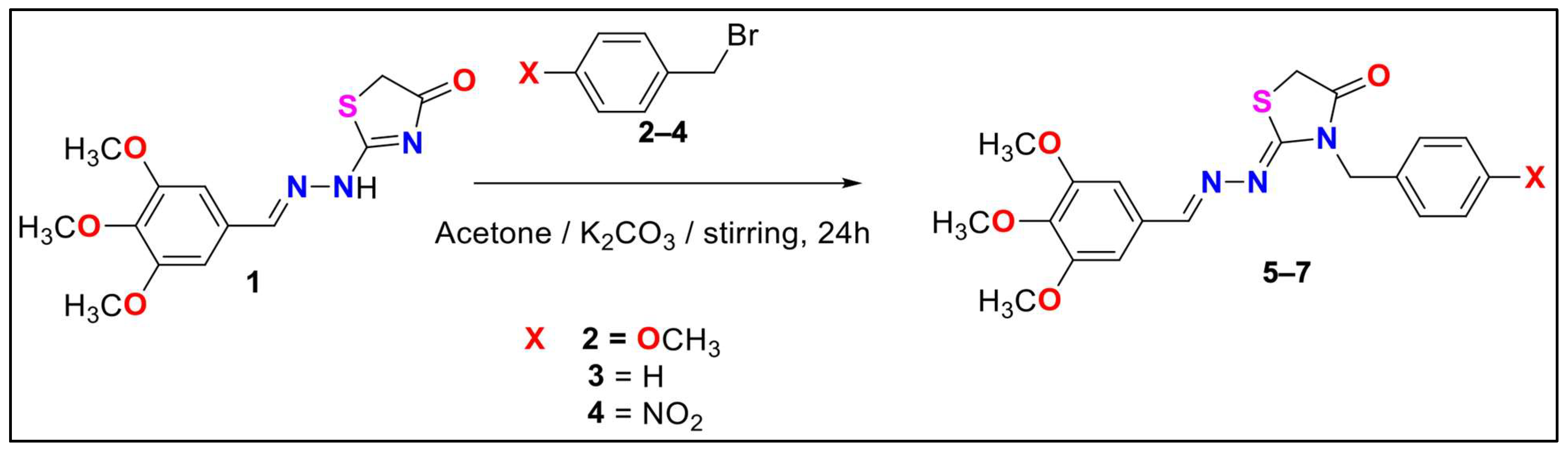
2.1. Chemical Profile of Thiazolidinone Derivative
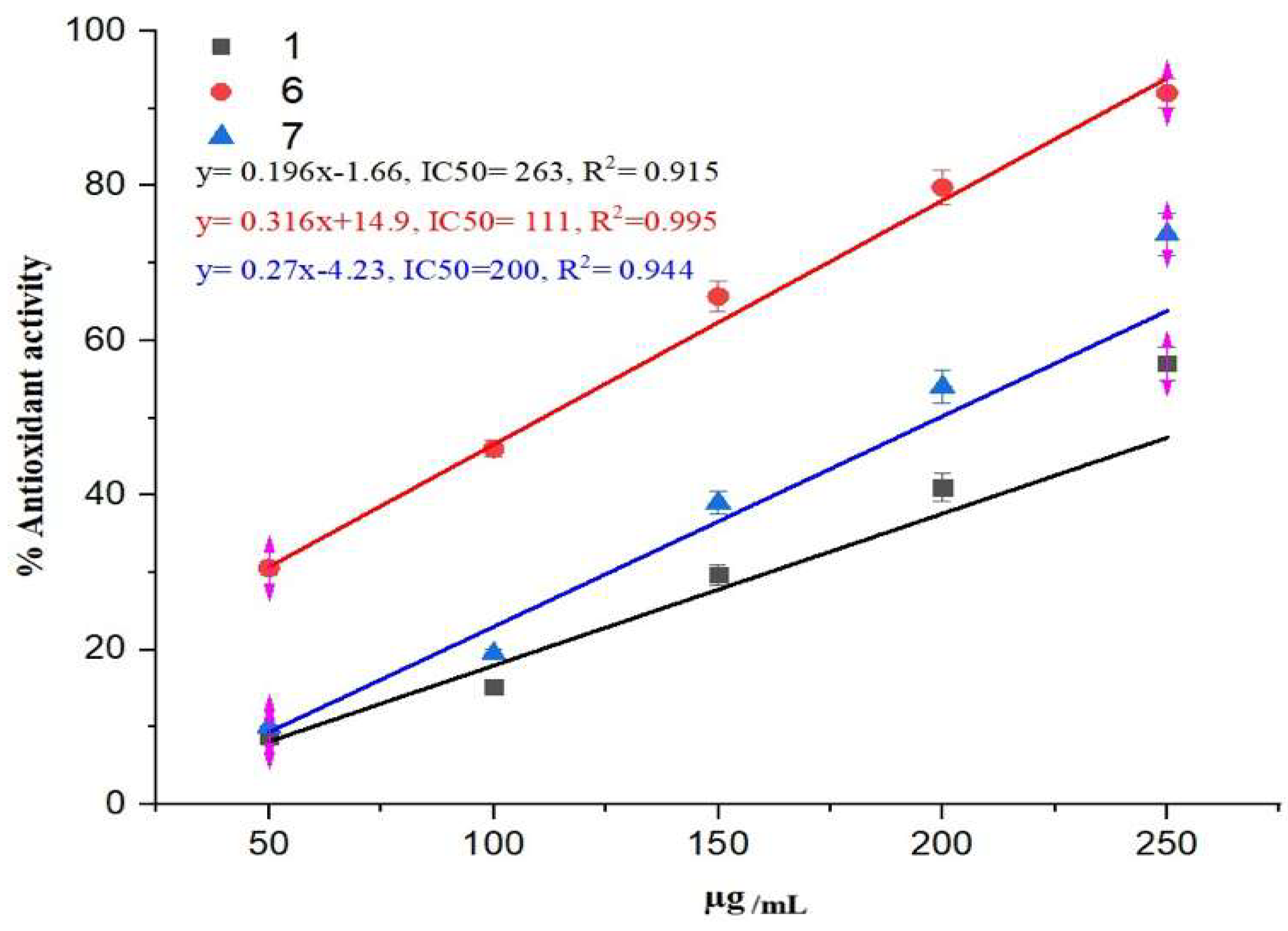
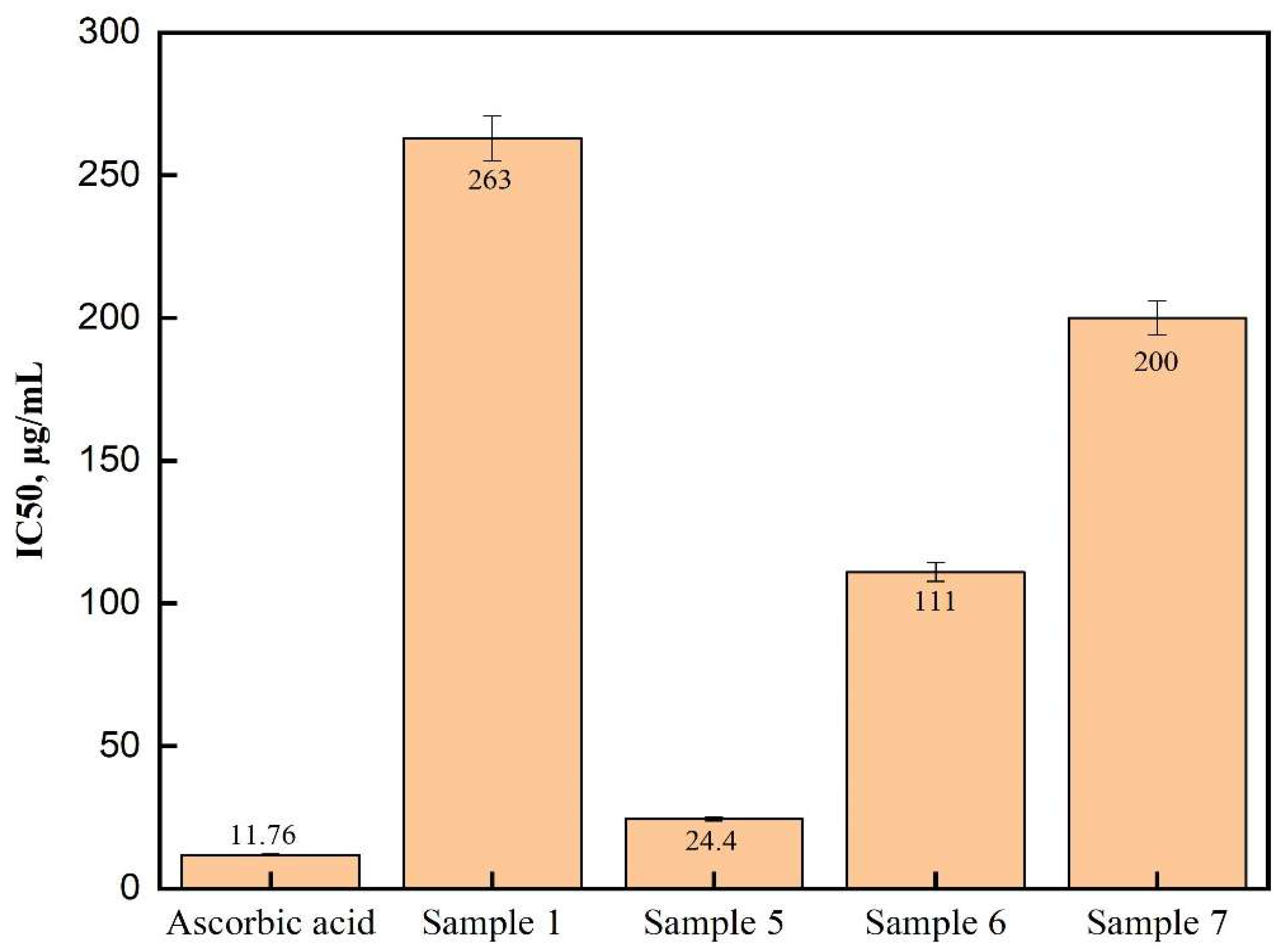
2.2. Antioxidant Finding
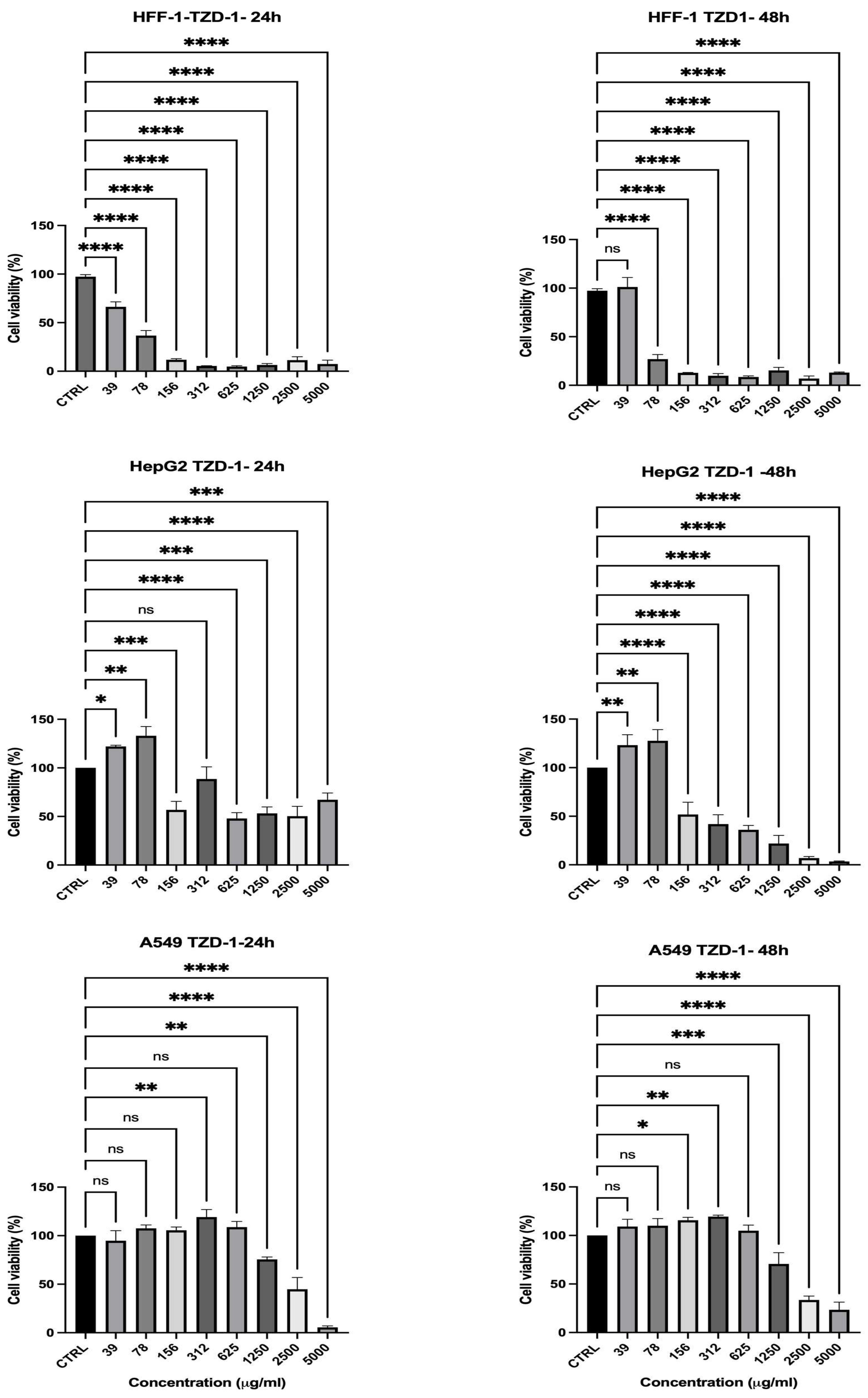
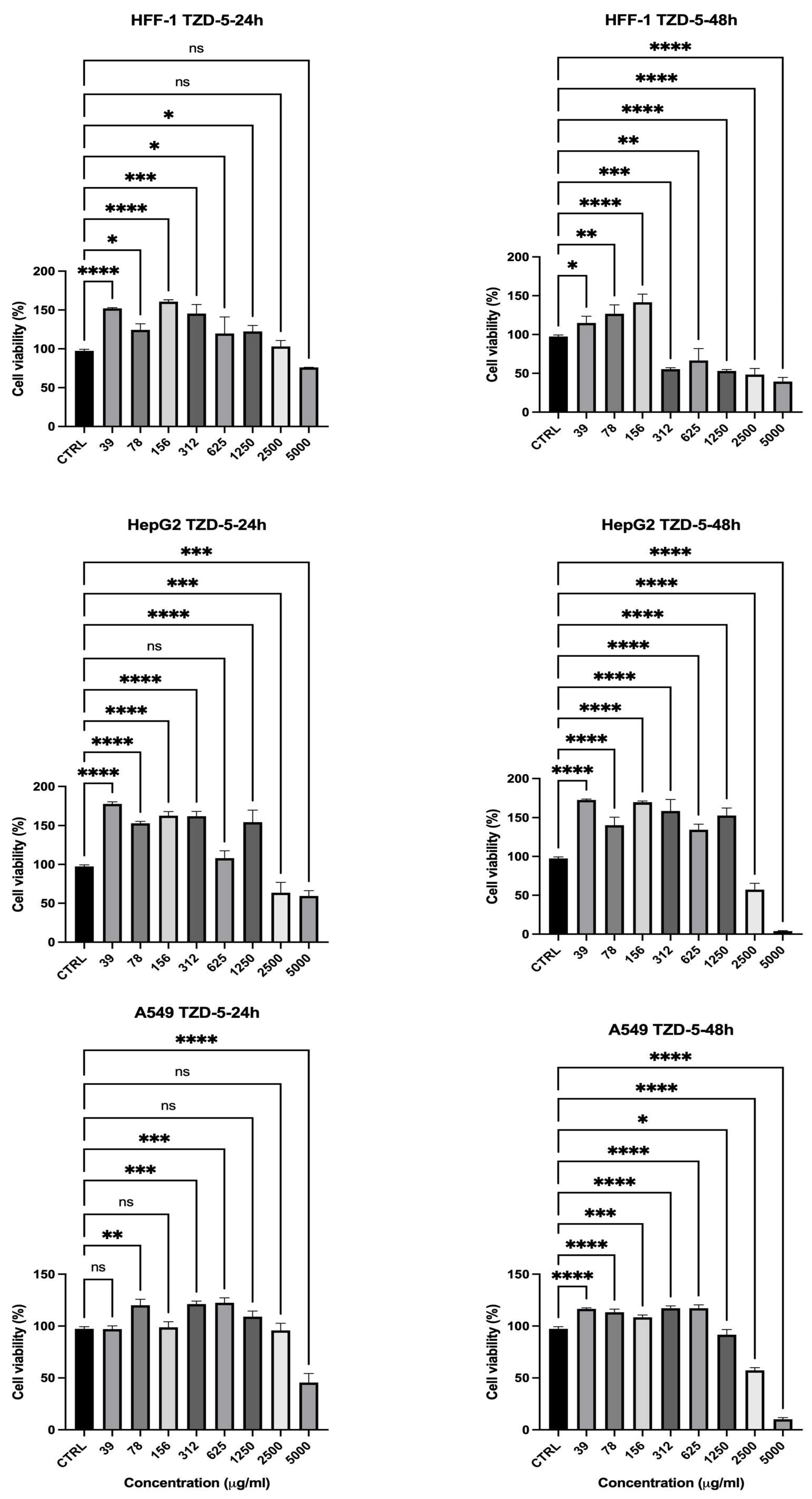
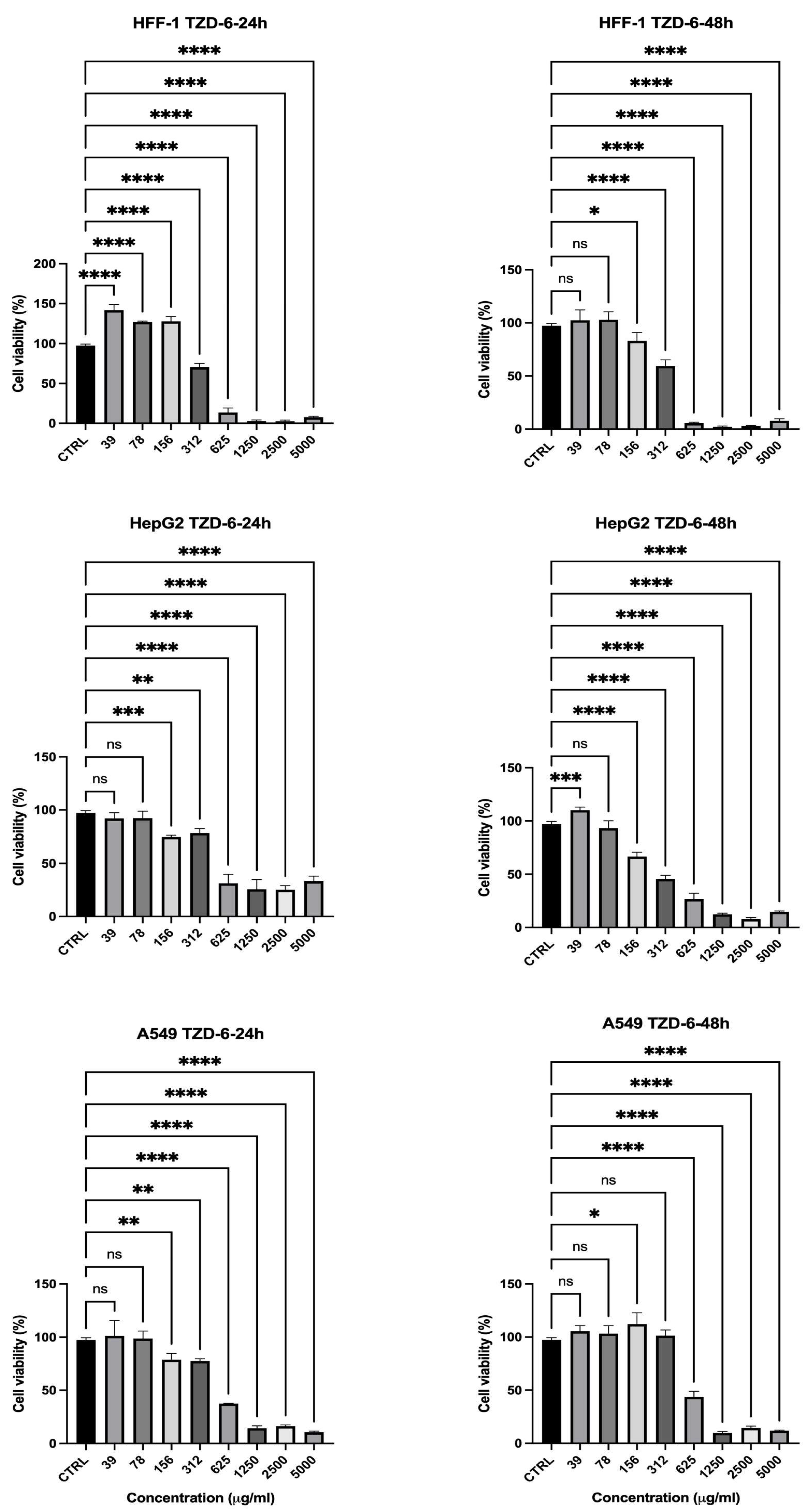
2.3. Cell Viability Assessment
3. Materials and Methods
3.1. Materials
3.2. Chemistry
3.2.1. Synthesis of 3-(4-methoxybenzyl)-2-((3,4,5-trimethoxybenzylidene)hydrazineylidene) thiazolidin-4-one (5)
3.2.2. Nuclear Magnetic Resonance (NMR) Analysis
3.2.3. Fourier-Transform Infrared Spectroscopy (FTIR) Analysis
3.2.4. Antioxidant Activity
3.3. Cell Viability Assay
3.4. Statistical Analysis
4. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Panda, S.K.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.B. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm. Sin. B 2025, 15, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.-J.; Won, Y.-S.; Kim, E.-K.; Park, S.-I.; Lee, S.J. Free radicals and their impact on health and antioxidant defenses: A review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Shaikh, S.A.; Labhade, S.R.; Kale, R.R.; Pachorkar, P.Y.; Meshram, R.J.; Jain, K.S.; Labhade, H.S.; Bhanushali, D.D.; More, R.A.; Nerkar, C.K.; et al. Thiadiazole-thiazole derivatives as potent anti-tubercular agents: Synthesis, biological evaluation, and In silico docking studies. Eur. J. Med. Chem. Rep. 2024, 12, 100183. [Google Scholar] [CrossRef]
- Mohanty, P.; Behera, S.; Behura, R.; Shubhadarshinee, L.; Mohapatra, P.; Barick, A.K.; Jali, B.R. Antibacterial activity of Thiazole and its derivatives: A. Biointerface Res. Appl. Chem. 2021, 12, 2171–2195. [Google Scholar] [CrossRef]
- Sadek, K.U.; Mekheimer, R.A.; Abd-Elmonem, M.; Abo-Elsoud, F.A.; Hayallah, A.M.; Mostafa, S.M.; Abdellattif, M.H.; Abourehab, M.A.S.; Farghaly, T.A.; Elkamhawy, A. Recent developments in the synthesis of hybrid heterocycles, a promising approach to develop multi-target antibacterial agents. J. Mol. Struct. 2023, 1286, 135616. [Google Scholar] [CrossRef]
- Hussain, R.; Rehman, W.; Khan, S.; Maalik, A.; Hefnawy, M.; Alanazi, A.S.; Khan, Y.; Rasheed, L. Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach. Pharmaceuticals 2023, 16, 1288. [Google Scholar] [CrossRef]
- Rana, R.; Kumar, N.; Gulati, H.K.; Sharma, A.; Khanna, A.; Pooja; Badhwar, R.; Dhir, M.; Jyoti; Singh, J.V.; et al. A comprehensive review on thiazole based conjugates as anti-cancer agents. J. Mol. Struct. 2023, 1292, 136194. [Google Scholar] [CrossRef]
- Farghaly, T.A.; Alsaedi, A.M.R.; Alenazi, N.A.; Harras, M.F. Anti-viral activity of thiazole derivatives: An updated patent review. Expert Opin. Ther. Pat. 2022, 32, 791–815. [Google Scholar] [CrossRef]
- Seboletswe, P.; Cele, N.; Singh, P. Thiazolidinone-Heterocycle Frameworks: A Concise Review of Their Pharmacological Significance. ChemMedChem 2023, 18, e202200618. [Google Scholar] [CrossRef]
- Mech, D.; Kurowska, A.; Trotsko, N. The Bioactivity of Thiazolidin-4-Ones: A Short Review of the Most Recent Studies. Int. J. Mol. Sci. 2021, 22, 11533. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Deep, A.; Marwaha, R.K. Design, synthesis, in silico studies and biological evaluation of 5-((E)-4-((E)-(substituted aryl/alkyl)methyl)benzylidene)thiazolidine-2,4-dione derivatives. BMC Chem. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Gor, D.; Gerber, B.S.; Walton, S.M.; Lee, T.A.; Nutescu, E.A.; Touchette, D.R. Antidiabetic drug use trends in patients with type 2 diabetes mellitus and chronic kidney disease: A cross-sectional analysis of the National Health and Nutrition Examination Survey. J. Diabetes 2020, 12, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, D.; Chen, T.; Wang, Y.; Miao, Z.; Xu, Y.; Chen, W.; Wang, X.; Li, Y.; Du, Z.; et al. Fragment-Based Drug Discovery of 2-Thiazolidinones as Inhibitors of the Histone Reader BRD4 Bromodomain. J. Med. Chem. 2013, 56, 3833–3851. [Google Scholar] [CrossRef]
- Nirwan, S.; Chahal, V.; Kakkar, R. Thiazolidinones: Synthesis, Reactivity, and Their Biological Applications. J. Heterocycl. Chem. 2019, 56, 1239–1253. [Google Scholar] [CrossRef]
- Aqlan, F.M.; Al-Bogami, A.S.; Alqahtani, N.F.; Wani, M.Y.; Khan, S.A. Thiazolidinone: A structural motif of great synthetic and biological importance. J. Mol. Struct. 2022, 1250, 131771. [Google Scholar] [CrossRef]
- Al Zahrani, N.A.; Al-Ghamdi, H.A.; El-Shishtawy, R.M. Synthesis and antioxidant properties of novel hybrid molecules containing gallic acid and thiazolidinone moieties. J. Mol. Struct. 2024, 1318, 139348. [Google Scholar] [CrossRef]
- Lončarić, M.; Strelec, I.; Pavić, V.; Rastija, V.; Karnaš, M.; Molnar, M. Green Synthesis of Thiazolidine-2,4-dione Derivatives and Their Lipoxygenase Inhibition Activity with QSAR and Molecular Docking Studies. Front. Chem. 2022, 10, 912822. [Google Scholar] [CrossRef]
- Liu, X.; Xie, H.; Luo, C.; Tong, L.; Wang, Y.; Peng, T.; Ding, J.; Jiang, H.; Li, H. Discovery and SAR of Thiazolidine-2,4-dione Analogues as Insulin-like Growth Factor-1 Receptor (IGF-1R) Inhibitors via Hierarchical Virtual Screening. J. Med. Chem. 2010, 53, 2661–2665. [Google Scholar] [CrossRef]
- Deng, S.; Zhao, Y.; Guo, X.; Hong, X.; Li, G.; Wang, Y.; Li, Q.; Bu, M.; Wang, M. Thiazolidinone-Conjugated Lupeol Derivatives as Potent Anticancer Agents Through a Mitochondria-Mediated Apoptotic Pathway. Molecules 2024, 29, 4957. [Google Scholar] [CrossRef]
- Sethi, N.S.; Prasad, D.N.; Singh, R.K. Synthesis, Anticancer, and Antibacterial Studies of Benzylidene Bearing 5-substituted and 3,5-disubstituted-2,4-Thiazolidinone Derivatives. Med. Chem. 2021, 17, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, N.S.; Aziz, N.A.A.M.; El-Hddad, S.S.A.; Abdelgawad, M.A.; Ghoneim, M.M.; Dawood, A.F.; Mohamady, S.; El-Adl, K.; Ahmed, S. Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment. Arch. Pharm. 2023, 356, 2300137. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Kaproń, B.; Plech, T.; Paduch, R.; Trotsko, N.; Paneth, P. Combined In Silico and In Vitro Analyses to Assess the Anticancer Potential of Thiazolidinone–Thiosemicarbazone Hybrid Molecules. Int. J. Mol. Sci. 2023, 24, 17521. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Comp. Code. | Mol. Form | Mol. Wt. | Wight (gm) | Yield (%) | Rf | m.p. |
|---|---|---|---|---|---|---|---|
| 1 | 1 | C13H15N3O4S | 309.08 | 0.79 | 100 | - | 271 |
| 2 | 5 | C21H23N3O5S | 429.49 | 0.3 | 80 | 0.59 | 177 |
| 3 | 6 | C20H21N3O4S | 399.47 | 0.3 | 80 | 0.59 | 175.5 |
| 4 | 7 | C20H20N4O6S | 444.46 | 0.28 | 62 | 0.58 | 176 |
| Compound | Time (h) | Concentration (µg/mL) | HFF-1 Viability (%) | HepG2 Viability (%) | A549 Viability (%) | Selectivity Toward Cancer Cells |
|---|---|---|---|---|---|---|
| TZD-1 | 24 | 1250 | 6.45% | 53.16% | 75.58% | Low (high toxicity to normal cells) |
| 48 | 1250 | 15.34% | 21.91% | 70.64% | Low | |
| TZD-5 | 24 | 2500 | 97% | 63% | 95.8% | Moderate (better selectivity) |
| 48 | 2500 | 48% | 57% | 55% | Moderate | |
| TZD-6 | 24 | 1250 | 2% | 25% | 13% | Low (high toxicity to normal cells) |
| 48 | 1250 | 1–2% | 12% | 9% | Low | |
| TZD-7 | 24 | 1250 | 110% | 96.4% | 70% | Moderate (better selectivity) |
| 48 | 1250 | 94% | 23% | 82.3% | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahrani, N.A.A.; Alshabibi, M.A.; Bakr, A.A.; Almughem, F.A.; Alshehri, A.A.; Al-Ghamdi, H.A.; Tawfik, E.A.; Damiati, L.A. Molecular Hybrids of Thiazolidinone: Bridging Redox Modulation and Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 6529. https://doi.org/10.3390/ijms26136529
Zahrani NAA, Alshabibi MA, Bakr AA, Almughem FA, Alshehri AA, Al-Ghamdi HA, Tawfik EA, Damiati LA. Molecular Hybrids of Thiazolidinone: Bridging Redox Modulation and Cancer Therapy. International Journal of Molecular Sciences. 2025; 26(13):6529. https://doi.org/10.3390/ijms26136529
Chicago/Turabian StyleZahrani, Nourah A. Al, Manal A. Alshabibi, Abrar A. Bakr, Fahad A. Almughem, Abdullah A. Alshehri, Huda A. Al-Ghamdi, Essam A. Tawfik, and Laila A. Damiati. 2025. "Molecular Hybrids of Thiazolidinone: Bridging Redox Modulation and Cancer Therapy" International Journal of Molecular Sciences 26, no. 13: 6529. https://doi.org/10.3390/ijms26136529
APA StyleZahrani, N. A. A., Alshabibi, M. A., Bakr, A. A., Almughem, F. A., Alshehri, A. A., Al-Ghamdi, H. A., Tawfik, E. A., & Damiati, L. A. (2025). Molecular Hybrids of Thiazolidinone: Bridging Redox Modulation and Cancer Therapy. International Journal of Molecular Sciences, 26(13), 6529. https://doi.org/10.3390/ijms26136529