
Pharmacological Treatment of MASLD: Contemporary Treatment and Future Perspectives


Abstract
1. Introduction
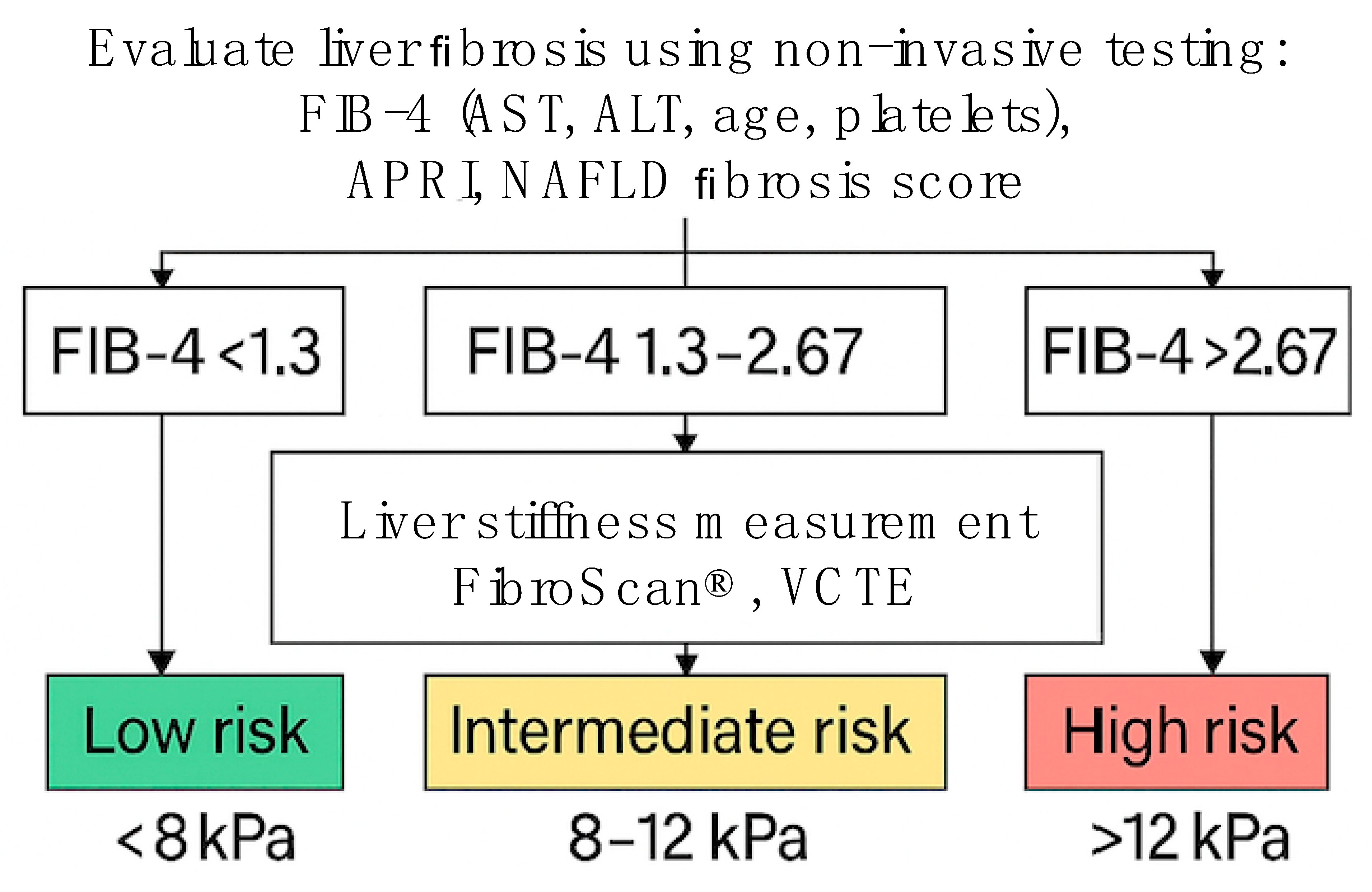
2. Contemporary Treatment Recommendations
3. Dietary Interventions in MASLD
4. Physical Activity and Lifestyle Modifications in MASLD
5. Pharmacological Treatment
- Pioglitazone (30–45 mg/day)
- 2.
- Vitamin E (800 IU/day, d-α-tocopherol)
- 3.
- Ursodeoxycholic Acid (UDCA, 13–15 mg/kg/day)
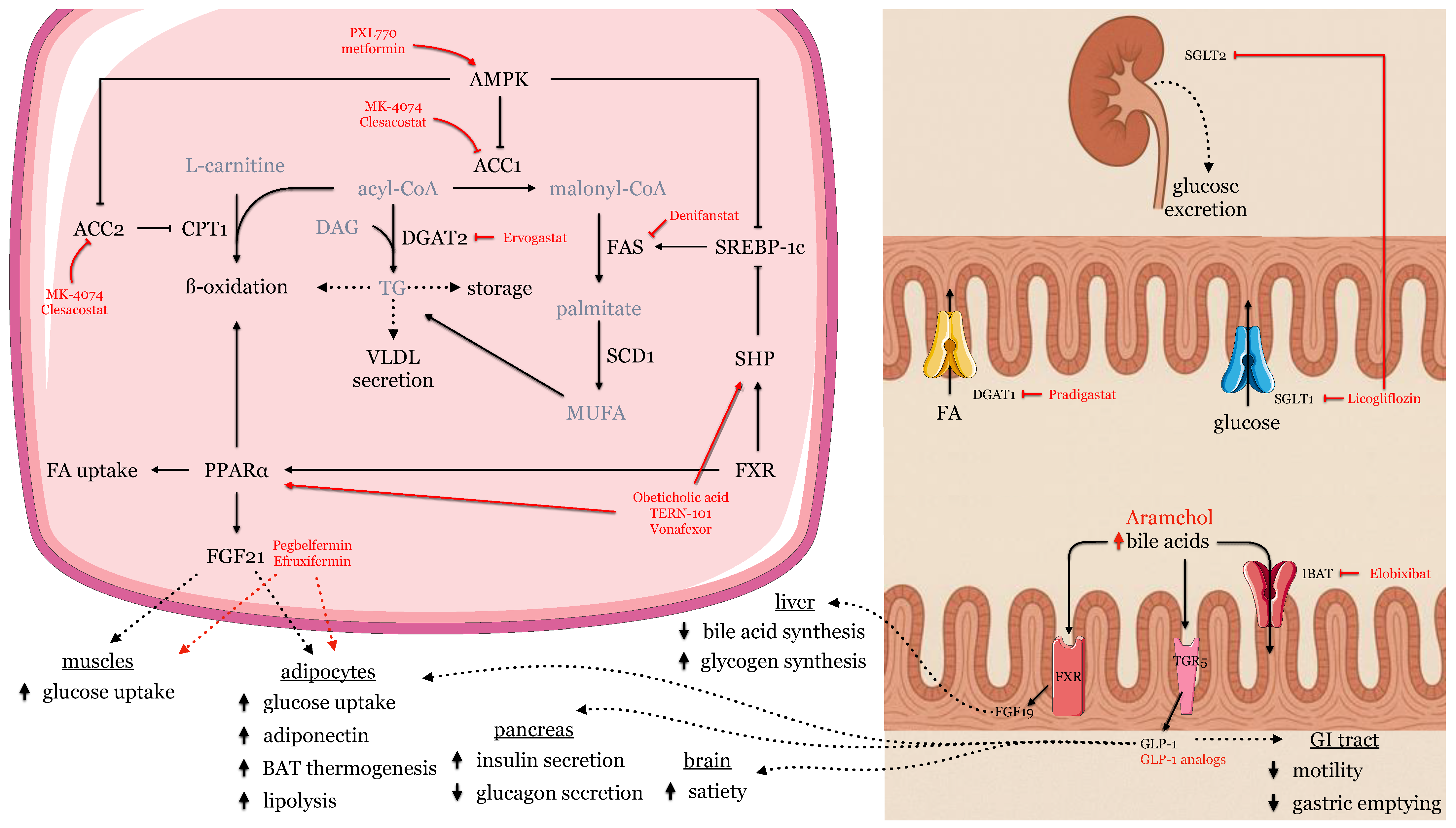
6. Review of New Drugs Currently Tested in RCTs
7. Acetyl-CoA Carboxylase (ACC) Inhibitors
8. Amino Acids, Carnitine, and N-Acetylcysteine
9. AMPK Activators
10. Bile Acid Metabolism
11. Diacylglycerol Acyltransferase Inhibitors
12. Fatty Acid Synthase Inhibitors
13. Free Fatty Acid Receptor 4 Agonist
14. Fibroblast Growth Factor 21 Signaling
15. Farnesoid X Receptor Agonists
16. GLP-1 and Dual GLP-1/GIP Agonists
17. Dual GLP-1/Glucagon Receptor Agonists
18. Ketohexokinase Inhibition
19. Mitochondrial Uncoupling
20. Nonsteroidal Anti-Inflammatory Drugs
21. PPAR Agonists
22. SGLT1 and SGLT2 Inhibitors
23. Testosterone
24. THR-β Agonists
25. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| ALP | Alkaline phosphatase |
| ALT | Alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| ASA | Acetylsalicylic acid |
| AST | Aspartate transaminase |
| ATP | Adenosine triphosphate |
| COX | Cyclooxygenase |
| CPT 1 | Carnitine palmitoyltransferase 1 |
| DGAT | Diacylglycerol acyltransferase |
| DNP | 2,4-dinitrophenol |
| FAS | Fatty acid synthase |
| FFAR | Free fatty acid receptor |
| FXR | Farnesoid X receptor |
| FGF | Fibroblast growth factor |
| GIP | Gastric inhibitory polypeptide |
| GLP-1 | Glucagon-like peptide-1 |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| IBAT | Ileal bile acid transporter |
| KHX | Ketohexokinase |
| KLB | Co-receptor β-Klotho |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| OCA | Obeticholic acid |
| PPAR | Peroxisome proliferator-activated receptor |
| RCT | Randomized clinical trial |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SHP | Small heterodimer partner |
| SREBP-1c | Sterol regulatory element-binding protein 1 |
| TGF-ß1 | Transforming growth factor-beta1 |
| TGR | Takeda G protein-coupled receptor |
| THR-ß | Thyroid hormone receptor ß |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Y.; Brandman, D. A Clinical Update on MASLD. JAMA Intern. Med. 2025, 185, 105. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular Pathways of Nonalcoholic Fatty Liver Disease Development and Progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Grabherr, F.; Tilg, H. Non-Alcoholic Fatty Liver Disease: Pathophysiological Concepts and Treatment Options. Cardiovasc. Res. 2023, 119, 1787–1798. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G. Molecular Mechanisms of Lipotoxicity in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a Mediator of Non-Alcoholic Fatty Liver Disease and Associated Atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.-P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic Fatty Liver Disease in Humans Is Associated with Increased Plasma Endotoxin and Plasminogen Activator Inhibitor 1 Concentrations and with Fructose Intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [CrossRef]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal Microbiota Determines Development of Non-Alcoholic Fatty Liver Disease in Mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the Clinical Assessment and Management of Nonalcoholic Fatty Liver Disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef]
- Kwong, A.J.; Kim, W.R.; Lake, J.R.; Schladt, D.P.; Schnellinger, E.M.; Gauntt, K.; McDermott, M.; Weiss, S.; Handarova, D.K.; Snyder, J.J.; et al. OPTN/SRTR 2022 Annual Data Report: Liver. Am. J. Transplant. 2024, 24, S176–S265. [Google Scholar] [CrossRef]
- Kanwal, F.; Shubrook, J.H.; Adams, L.A.; Pfotenhauer, K.; Wai-Sun Wong, V.; Wright, E.; Abdelmalek, M.F.; Harrison, S.A.; Loomba, R.; Mantzoros, C.S.; et al. Clinical Care Pathway for the Risk Stratification and Management of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2021, 161, 1657–1669. [Google Scholar] [CrossRef]
- Cusi, K.; Isaacs, S.; Barb, D.; Basu, R.; Caprio, S.; Garvey, W.T.; Kashyap, S.; Mechanick, J.I.; Mouzaki, M.; Nadolsky, K.; et al. American Association of Clinical Endocrinology Clinical Practice Guideline for the Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Primary Care and Endocrinology Clinical Settings. Endocr. Pract. 2022, 28, 528–562. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef]
- Belfort-DeAguiar, R.; Lomonaco, R.; Cusi, K. Approach to the Patient With Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2023, 108, 483–495. [Google Scholar] [CrossRef]
- Dansinger, M.L.; Gleason, J.A.; Griffith, J.L.; Selker, H.P.; Schaefer, E.J. Comparison of the Atkins, Ornish, Weight Watchers, and Zone Diets for Weight Loss and Heart Disease Risk Reduction. JAMA 2005, 293, 43. [Google Scholar] [CrossRef]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Long-Term Persistence of Hormonal Adaptations to Weight Loss. N. Engl. J. Med. 2011, 365, 1597–1604. [Google Scholar] [CrossRef]
- Rajewski, P.; Cieściński, J.; Rajewski, P.; Suwała, S.; Rajewska, A.; Potasz, M. Dietary Interventions and Physical Activity as Crucial Factors in the Prevention and Treatment of Metabolic Dysfunction-Associated Steatotic Liver Disease. Biomedicines 2025, 13, 217. [Google Scholar] [CrossRef]
- Plauth, M.; Bernal, W.; Dasarathy, S.; Merli, M.; Plank, L.D.; Schütz, T.; Bischoff, S.C. ESPEN Guideline on Clinical Nutrition in Liver Disease. Clin. Nutr. 2019, 38, 485–521. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Milanović, M.; Milić, N.; Luzza, F.; Giuffrè, A.M. Olive Oil Antioxidants and Non-Alcoholic Fatty Liver Disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Anania, C.; Perla, F.M.; Olivero, F.; Pacifico, L.; Chiesa, C. Mediterranean Diet and Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2018, 24, 2083–2094. [Google Scholar] [CrossRef]
- Xu, C.; Markova, M.; Seebeck, N.; Loft, A.; Hornemann, S.; Gantert, T.; Kabisch, S.; Herz, K.; Loske, J.; Ost, M.; et al. High-protein Diet More Effectively Reduces Hepatic Fat than Low-protein Diet despite Lower Autophagy and FGF21 Levels. Liver Int. 2020, 40, 2982–2997. [Google Scholar] [CrossRef]
- Choi, Y.J.; Jeon, S.-M.; Shin, S. Impact of a Ketogenic Diet on Metabolic Parameters in Patients with Obesity or Overweight and with or without Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Nutrients 2020, 12, 2005. [Google Scholar] [CrossRef]
- Belopolsky, Y.; Khan, M.Q.; Sonnenberg, A.; Davidson, D.J.; Fimmel, C.J. Ketogenic, Hypocaloric Diet Improves Nonalcoholic Steatohepatitis. J. Transl. Intern. Med. 2020, 8, 26–31. [Google Scholar] [CrossRef]
- Holmer, M.; Lindqvist, C.; Petersson, S.; Moshtaghi-Svensson, J.; Tillander, V.; Brismar, T.B.; Hagström, H.; Stål, P. Treatment of NAFLD with Intermittent Calorie Restriction or Low-Carb High-Fat Diet–A Randomised Controlled Trial. JHEP Rep. 2021, 3, 100256. [Google Scholar] [CrossRef]
- Johari, M.I.; Yusoff, K.; Haron, J.; Nadarajan, C.; Ibrahim, K.N.; Wong, M.S.; Hafidz, M.I.A.; Chua, B.E.; Hamid, N.; Arifin, W.N.; et al. A Randomised Controlled Trial on the Effectiveness and Adherence of Modified Alternate-Day Calorie Restriction in Improving Activity of Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 11232. [Google Scholar] [CrossRef] [PubMed]
- Trepanowski, J.F.; Kroeger, C.M.; Barnosky, A.; Klempel, M.; Bhutani, S.; Hoddy, K.K.; Rood, J.; Ravussin, E.; Varady, K.A. Effects of Alternate-Day Fasting or Daily Calorie Restriction on Body Composition, Fat Distribution, and Circulating Adipokines: Secondary Analysis of a Randomized Controlled Trial. Clin. Nutr. 2018, 37, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Lavallee, C.M.; Bruno, A.; Ma, C.; Raman, M. The Role of Intermittent Fasting in the Management of Nonalcoholic Fatty Liver Disease: A Narrative Review. Nutrients 2022, 14, 4655. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V. Aerobic Exercise in the Management of Metabolic Dysfunction Associated Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2021, 14, 3627–3645. [Google Scholar] [CrossRef]
- Bae, J.C.; Suh, S.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Kim, S.W.; Hur, K.Y.; Kim, J.H.; et al. Regular Exercise Is Associated with a Reduction in the Risk of NAFLD and Decreased Liver Enzymes in Individuals with NAFLD Independent of Obesity in Korean Adults. PLoS ONE 2012, 7, e46819. [Google Scholar] [CrossRef]
- Ryu, S.; Chang, Y.; Jung, H.-S.; Yun, K.E.; Kwon, M.-J.; Choi, Y.; Kim, C.-W.; Cho, J.; Suh, B.-S.; Cho, Y.K.; et al. Relationship of Sitting Time and Physical Activity with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 63, 1229–1237. [Google Scholar] [CrossRef]
- Byambasukh, O.; Zelle, D.; Corpeleijn, E. Physical Activity, Fatty Liver, and Glucose Metabolism Over the Life Course: The Lifelines Cohort. Am. J. Gastroenterol. 2019, 114, 907–915. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis. JAMA Intern. Med. 2017, 177, 633. [Google Scholar] [CrossRef]
- Wen, H.; Deng, H.; Yang, L.; Li, L.; Lin, J.; Zheng, P.; Bjelakovic, M.; Ji, G. Vitamin E for People with Non-Alcoholic Fatty Liver Disease. Cochrane Database Syst. Rev. 2024, 2024, CD015033. [Google Scholar] [CrossRef]
- Chee, N.M.; Sinnanaidu, R.P.; Chan, W. Vitamin E Improves Serum Markers and Histology in Adults with Metabolic Dysfunction-associated Steatotic Liver Disease: Systematic Review and Meta-analysis. J. Gastroenterol. Hepatol. 2024, 39, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Schölmerich, J.; Becher, M.-S.; Schmidt, K.; Schubert, R.; Kremer, B.; Feldhaus, S.; Gerok, W. Influence of Hydroxylation and Conjugation of Bile Salts on Their Membrane-Damaging Properties-Studies on Isolated Hepatocytes and Lipid Membrane Vesicles. Hepatology 1984, 4, 661–666. [Google Scholar] [CrossRef]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, E.M.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-Dose Ursodeoxycholic Acid for the Treatment of Primary Sclerosing Cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Chen, Y.; Ma, K.; Ye, Y.; Zheng, L.; Yang, Y.; Li, Y.; Jin, X. The Role of Ursodeoxycholic Acid in Non-Alcoholic Steatohepatitis: A Systematic Review. BMC Gastroenterol. 2013, 13, 140. [Google Scholar] [CrossRef]
- Lindor, K.D.; Kowdley, K.V.; Heathcote, J.E.; Harrison, E.M.; Jorgensen, R.; Angulo, P.; Lymp, J.F.; Burgart, L.; Colin, P. Ursodeoxycholic Acid for Treatment of Nonalcoholic Steatohepatitis: Results of a Randomized Trial. Hepatology 2004, 39, 770–778. [Google Scholar] [CrossRef]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic Acid or Clofibrate in the Treatment of Non–Alcohol–Induced Steatohepatitis: A Pilot Study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef]
- Zhang, W.; Tang, Y.; Huang, J.; Hu, H. Efficacy of Ursodeoxycholic Acid in Nonalcoholic Fatty Liver Disease: An Updated Meta-Analysis of Randomized Controlled Trials. Asia Pac. J. Clin. Nutr. 2020, 29, 696–705. [Google Scholar]
- Kim, C.-W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef]
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC Inhibitor Alone or Co-Administered with a DGAT2 Inhibitor in Patients with Non-Alcoholic Fatty Liver Disease: Two Parallel, Placebo-Controlled, Randomized Phase 2a Trials. Nat. Med. 2021, 27, 1836–1848. [Google Scholar] [CrossRef]
- Sivakumar, P.; Saul, M.; Robinson, D.; King, L.E.; Amin, N.B. SomaLogic Proteomics Reveals New Biomarkers and Provides Mechanistic, Clinical Insights into Acetyl CoA Carboxylase (ACC) Inhibition in Non-Alcoholic Steatohepatitis (NASH). Sci. Rep. 2024, 14, 17072. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.B.; Darekar, A.; Anstee, Q.M.; Wong, V.W.-S.; Tacke, F.; Vourvahis, M.; Lee, D.S.; Charlton, M.; Alkhouri, N.; Nakajima, A.; et al. Efficacy and Safety of an Orally Administered DGAT2 Inhibitor Alone or Coadministered with a Liver-Targeted ACC Inhibitor in Adults with Non-Alcoholic Steatohepatitis (NASH): Rationale and Design of the Phase II, Dose-Ranging, Dose-Finding, Randomised, Placebo-Controlled MIRNA (Metabolic Interventions to Resolve NASH with Fibrosis) Study. BMJ Open 2022, 12, e056159. [Google Scholar] [CrossRef]
- Harrison, S.A.; Baum, S.J.; Gunn, N.T.; Younes, Z.H.; Kohli, A.; Patil, R.; Koziel, M.J.; Chera, H.; Zhao, J.; Chakravarthy, M.V. Safety, Tolerability, and Biologic Activity of AXA1125 and AXA1957 in Subjects With Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2021, 116, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Alkhouri, N.; Harrison, S.A.; Fouqueray, P.; Moller, D.E.; Hallakou-Bozec, S.; Bolze, S.; Grouin, J.-M.; Jeannin Megnien, S.; Dubourg, J.; et al. Efficacy and Safety of PXL770, a Direct AMP Kinase Activator, for the Treatment of Non-Alcoholic Fatty Liver Disease (STAMP-NAFLD): A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Study. Lancet Gastroenterol. Hepatol. 2021, 6, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lutchman, G.; Kleiner, D.E.; Ricks, M.; Feld, J.J.; Borg, B.B.; Modi, A.; Nagabhyru, P.; Sumner, A.E.; Liang, T.J.; et al. Clinical Trial: Pilot Study of Metformin for the Treatment of Non-alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2009, 29, 172–182. [Google Scholar] [CrossRef]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in Patients with Nonalcoholic Steatohepatitis: A Randomized, Double-Blind, Placebo-Controlled Phase 2b Trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef]
- Loomba, R.; Mohseni, R.; Lucas, K.J.; Gutierrez, J.A.; Perry, R.G.; Trotter, J.F.; Rahimi, R.S.; Harrison, S.A.; Ajmera, V.; Wayne, J.D.; et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial. Gastroenterology 2021, 161, 1475–1486. [Google Scholar] [CrossRef]
- Harrison, S.A.; Alkhouri, N.; Ortiz-Lasanta, G.; Rudraraju, M.; Tai, D.; Wack, K.; Shah, A.; Besuyen, R.; Steineger, H.H.; Fraser, D.A.; et al. A Phase IIb Randomised-Controlled Trial of the FFAR1/FFAR4 Agonist Icosabutate in MASH. J. Hepatol. 2025, in press. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in Non-Alcoholic Steatohepatitis: A Randomized, Double-Blind, Placebo-Controlled, Phase 2a Trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.; Neff, G.; Patil, R.; Behling, C.; Hu, C.; Shringarpure, R.; de Temple, B.; Fong, E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase IIa Trial of Efruxifermin for Patients with Compensated NASH Cirrhosis. JHEP Rep. 2023, 5, 100563. [Google Scholar] [CrossRef]
- Alkhouri, N.; Beyer, C.; Shumbayawonda, E.; Andersson, A.; Yale, K.; Rolph, T.; Chung, R.T.; Vuppalanchi, R.; Cusi, K.; Loomba, R.; et al. Decreases in CT1 and Liver Fat Content Reflect Treatment-Induced Histological Improvements in MASH. J. Hepatol. 2025, 82, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Decato, B.E.; Charles, E.D.; Shevell, D.E.; McNaney, C.; Shipkova, P.; Apfel, A.; Tirucherai, G.S.; Sanyal, A.J. Pegbelfermin Selectively Reduces Secondary Bile Acid Concentrations in Patients with Non-Alcoholic Steatohepatitis. JHEP Rep. 2022, 4, 100392. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated Fibroblast Growth Factor 21 Analogue, in Patients with Non-Alcoholic Steatohepatitis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Trial. Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef] [PubMed]
- Raji, A.; Gantz, I.; Crutchlow, M.; Flynn, H.; Xu, L.; Rodgers, A.J.; Krishnan, R.; Rizk, M.L.; Hu, S.; Kaufman, K.D.; et al. Clinical Trial: A Phase 2b Study to Evaluate the Efficacy and Safety of MK-3655 in Individuals With Pre-Cirrhotic MASH. Aliment. Pharmacol. Ther. 2025, 61, 1152–1162. [Google Scholar] [CrossRef]
- Hameed, B.; Terrault, N.A.; Gill, R.M.; Loomba, R.; Chalasani, N.; Hoofnagle, J.H.; Van Natta, M.L. Clinical and Metabolic Effects Associated with Weight Changes and Obeticholic Acid in Non-alcoholic Steatohepatitis. Aliment. Pharmacol. Ther. 2018, 47, 645–656. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of Obeticholic Acid on the Lipoprotein Profile in Patients with Non-Alcoholic Steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef]
- Shen, W.; Middleton, M.S.; Cunha, G.M.; Delgado, T.I.; Wolfson, T.; Gamst, A.; Fowler, K.J.; Alazraki, A.; Trout, A.T.; Ohliger, M.A.; et al. Changes in Abdominal Adipose Tissue Depots Assessed by MRI Correlate with Hepatic Histologic Improvement in Non-Alcoholic Steatohepatitis. J. Hepatol. 2023, 78, 238–246. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Loustaud-Ratti, V.; Bureau, C.; Lawitz, E.; Abdelmalek, M.; Alkhouri, N.; Francque, S.; Girma, H.; Darteil, R.; et al. Hepatic and Renal Improvements with FXR Agonist Vonafexor in Individuals with Suspected Fibrotic NASH. J. Hepatol. 2023, 78, 479–492. [Google Scholar] [CrossRef]
- Ratziu, V.; Rinella, M.E.; Neuschwander-Tetri, B.A.; Lawitz, E.; Denham, D.; Kayali, Z.; Sheikh, A.; Kowdley, K.V.; Desta, T.; Elkhashab, M.; et al. EDP-305 in Patients with NASH: A Phase II Double-Blind Placebo-Controlled Dose-Ranging Study. J. Hepatol. 2022, 76, 506–517. [Google Scholar] [CrossRef]
- Loomba, R.; Abdelmalek, M.F.; Armstrong, M.J.; Jara, M.; Kjær, M.S.; Krarup, N.; Lawitz, E.; Ratziu, V.; Sanyal, A.J.; Schattenberg, J.M.; et al. Semaglutide 2·4 Mg Once Weekly in Patients with Non-Alcoholic Steatohepatitis-Related Cirrhosis: A Randomised, Placebo-Controlled Phase 2 Trial. Lancet Gastroenterol. Hepatol. 2023, 8, 511–522. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okanoue, T.; Sundby Palle, M.; Sejling, A.; Tawfik, M.; Roden, M. Similar Weight Loss with Semaglutide Regardless of Diabetes and Cardiometabolic Risk Parameters in Individuals with Metabolic Dysfunction-associated Steatotic Liver Disease: Post Hoc Analysis of Three Randomised Controlled Trials. Diabetes Obes. Metab. 2025, 27, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Ditzenberger, G.L.; Lake, J.E.; Kitch, D.W.; Kantor, A.; Muthupillai, R.; Moser, C.; Belaunzaran-Zamudio, P.F.; Brown, T.T.; Corey, K.; Landay, A.L.; et al. Effects of Semaglutide on Muscle Structure and Function in the SLIM LIVER Study. Clin. Infect. Dis. 2025, 80, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gómez, M.; Lawitz, E.; Shankar, R.R.; Chaudhri, E.; Liu, J.; Lam, R.L.H.; Kaufman, K.D.; Engel, S.S.; Bruzone, S.O.; Coronel, M.J.; et al. A Phase IIa Active-Comparator-Controlled Study to Evaluate the Efficacy and Safety of Efinopegdutide in Patients with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2023, 79, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Hartman, M.L.; Lawitz, E.J.; Vuppalanchi, R.; Boursier, J.; Bugianesi, E.; Yoneda, M.; Behling, C.; Cummings, O.W.; Tang, Y.; et al. Tirzepatide for Metabolic Dysfunction–Associated Steatohepatitis with Liver Fibrosis. N. Engl. J. Med. 2024, 391, 299–310. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Bedossa, P.; Fraessdorf, M.; Neff, G.W.; Lawitz, E.; Bugianesi, E.; Anstee, Q.M.; Hussain, S.A.; Newsome, P.N.; Ratziu, V.; et al. A Phase 2 Randomized Trial of Survodutide in MASH and Fibrosis. N. Engl. J. Med. 2024, 391, 311–319. [Google Scholar] [CrossRef]
- Shankar, S.S.; Daniels, S.J.; Robertson, D.; Sarv, J.; Sánchez, J.; Carter, D.; Jermutus, L.; Challis, B.; Sanyal, A.J. Safety and Efficacy of Novel Incretin Co-Agonist Cotadutide in Biopsy-Proven Noncirrhotic MASH With Fibrosis. Clin. Gastroenterol. Hepatol. 2024, 22, 1847–1857.e11. [Google Scholar] [CrossRef]
- Kazierad, D.J.; Chidsey, K.; Somayaji, V.R.; Bergman, A.J.; Birnbaum, M.J.; Calle, R.A. Inhibition of Ketohexokinase in Adults with NAFLD Reduces Liver Fat and Inflammatory Markers: A Randomized Phase 2 Trial. Med 2021, 2, 800–813.e3. [Google Scholar] [CrossRef]
- Saxena, A.R.; Lyle, S.; Khavandi, K.; Qiu, R.; Whitlock, M.; Esler, W.P.; Kim, A.M. A Phase 2a, Randomized, Double-blind, Placebo-controlled, Three-arm, Parallel-group Study to Assess the Efficacy, Safety, Tolerability and Pharmacodynamics of PF-06835919 in Patients with Non-alcoholic Fatty Liver Disease and Type 2 Diabetes. Diabetes Obes. Metab. 2023, 25, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Khan, S.; Portell, F.; Jorkasky, D.; Dennis, J.; Khan, O.; Johansson, L.; Johansson, E.; Sanyal, A.J. Safety and Efficacy of Once-Daily HU6 versus Placebo in People with Non-Alcoholic Fatty Liver Disease and High BMI: A Randomised, Double-Blind, Placebo-Controlled, Phase 2a Trial. Lancet Gastroenterol. Hepatol. 2023, 8, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Wilechansky, R.M.; Stoyanova, S.; Grossman, A.; Dichtel, L.E.; Lauer, G.M.; Miller, K.K.; Hoshida, Y.; Corey, K.E.; Loomba, R.; et al. Aspirin for Metabolic Dysfunction–Associated Steatotic Liver Disease Without Cirrhosis. JAMA 2024, 331, 920. [Google Scholar] [CrossRef]
- Harrison, S.A.; Thang, C.; Bolze, S.; Dewitt, S.; Hallakou-Bozec, S.; Dubourg, J.; Bedossa, P.; Cusi, K.; Ratziu, V.; Grouin, J.-M.; et al. Evaluation of PXL065–Deuterium-Stabilized (R)-Pioglitazone in Patients with NASH: A Phase II Randomized Placebo-Controlled Trial (DESTINY-1). J. Hepatol. 2023, 78, 914–925. [Google Scholar] [CrossRef]
- Monternier, P.; Singh, J.; Parasar, P.; Theurey, P.; DeWitt, S.; Jacques, V.; Klett, E.; Kaur, N.; Nagaraja, T.N.; Moller, D.E.; et al. Therapeutic Potential of Deuterium-stabilized (R)-pioglitazone—PXL065—For X-linked Adrenoleukodystrophy. J. Inherit. Metab. Dis. 2022, 45, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Parmar, D.; Sheikh, F.; Sarin, S.K.; Cisneros, L.; Gawrieh, S.; Momin, T.; Duseja, A.; Sanyal, A.J. Saroglitazar, a Dual PPAR α/γ Agonist, Improves Atherogenic Dyslipidemia in Patients With Non-Cirrhotic Nonalcoholic Fatty Liver Disease: A Pooled Analysis. Clin. Gastroenterol. Hepatol. 2023, 21, 2597–2605.e2. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Cooreman, M.P.; Butler, J.; Giugliano, R.P.; Zannad, F.; Dzen, L.; Huot-Marchand, P.; Baudin, M.; Beard, D.R.; Junien, J.-L.; Broqua, P.; et al. The Pan-PPAR Agonist Lanifibranor Improves Cardiometabolic Health in Patients with Metabolic Dysfunction-Associated Steatohepatitis. Nat. Commun. 2024, 15, 3962. [Google Scholar] [CrossRef]
- Pierre, B.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; Jean-Louis, A. A Randomised, Double-Blind, Placebo-Controlled, Multi-Centre, Dose-Range, Proof-of-Concept, 24-Week Treatment Study of Lanifibranor in Adult Subjects with Non-Alcoholic Steatohepatitis: Design of the NATIVE Study. Contemp. Clin. Trials 2020, 98, 106170. [Google Scholar] [CrossRef]
- Barb, D.; Kalavalapalli, S.; Godinez Leiva, E.; Bril, F.; Huot-Marchand, P.; Dzen, L.; Rosenberg, J.T.; Junien, J.-L.; Broqua, P.; Rocha, A.O.; et al. Pan-PPAR Agonist Lanifibranor Improves Insulin Resistance and Hepatic Steatosis in Patients with T2D and MASLD. J. Hepatol. 2025, 82, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Manghi, F.P.; Smith, W.B.; Alpenidze, D.; Aizenberg, D.; Klarenbeek, N.; Chen, C.-Y.; Zuckerman, E.; Ravussin, E.; Charatcharoenwitthaya, P.; et al. Licogliflozin for Nonalcoholic Steatohepatitis: A Randomized, Double-Blind, Placebo-Controlled, Phase 2a Study. Nat. Med. 2022, 28, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Albhaisi, S.; Kim, K.; Baker, J.; Chidambaram, N.; Patel, M.V.; Charlton, M.; Sanyal, A.J. LPCN 1144 Resolves NAFLD in Hypogonadal Males. Hepatol. Commun. 2020, 4, 1430–1440. [Google Scholar] [CrossRef]
- Mateo-Marín, M.A.; Alves-Bezerra, M. Targeting Acetyl-CoA Carboxylases for the Treatment of MASLD. J. Lipid Res. 2024, 65, 100676. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, A.; Szarmach, I.; Żendzian-Piotrowska, M.; Maciejczyk, M. The Effect of N-Acetylcysteine on Respiratory Enzymes, ADP/ATP Ratio, Glutathione Metabolism, and Nitrosative Stress in the Salivary Gland Mitochondria of Insulin Resistant Rats. Nutrients 2020, 12, 458. [Google Scholar] [CrossRef]
- Zalewska, A.; Zięba, S.; Kostecka-Sochoń, P.; Kossakowska, A.; Żendzian-Piotrowska, M.; Matczuk, J.; Maciejczyk, M. NAC Supplementation of Hyperglycemic Rats Prevents the Development of Insulin Resistance and Improves Antioxidant Status but Only Alleviates General and Salivary Gland Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 8831855. [Google Scholar] [CrossRef]
- Liu, A.; Cai, Y.; Yuan, Y.; Liu, M.; Zhang, Z.; Xu, Y.; Jiao, P. Efficacy and Safety of Carnitine Supplementation on NAFLD: A Systematic Review and Meta-Analysis. Syst. Rev. 2023, 12, 74. [Google Scholar] [CrossRef]
- Savic, D.; Hodson, L.; Neubauer, S.; Pavlides, M. The Importance of the Fatty Acid Transporter L-Carnitine in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 2178. [Google Scholar] [CrossRef]
- Op den Kamp-Bruls, Y.M.H.; Op den Kamp, Y.J.M.; Veeraiah, P.; Zapata Perez, R.; Phielix, E.; Havekes, B.; Schaart, G.; Kornips, E.; Berendsen, B.R.B.; Virmani, A.; et al. Carnitine Supplementation Improves Insulin Sensitivity and Skeletal Muscle Acetylcarnitine Formation in Patients with Type 2 Diabetes. Diabetes Obes. Metab. 2025, 27, 2864–2877. [Google Scholar] [CrossRef]
- Yin, H.; Yuan, F.; Jiao, F.; Niu, Y.; Jiang, X.; Deng, J.; Guo, Y.; Chen, S.; Zhai, Q.; Hu, C.; et al. Intermittent Leucine Deprivation Produces Long-Lasting Improvement in Insulin Sensitivity by Increasing Hepatic Gcn2 Expression. Diabetes 2022, 71, 206–218. [Google Scholar] [CrossRef]
- De Bandt, J.-P.; Coumoul, X.; Barouki, R. Branched-Chain Amino Acids and Insulin Resistance, from Protein Supply to Diet-Induced Obesity. Nutrients 2022, 15, 68. [Google Scholar] [CrossRef]
- Viollet, B. Targeting the AMPK Pathway for the Treatment of Type 2 Diabetes. Front. Biosci. 2009, 14, 3380. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK Inhibition in Health and Disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276–295. [Google Scholar] [CrossRef] [PubMed]
- Simbrunner, B.; Trauner, M.; Reiberger, T. Review Article: Therapeutic Aspects of Bile Acid Signalling in the Gut-liver Axis. Aliment. Pharmacol. Ther. 2021, 54, 1243–1262. [Google Scholar] [CrossRef] [PubMed]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernández-Ramos, D.; Martínez-Arranz, I.; Delgado, T.C.; Simon, J.; Gutiérrez-de Juan, V.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in Steatohepatitis and Fibrosis in Mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Basta, B.; Mato, J.M.; Craig, A.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Tsvirkun, D.; Hayardeny, L.; Chandar, V.; Schwartz, R.E.; et al. Aramchol Downregulates Stearoyl CoA-Desaturase 1 in Hepatic Stellate Cells to Attenuate Cellular Fibrogenesis. JHEP Rep. 2021, 3, 100237. [Google Scholar] [CrossRef]
- Fernández-Ramos, D.; Lopitz-Otsoa, F.; Delacruz-Villar, L.; Bilbao, J.; Pagano, M.; Mosca, L.; Bizkarguenaga, M.; Serrano-Macia, M.; Azkargorta, M.; Iruarrizaga-Lejarreta, M.; et al. Arachidyl Amido Cholanoic Acid Improves Liver Glucose and Lipid Homeostasis in Nonalcoholic Steatohepatitis via AMPK and MTOR Regulation. World J. Gastroenterol. 2020, 26, 5101–5117. [Google Scholar] [CrossRef]
- Wang, W.; Gao, X.; Niu, W.; Yin, J.; He, K. Targeting Metabolism: Innovative Therapies for MASLD Unveiled. Int. J. Mol. Sci. 2025, 26, 4077. [Google Scholar] [CrossRef] [PubMed]
- Aseem, S.O.; Way, G.; Wang, J.; Zhao, D.; Tai, Y.; Gurley, E.; Zeng, J.; Wang, X.; Hylemon, P.B.; Huebert, R.C.; et al. Aramchol Attenuates Fibrosis in Mouse Models of Biliary Fibrosis and Blocks the TGFβ-Induced Fibroinflammatory Mediators in Cholangiocytes. bioRxiv 2024, bioRxiv:2024.11.06.621880. [Google Scholar]
- Meyers, C.D.; Serrano-Wu, M.; Amer, A.; Chen, J.; Erik, R.; Commerford, R.; Hubbard, B.; Brousseau, M.; Li, L.; Meihui, P.; et al. The DGAT1 Inhibitor Pradigastat Decreases Chylomicron Secretion and Prevents Postprandial Triglyceride Elevation in Humans. J. Clin. Lipidol. 2013, 7, 285. [Google Scholar] [CrossRef]
- Meyers, C.D.; Tremblay, K.; Amer, A.; Chen, J.; Jiang, L.; Gaudet, D. Effect of the DGAT1 Inhibitor Pradigastat on Triglyceride and ApoB48 Levels in Patients with Familial Chylomicronemia Syndrome. Lipids Health Dis. 2015, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Buhman, K.K.; Smith, S.J.; Stone, S.J.; Repa, J.J.; Wong, J.S.; Knapp, F.F.; Burri, B.J.; Hamilton, R.L.; Abumrad, N.A.; Farese, R.V. DGAT1 Is Not Essential for Intestinal Triacylglycerol Absorption or Chylomicron Synthesis. J. Biol. Chem. 2002, 277, 25474–25479. [Google Scholar] [CrossRef]
- O’Farrell, M.; Duke, G.; Crowley, R.; Buckley, D.; Martins, E.B.; Bhattacharya, D.; Friedman, S.L.; Kemble, G. FASN Inhibition Targets Multiple Drivers of NASH by Reducing Steatosis, Inflammation and Fibrosis in Preclinical Models. Sci. Rep. 2022, 12, 15661. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, T.; Yagi, T.; Uchida, T.; Sakai, M.; Mitsushima, M.; Naganuma, T.; Yano, H.; Inaba, Y.; Inoue, H.; Yanagida, K.; et al. Hepatic FASN Deficiency Differentially Affects Nonalcoholic Fatty Liver Disease and Diabetes in Mouse Obesity Models. JCI Insight 2023, 8, e161282. [Google Scholar] [CrossRef]
- Kiepura, A.; Stachyra, K.; Olszanecki, R. Anti-Atherosclerotic Potential of Free Fatty Acid Receptor 4 (FFAR4). Biomedicines 2021, 9, 467. [Google Scholar] [CrossRef]
- Szukiewicz, D. Potential Therapeutic Exploitation of G Protein-Coupled Receptor 120 (GPR120/FFAR4) Signaling in Obesity-Related Metabolic Disorders. Int. J. Mol. Sci. 2025, 26, 2501. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy Deficiency Leads to Protection from Obesity and Insulin Resistance by Inducing Fgf21 as a Mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A Critical Role for ChREBP-Mediated FGF21 Secretion in Hepatic Fructose Metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef]
- Lee, H.A.; Kim, H.Y. Therapeutic Mechanisms and Clinical Effects of Glucagon-like Peptide 1 Receptor Agonists in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 9324. [Google Scholar] [CrossRef]
- Yabut, J.M.; Drucker, D.J. Glucagon-like Peptide-1 Receptor-Based Therapeutics for Metabolic Liver Disease. Endocr. Rev. 2023, 44, 14–32. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon Stimulates Gluconeogenesis by INSP3R1-Mediated Hepatic Lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef]
- Kim, T.; Nason, S.; Holleman, C.; Pepin, M.; Wilson, L.; Berryhill, T.F.; Wende, A.R.; Steele, C.; Young, M.E.; Barnes, S.; et al. Glucagon Receptor Signaling Regulates Energy Metabolism via Hepatic Farnesoid X Receptor and Fibroblast Growth Factor 21. Diabetes 2018, 67, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Qiu, X.; Li, J.; Liang, J.; Li, W.; Zhang, C.; Zhang, Z.-N.; Luan, B. Glucagon-Induced Extracellular CAMP Regulates Hepatic Lipid Metabolism. J. Endocrinol. 2017, 234, 73–87. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic Inhibition of Ketohexokinase Prevents Fructose-Induced Metabolic Dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef] [PubMed]
- Helsley, R.N.; Park, S.H.; Vekaria, H.J.; Sullivan, P.G.; Conroy, L.R.; Sun, R.C.; del Mar Romero, M.; Herrero, L.; Bons, J.; King, C.D.; et al. Ketohexokinase-C Regulates Global Protein Acetylation to Decrease Carnitine Palmitoyltransferase 1a-Mediated Fatty Acid Oxidation. J. Hepatol. 2023, 79, 25–42. [Google Scholar] [CrossRef]
- Futatsugi, K.; Smith, A.C.; Tu, M.; Raymer, B.; Ahn, K.; Coffey, S.B.; Dowling, M.S.; Fernando, D.P.; Gutierrez, J.A.; Huard, K.; et al. Discovery of PF-06835919: A Potent Inhibitor of Ketohexokinase (KHK) for the Treatment of Metabolic Disorders Driven by the Overconsumption of Fructose. J. Med. Chem. 2020, 63, 13546–13560. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Zhang, X.-M.; Boyer, J.L.; Shulman, G.I. Controlled-Release Mitochondrial Protonophore Reverses Diabetes and Steatohepatitis in Rats. Science 2015, 347, 1253–1256. [Google Scholar] [CrossRef]
- Vlasova, K.Y.; Ostroverkhov, P.; Vedenyapina, D.; Yakimova, T.; Trusova, A.; Lomakina, G.Y.; Vodopyanov, S.S.; Grin, M.; Klyachko, N.; Chekhonin, V.; et al. Liposomal Form of 2,4-Dinitrophenol Lipophilic Derivatives as a Promising Therapeutic Agent for ATP Synthesis Inhibition. Nanomaterials 2022, 12, 2162. [Google Scholar] [CrossRef]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.-W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic Platelet-Derived Growth Factor-β, Delivered by Platelets, Promotes Activation of Hepatic Stellate Cells and Biliary Fibrosis in Mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cai, W.; Chu, E.S.H.; Tang, J.; Wong, C.-C.; Wong, S.H.; Sun, W.; Liang, Q.; Fang, J.; Sun, Z.; et al. Hepatic Cyclooxygenase-2 Overexpression Induced Spontaneous Hepatocellular Carcinoma Formation in Mice. Oncogene 2017, 36, 4415–4426. [Google Scholar] [CrossRef]
- Paik, Y.-H.; Kim, J.K.; Lee, J.I.; Kang, S.H.; Kim, D.Y.; An, S.H.; Lee, S.J.; Lee, D.K.; Han, K.-H.; Chon, C.Y.; et al. Celecoxib Induces Hepatic Stellate Cell Apoptosis through Inhibition of Akt Activation and Suppresses Hepatic Fibrosis in Rats. Gut 2009, 58, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα Is a Mediator and Potential Interventional Target for NASH and Subsequent Liver Cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef]
- Raj, H.; Durgia, H.; Palui, R.; Kamalanathan, S.; Selvarajan, S.; Kar, S.S.; Sahoo, J. SGLT-2 Inhibitors in Non-Alcoholic Fatty Liver Disease Patients with Type 2 Diabetes Mellitus: A Systematic Review. World J. Diabetes 2019, 10, 114–132. [Google Scholar] [CrossRef]
- Hsiang, J.C.; Wong, V.W.-S. SGLT2 Inhibitors in Liver Patients. Clin. Gastroenterol. Hepatol. 2020, 18, 2168–2172.e2. [Google Scholar] [CrossRef] [PubMed]
- Mammi, C.; Calanchini, M.; Antelmi, A.; Cinti, F.; Rosano, G.M.C.; Lenzi, A.; Caprio, M.; Fabbri, A. Androgens and Adipose Tissue in Males: A Complex and Reciprocal Interplay. Int. J. Endocrinol. 2012, 2012, 789653. [Google Scholar] [CrossRef]
- Alemany, M. Steroid Hormones Interrelationships in the Metabolic Syndrome: An Introduction to the Ponderostat Hypothesis. Hormones 2012, 11, 272–289. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 294278. [Google Scholar] [CrossRef]
- Sarkar, M.; Yates, K.; Suzuki, A.; Lavine, J.; Gill, R.; Ziegler, T.; Terrault, N.; Dhindsa, S. Low Testosterone Is Associated With Nonalcoholic Steatohepatitis and Fibrosis Severity in Men. Clin. Gastroenterol. Hepatol. 2021, 19, 400–402.e2. [Google Scholar] [CrossRef]
- Karim, G.; Bansal, M.B. Resmetirom: An Orally Administered, Small-Molecule, Liver-Directed, β-Selective THR Agonist for the Treatment of Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Eur. Endocrinol. 2023, 19, 60. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Drug | Dose | Mechanism of Action | Histological Effect | Notable Side Effects |
|---|---|---|---|---|
| Pioglitazone [13,15,38,39,40] | 30–45 mg/day | PPAR-γ agonist; improves insulin sensitivity, reduces hepatic lipotoxicity | Improves steatosis, inflammation, and hepatocyte ballooning | Weight gain, fluid retention, possible increased risk of bladder cancer |
| Vitamin E [15,38,41,42] | 800 IU/day | Antioxidant; neutralizes ROS, reduces oxidative stress and hepatocyte injury | Improves steatohepatitis, no effect on fibrosis | Possible increased risk of prostate cancer |
| Ursodeoxycholic acid [43,44,45,46,47,48] | 13–15 mg/kg/day | Hydrophilic bile acid; cytoprotective, anti-inflammatory, reduces bile acid toxicity | Uncertain; a small number of studies show biochemical but not histological improvement | Diarrhea |
| Name | Reference | NCT | Mechanism of Action | Duration/Phase | Results Posted | Effect | Population |
|---|---|---|---|---|---|---|---|
| MK-4074 | [49] | NCT01431521 | Dual acetyl-CoA carboxylase (ACC1 and -2) inhibitor | 4 weeks, 1 | 2016 | ↓ steatosis; no effect; AST, ALT | Obese, NAFLD |
| Clesacostat PF-05221304 | [50] | NCT03248882 | Dual acetyl-CoA carboxylase (ACC1 and -2) inhibitor | 16 weeks, 2a | 2020 | ↓ steatosis, ALT | Overweight/obese, NAFLD, or NASH |
| Clesacostat PF-05221304 | [51] | NCT03776175 | Dual acetyl-CoA carboxylase (ACC1 and -2) inhibitor | 6 weeks, 2a | 2020 | ↓ steatosis | Overweight/obese NAFLD, T2DM, or other metabolic syndrome comorbidities |
| Clesacostat PF-05221304 | [52] | NCT04399538 | Dual acetyl-CoA carboxylase (ACC1 and -2) inhibitor | 6 weeks, 2a | 2023 | ↓ steatosis; ↑ TG | Overweight/obese NAFLD |
| AXA1125 & AXA1957 | [53] | NCT04073368 | Amino acids and NAC | 16 weeks, N/A | 2021 | ↓ steatosis, ALT; ↑ AE; no effect: AST, HOMA-IR | NASH |
| PXL770 | [54] | NCT03763877 | AMPK activator | 12 weeks, 2 | 2021 | ↓ steatosis, ALT, AST, HbA1c, Fib4; no effect: lipid profile, weight | Overweight/obese NAFLD |
| Metformin | [55] | NCT00063232 | AMPK activator | 48 weeks, 2 | 2011 | ↓ inflammation, ALT, HOMA-IR | NASH |
| Aramchol | [56] | NCT02279524 | Bile salt fatty acid conjugate | 52 weeks, 2b | 2021 | ↓ steatosis, fibrosis, ALT, AST, HbA1c | Overweight/obese NASH |
| Elobixibat | NCT04006145 | Inhibitor of the ileal bile acid transporter | 16 weeks, 2 | 2021 | ↓ LDL | NAFLD/NASH | |
| Pradigastat LCQ908 | NCT01811472 | DGAT1 Inhibitor | 24 weeks, 2 | 2016 | ↓ steatosis, ALT, weight, WC; no effect: AST, GGTP, TG | NAFLD | |
| Ervogastat PF-06865571 | [51] | NCT03776175 | DGAT2 inhibitor | 6 weeks, 2a | 2020 | ↓ steatosis | Overweight/obese NAFLD, T2DM, or other metabolic syndrome comorbidities |
| Ervogastat PF-06865571 | [52] | NCT04399538 | DGAT2 inhibitor | 6 weeks, 2a | 2023 | ↓ steatosis; ↑ TG | Overweight/obese NAFLD |
| Denifanstat TVB-2640 | [57] | NCT03938246 | FAS inhibitor | 12 weeks, 2 | 2024 | ↓ steatosis, ALT | Overweight/obese NAFLD with stage 1-3 fibrosis |
| Icosabutate | [58] | NCT04052516 | Free fatty acid receptor 4 (FFAR4) agonist | 52 weeks, 2b | 2025 | ↓ ALT, GGTP, AST, HOMA-IR, bilirubin, hsCRP, pro-C3; no effect: steatosis | NASH |
| Efruxifermin | [59,60,61] | NCT03976401 | FGF21 analog | 12 weeks, 2a | 2022 | ↓ steatosis, ALT, liver stiffness, pro-C3 | Overweight/obese, NAFLD |
| Pegbelfermin BMS-986036 | [62,63] | NCT02413372 | Pegylated FGF21 | 16 weeks, 2 | 2020 | ↓ steatosis | Overweight/obese, NASH |
| MK-3655 | [64] | NCT04583423 | Humanized mAb that binds KLB FGFR1c/β-klotho activator | 16 weeks, 2 | 2024 | No effect | Overweight/obese, NASH |
| Obeticholic acid INT-747 | [65,66,67,68] | NCT01265498 | FXR agonist | 72 weeks, 2 | 2015 | ↓ histological steatosis, ALT, AST, GGTP, total bilirubin, HDL, body weight, BMI; ↑ alkaline phosphatase, HOMA-IR, total cholesterol, LDL; no effect: TG, HbA1c, WC, blood pressure | NASH |
| Obeticholic acid INT-747 | [69] | NCT00501592 | FXR agonist | 6 weeks, 2 | 2012 | ↓ ALT, GGTP | T2DM, NAFLD |
| TERN-101 | NCT04328077 | FXR agonist | 12 weeks, 2 | 2022 | ↓ ALT | Overweight/obese, NASH | |
| Vonafexor | [70] | NCT03812029 | FXR Agonist | 12 weeks, 2a | 2023 | ↓ steatosis, ALT, GGTP, body weight, WC | F2/F3 fibrosis, NASH |
| EDP-305 | [71] | NCT03421431 | FXR agonist | 12 weeks, 2 | 2021 | ↓ steatosis, ALT, increase ApoA1; no effect: lipid profile, AboB, ApoC3, HOMA-IR, HbA1c | NASH |
| Semaglutide | [72,73] | NCT03987451 | GLP-1 agonist | 48 weeks, 2 | 2024 | ↓ steatosis, ALT, AST, GGTP, HbA1c, HOMA-IR, body weight, BMI | Overweight/obese, NASH |
| Semaglutide | [74] | NCT02970942 | GLP-1 agonist | 72 weeks, 2 | 2021 | ↓ steatosis, ALT, AST, GGTP, HbA1c, body weight | NASH |
| Semaglutide | [75] | NCT04216589 | GLP-1 agonist | 24 weeks, 2 | 2024 | ↓ steatosis, HbA1c, HOMA-IR, body weight, BMI, WC | NAFLD, HIV infection |
| Semaglutide | [76] | NCT04944992 | GLP-1 agonist | 24 weeks, 2 | 2023 | ↓ steatosis, weight, tot. chol. LDL, TG, apoB; ↑ HDL | Overweight/obese, NAFLD |
| Tirzepatide | [77] | NCT04166773 | Dual GLP-1/GIP agonist | 56 weeks, 2 | 2025 | ↓ steatosis, fibrosis, body weight | Overweight/obese, NASH |
| Efinopegdutide | [76] | NCT04944992 | Dual GLP-1/glucagon receptor agonist | 24 weeks, 2 | 2023 | ↓ steatosis, weight, tot. chol. LDL, TG, apoB, HDL | Overweight/obese, NAFLD |
| Survodutide BI456906 | [78] | NCT04771273 | Dual GLP-1/glucagon receptor agonist | 48 weeks, 2 | 2024 | ↓ steatosis, fibrosis | NASH or NAFLD with fibrosis stages F1-F3 |
| Cotadutide MEDI0382 | [79] | NCT04019561 | Dual GLP-1/glucagon receptor agonist | 23 days, 2 | 2022 | ↓ steatosis, ALT, AST, body weight, BMI | Overweight/obese, NAFLD/NASH |
| PF-06835919 | [80,81] | NCT03969719 | Ketohexokinase inhibitor | 16 weeks, 2a | 2022 | ↓ steatosis, ALT, HbA1c; no change: HOMA-IR | NAFLD, T2DM |
| HU6 | [82] | NCT04874233 | Mitochondrial uncoupler, prodrug of 2,4-dinitrophenol (DNP) | 61 days, 2a | 2024 | ↓ steatosis | Overweight/obese, NAFLD |
| Aspirin | [83] | NCT04031729 | Nonsteroidal anti-inflammatory drug (NSAID) | 6 months,1/2 | 2024 | ↓ steatosis | NAFLD |
| dR-pioglitazone PXL065 | [84,85] | NCT04321343 | Deuterium-stabilized R-stereoisomer of pioglitazone (PPARγ agonist) | 36 weeks, 2 | 2023 | ↓ steatosis, fibrosis, ALT, AST, GGTP, HbA1c, HOMA-IR | NAFLD and fibrosis score F1, F2, or F3 |
| Saroglitazar | [86] | NCT03061721 | Dual PPAR α/γ agonist | 16 weeks, 2 | 2024 | ↓ steatosis, fibrosis, ALT | Overweight/obese, NAFLD |
| Saroglitazar | [87] | NCT03863574 | Dual PPAR α/γ agonist | 24 weeks, 2 | 2024 | ↓ ALT, GGTP, AST, TG | Overweight/obese, NASH |
| Lanifibranor IVA337 | [88,89,90,91] | NCT03008070 | Pan-PPAR agonist | 24 weeks, 2 | 2021 | ↓ fibrosis, ALT, AST, GGTP, hsCRP, HOMA, HbA1c, TG, ApoA1; no effect: total cholesterol and LDL; ↑ HDL, adiponectine | NASH |
| Lanifibranor IVA337 | [88,89,90,91] | NCT03459079 | Pan-PPAR agonist | 24 weeks, 2 | 2024 | ↓ steatosis, fibrosis, HbA1c, hepatic IR, fibrosis, ↑ HDL | NAFLD |
| Licogliflozin LIK066 | [92] | NCT03205150 | SGLT1 and SGLT2 inhibitor | 12 weeks, 2 | 2021 | ↓ steatosis, ALT, AST, body weight | NASH |
| Lipocine LPCN 1144 | [93] | NCT04134091 | Prodrug of testosterone | 36 weeks, 2 | 2023 | ↓ steatosis, fibrosis, ALT, AST, ALP, GGTP | NASH |
| TERN-501 | NCT05415722 | THR-β agonist | 12 weeks, 2 | 2025 | ↓ steatosis | Overweight/obese, NASH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drygalski, K. Pharmacological Treatment of MASLD: Contemporary Treatment and Future Perspectives. Int. J. Mol. Sci. 2025, 26, 6518. https://doi.org/10.3390/ijms26136518
Drygalski K. Pharmacological Treatment of MASLD: Contemporary Treatment and Future Perspectives. International Journal of Molecular Sciences. 2025; 26(13):6518. https://doi.org/10.3390/ijms26136518
Chicago/Turabian StyleDrygalski, Krzysztof. 2025. "Pharmacological Treatment of MASLD: Contemporary Treatment and Future Perspectives" International Journal of Molecular Sciences 26, no. 13: 6518. https://doi.org/10.3390/ijms26136518
APA StyleDrygalski, K. (2025). Pharmacological Treatment of MASLD: Contemporary Treatment and Future Perspectives. International Journal of Molecular Sciences, 26(13), 6518. https://doi.org/10.3390/ijms26136518