
Apoptosis in Cardiac Conditions Including Cirrhotic Cardiomyopathy


Abstract
1. Introduction
2. Disordered Cardiovascular (CV) System in Cirrhosis
3. Apoptosis as a Factor in Cardiac Dysfunction
4. Other Types of Cell Death
5. Heart Failure
6. Myocardial Infarction
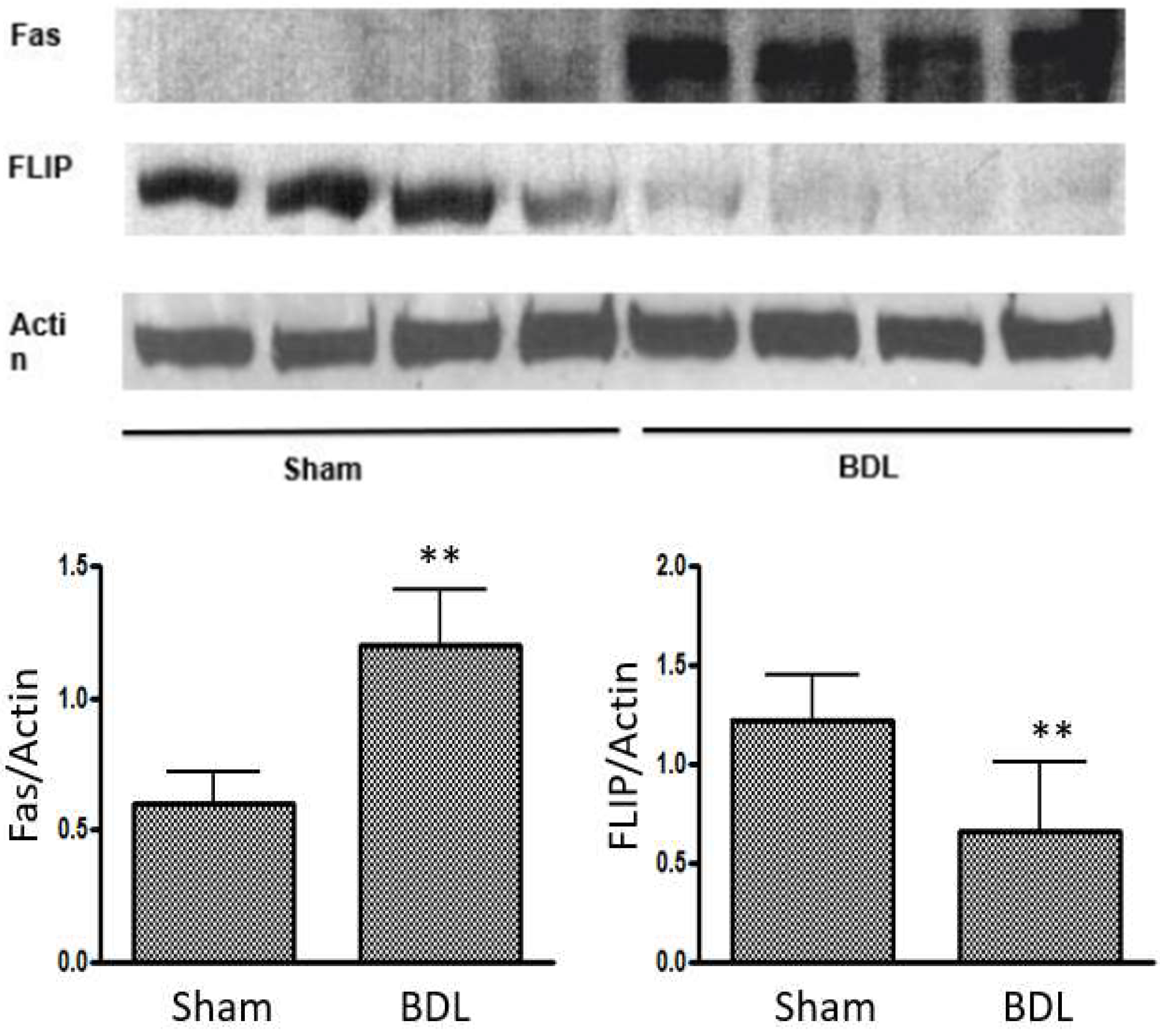
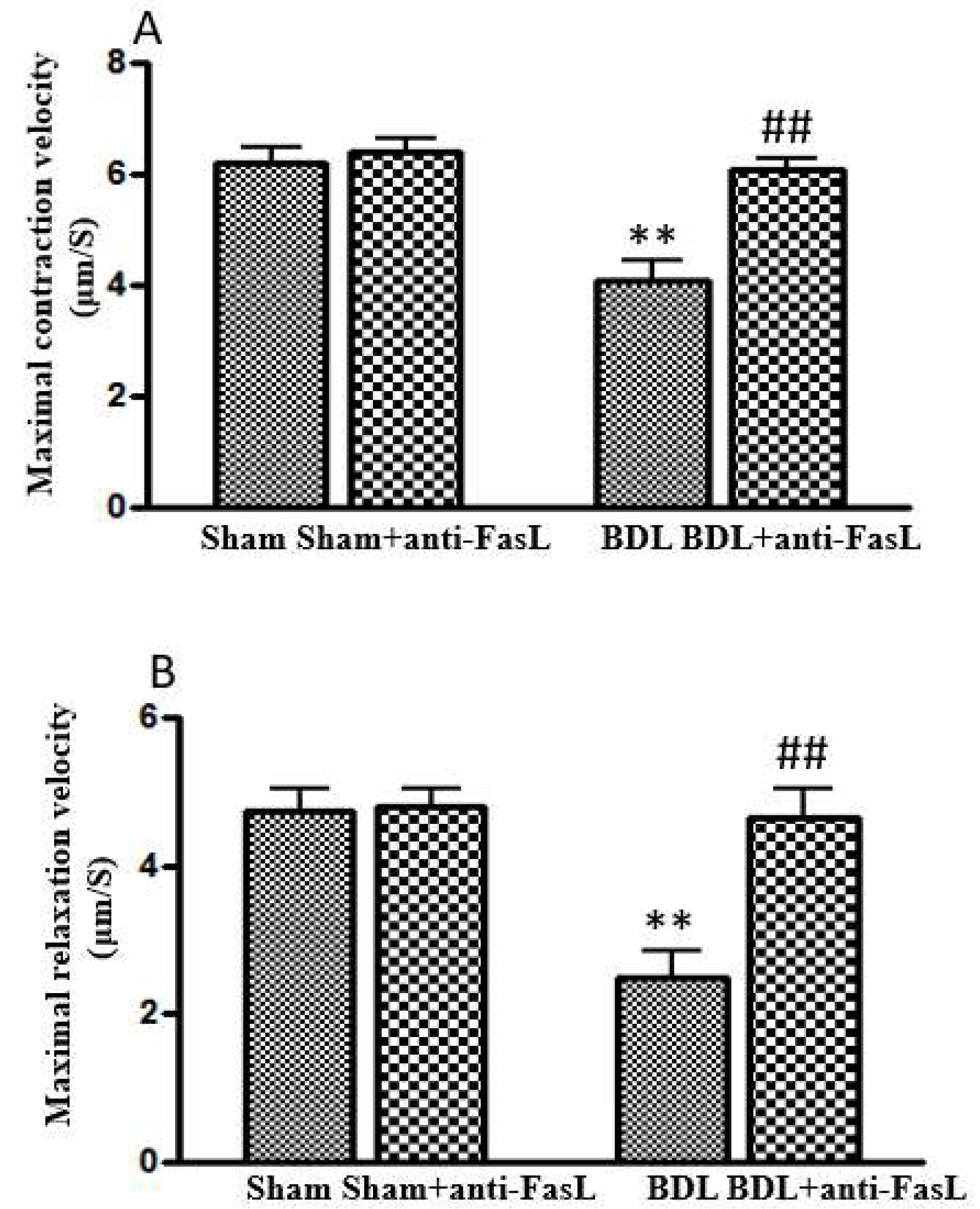
7. Role of Apoptosis in Cirrhotic Cardiomyopathy
8. Oxidative Stress and Apoptosis
9. Inflammation
10. TNFα
11. Galectin-3
12. Potential Therapies to Mitigate Apoptosis in Cirrhotic Cardiomyopathy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, Y.-J.; Hopkins, R.G.; Loo, G. Increased GADD153 gene expression during iron chelation-induced apoptosis in Jurkat T-lymphocytes. Biochim. Biophys. Acta (BBA)–Mol. Cell Res. 2004, 1691, 41–50. [Google Scholar] [CrossRef]
- Fietta, P. Many ways to die: Passive and active cell death styles. Riv. Biol. 2006, 99, 69–83. [Google Scholar]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Boonyong, C.; Angkhasirisap, W.; Kengkoom, K.; Jianmongkol, S. Different protective capability of chlorogenic acid and quercetin against indomethacin-induced gastrointestinal ulceration. J. Pharm. Pharmacol. 2023, 75, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xu, W.; Li, Y.; Wang, Z.; Wang, S.; Liu, Y.; Bai, M.; Lou, Y.; Yang, Q. SIRT1 regulates endoplasmic reticulum stress-related organ damage. Acta Histochem. 2024, 126, 152134. [Google Scholar] [CrossRef]
- Liu, H.; Jayakumar, S.; Traboulsi, M.; Lee, S.S. Cirrhotic cardiomyopathy: Implications for liver transplantation. Liver Transpl. 2017, 23, 826–835. [Google Scholar] [CrossRef]
- Gulcicegi, D.E.; Goeser, T.; Kasper, P. Prognostic assessment of liver cirrhosis and its complications: Current concepts and future perspectives. Front. Med. 2023, 10, 1268102. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Z.; Lee, S.S. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology 2000, 118, 937–944. [Google Scholar] [CrossRef]
- Liu, H.; Song, D.; Lee, S.S. Role of heme oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G68–G74. [Google Scholar] [CrossRef]
- Song, D.; Liu, H.; Sharkey, K.A.; Lee, S.S. Hyperdynamic circulation in portal-hypertensive rats is dependent on central c-fos gene expression. Hepatology 2002, 35, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Naser, J.A.; Lin, G.; Lee, S.S. Cardiomyopathy in cirrhosis: From pathophysiology to clinical care. JHEP Rep. 2024, 6, 100911. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Perello, A.; Agullo, L.; Ruiz-Meana, M.; Schluter, K.D.; Escalona, N.; Graupera, M.; Bosch, J.; Garcia-Dorado, D. Left ventricular hypertrophy in rats with biliary cirrhosis. Hepatology 2003, 38, 589–598. [Google Scholar] [CrossRef]
- Gaskari, S.A.; Liu, H.; D’Mello, C.; Kunos, G.; Lee, S.S. Blunted cardiac response to hemorrhage in cirrhotic rats is mediated by local macrophage-released endocannabinoids. J. Hepatol. 2015, 62, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Liu, H.; Nam, S.W.; Kunos, G.; Lee, S.S. Mechanisms of TNFalpha-induced cardiac dysfunction in cholestatic bile duct-ligated mice: Interaction between TNFalpha and endocannabinoids. J. Hepatol. 2010, 53, 298–306. [Google Scholar] [CrossRef]
- Liu, L.; Liu, H.; Nam, S.W.; Lee, S.S. Protective effects of erythropoietin on cirrhotic cardiomyopathy in rats. Dig. Liver Dis. 2012, 44, 1012–1017. [Google Scholar] [CrossRef]
- Nam, S.W.; Liu, H.; Wong, J.Z.; Feng, A.Y.; Chu, G.; Merchant, N.; Lee, S.S. Cardiomyocyte apoptosis contributes to pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated mice. Clin. Sci. 2014, 127, 519–526. [Google Scholar] [CrossRef]
- Wang, R.; Luo, X.; Li, S.; Wen, X.; Zhang, X.; Zhou, Y.; Xie, W. A bibliometric analysis of cardiomyocyte apoptosis from 2014 to 2023: A review. Medicine 2023, 102, e35958. [Google Scholar] [CrossRef]
- Sheng, S.Y.; Li, J.M.; Hu, X.Y.; Wang, Y. Regulated cell death pathways in cardiomyopathy. Acta Pharmacol. Sin. 2023, 44, 1521–1535. [Google Scholar] [CrossRef]
- Sun, M.; Yin, F.; Wu, X.; Sun, S.; An, Y.; Zhu, M.; Li, X.; Liu, W. Effects of ivabradine on myocardial autophagia and apoptosis in isoprenaline-induced heart failure in mice. Iran. J. Basic Med. Sci. 2024, 27, 107–113. [Google Scholar] [CrossRef]
- Qin, F.; Shite, J.; Mao, W.; Liang, C.S. Selegiline attenuates cardiac oxidative stress and apoptosis in heart failure: Association with improvement of cardiac function. Eur. J. Pharmacol. 2003, 461, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Medali, T.; Couchie, D.; Mougenot, N.; Mihoc, M.; Bergmann, O.; Derks, W.; Szweda, L.I.; Yacoub, M.; Soliman, S.; Aguib, Y.; et al. Thioredoxin-1 and its mimetic peptide improve systolic cardiac function and remodeling after myocardial infarction. FASEB J. 2024, 38, e23291. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, J.; Matilla, L.; Martinez-Martinez, E.; Gueret, A.; Fernandez-Celis, A.; Henry, J.P.; Nicol, L.; Jaisser, F.; Mulder, P.; Ouvrard-Pascaud, A.; et al. Myocardial Injury After Ischemia/Reperfusion Is Attenuated By Pharmacological Galectin-3 Inhibition. Sci. Rep. 2019, 9, 9607. [Google Scholar] [CrossRef]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef]
- Corbic, M.; Sretenovic, J.; Zivkovic, V.; Jakovljevic, V.; Nikolic Turnic, T. Phosphodiesterase-5 Inhibitors as Therapeutics for Cardiovascular Diseases: A Brief Review. Iran. J. Public Health 2023, 52, 870–879. [Google Scholar] [CrossRef]
- Axelrod, J.L.; Pekson, R.; Kitsis, R.N. Introduction to review series: Regulated necrosis programs in heart disease. J. Mol. Cell. Cardiol. 2023, 182, 73–74. [Google Scholar] [CrossRef]
- Grisanti, L.A. TRAIL and its receptors in cardiac diseases. Front. Physiol. 2023, 14, 1256852. [Google Scholar] [CrossRef] [PubMed]
- Szobi, A.; Goncalvesova, E.; Varga, Z.V.; Leszek, P.; Kusmierczyk, M.; Hulman, M.; Kyselovic, J.; Ferdinandy, P.; Adameova, A. Analysis of necroptotic proteins in failing human hearts. J. Transl. Med. 2017, 15, 86. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Li, L.; Chen, Y.; Li, J.; Doan, J.; Steinmetz, R.; Liu, Q. Cardioprotective Role of Tumor Necrosis Factor Receptor-Associated Factor 2 by Suppressing Apoptosis and Necroptosis. Circulation 2017, 136, 729–742. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Z.; Wang, S.; Wang, X.; Mao, J. Mechanism of ferroptosis in heart failure: The role of the RAGE/TLR4-JNK1/2 pathway in cardiomyocyte ferroptosis and intervention strategies. Ageing Res. Rev. 2025, 109, 102770. [Google Scholar] [CrossRef]
- Fonseka, O.; Gare, S.R.; Chen, X.; Zhang, J.; Alatawi, N.H.; Ross, C.; Liu, W. Molecular Mechanisms Underlying Heart Failure and Their Therapeutic Potential. Cells 2025, 14, 324. [Google Scholar] [CrossRef]
- Qin, J.; Yang, Q.; Wang, Y.; Shi, M.; Zhao, X.; Zhou, Y. The role of pyroptosis in heart failure and related traditional chinese medicine treatments. Front. Pharmacol. 2024, 15, 1377359. [Google Scholar] [CrossRef]
- Habimana, O.; Modupe Salami, O.; Peng, J.; Yi, G.H. Therapeutic implications of targeting pyroptosis in Cardiac-related etiology of heart failure. Biochem. Pharmacol. 2022, 204, 115235. [Google Scholar] [CrossRef]
- Redfield, M.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction: A Review. JAMA 2023, 329, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, B.K.; Ruppert, M.; Tokodi, M.; Olah, A.; Braun, S.; Karime, C.; Ladanyi, Z.; Sayour, A.A.; Barta, B.A.; Merkely, B.; et al. Myocardial work index: A marker of left ventricular contractility in pressure- or volume overload-induced heart failure. ESC Heart Fail 2021, 8, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, F.; Wang, M.; Tao, M.; Liu, Z.; Hai, Z. Caspase-3-Responsive Fluorescent/Photoacoustic Imaging of Tumor Apoptosis. Anal. Chem. 2023, 95, 9404–9408. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Ambrose, J.A. Understanding myocardial infarction. F1000Res 2018, 7, F1000–Faculty. [Google Scholar] [CrossRef]
- Mohammed, O.A.; Alghamdi, M.; Alfaifi, J.; Alamri, M.M.S.; Al-Shahrani, A.M.; Alharthi, M.H.; Alshahrani, A.M.; Alhalafi, A.H.; Adam, M.I.E.; Bahashwan, E.; et al. The emerging role of miRNAs in myocardial infarction: From molecular signatures to therapeutic targets. Pathol. Res. Pract. 2024, 253, 155087. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.; Lai, H.; Shen, J.; Gao, F. Inhibiting MiR-320a promotes myocardial apoptosis in myocardial infarction rats through activating VEGF pathway. Cell. Mol. Biol. 2023, 69, 170–175. [Google Scholar] [CrossRef]
- Seropian, I.M.; Cassaglia, P.; Miksztowicz, V.; Gonzalez, G.E. Unraveling the role of galectin-3 in cardiac pathology and physiology. Front. Physiol. 2023, 14, 1304735. [Google Scholar] [CrossRef]
- Li, X.; Tang, X.; Lu, J.; Yuan, S. Therapeutic inhibition of galectin-3 improves cardiomyocyte apoptosis and survival during heart failure. Mol. Med. Rep. 2018, 17, 4106–4112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cheng, K.; Chen, H.; Tu, J.; Shen, Y.; Pang, L.; Wu, W. Galectin-3 knock down inhibits cardiac ischemia-reperfusion injury through interacting with bcl-2 and modulating cell apoptosis. Arch. Biochem. Biophys. 2020, 694, 108602. [Google Scholar] [CrossRef]
- Chahal, D.; Liu, H.; Shamatutu, C.; Sidhu, H.; Lee, S.S.; Marquez, V. Review article: Comprehensive analysis of cirrhotic cardiomyopathy. Aliment. Pharmacol. Ther. 2021, 53, 985–998. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals and antioxidants—quo vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial damage induced by conditions of oxidative stress. Free Radic. Biol. Med. 1999, 26, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, M.; Anjorin, O.; Yoniles, A.; Liu, J.; Wang, M. Importance of Per2 in cardiac mitochondrial protection during stress. Sci. Rep. 2024, 14, 1290. [Google Scholar] [CrossRef]
- Moradi, A.; Aslani, M.R.; Mirshekari Jahangiri, H.; Naderi, N.; Aboutaleb, N. Protective effects of 4-methylumbelliferone on myocardial ischemia/reperfusion injury in rats through inhibition of oxidative stress and downregulation of TLR4/NF-kappaB/NLRP3 signaling pathway. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 5015–5027. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Sharov, V.G. Apoptosis in heart failure. Prog. Cardiovasc. Dis. 1998, 40, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Nguyen, H.H.; Hwang, S.Y.; Lee, S.S. Oxidative Mechanisms and Cardiovascular Abnormalities of Cirrhosis and Portal Hypertension. Int. J. Mol. Sci. 2023, 24, 16850. [Google Scholar] [CrossRef]
- Liu, H.; Alhassan, N.; Yoon, K.T.; Almutlaq, L.; Lee, S.S. Oxidative stress triggers hyperdynamic circulation via central neural activation in portal hypertensive rats. Hepatol. Int. 2023, 17, 689–697. [Google Scholar] [CrossRef]
- Mabatha, K.C.; Letuka, P.; Aremu, O.; Zulu, M.Z. Macrophages of the Heart: Homeostasis and Disease. Biomed. J. 2025, 100867. [Google Scholar] [CrossRef] [PubMed]
- De Biase, L.; Pignatelli, P.; Lenti, L.; Tocci, G.; Piccioni, F.; Riondino, S.; Pulcinelli, F.M.; Rubattu, S.; Volpe, M.; Violi, F. Enhanced TNF alpha and oxidative stress in patients with heart failure: Effect of TNF alpha on platelet O2- production. Thromb. Haemost. 2003, 90, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Staniek, J.; Lorenzetti, R.; Heller, B.; Janowska, I.; Schneider, P.; Unger, S.; Warnatz, K.; Seidl, M.; Venhoff, N.; Thiel, J.; et al. TRAIL-R1 and TRAIL-R2 Mediate TRAIL-Dependent Apoptosis in Activated Primary Human B Lymphocytes. Front. Immunol. 2019, 10, 951. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Wang, Z.; Wei, Y.; Wang, M.; Liu, M.; Wang, X.; Jiang, Y.; Shi, G.; Zhao, D.; et al. Blocking the death checkpoint protein TRAIL improves cardiac function after myocardial infarction in monkeys, pigs, and rats. Sci. Transl. Med. 2020, 12, eaaw3172. [Google Scholar] [CrossRef]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Ferrari, R. The role of TNF in cardiovascular disease. Pharmacol. Res. 1999, 40, 97–105. [Google Scholar] [CrossRef]
- Bryant, D.; Becker, L.; Richardson, J.; Shelton, J.; Franco, F.; Peshock, R.; Thompson, M.; Giroir, B. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998, 97, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Engel Sallberg, A.; Helleberg, S.; Ahmed, S.; Ahmed, A.; Radegran, G. Plasma tumour necrosis factor-alpha-related proteins in prognosis of heart failure with pulmonary hypertension. ESC Heart Fail. 2023, 10, 3582–3591. [Google Scholar] [CrossRef] [PubMed]
- Moe, G.W.; Marin-Garcia, J.; Konig, A.; Goldenthal, M.; Lu, X.; Feng, Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1813–H1820. [Google Scholar] [CrossRef] [PubMed]
- Moe, G.W.; Armstrong, P. Pacing-induced heart failure: A model to study the mechanism of disease progression and novel therapy in heart failure. Cardiovasc. Res. 1999, 42, 591–599. [Google Scholar] [CrossRef]
- Yoon, K.T.; Liu, H.; Zhang, J.; Han, S.; Lee, S.S. Galectin-3 inhibits cardiac contractility via a tumor necrosis factor alpha-dependent mechanism in cirrhotic rats. Clin. Mol. Hepatol. 2022, 28, 232–241. [Google Scholar] [CrossRef]
- Wanninger, J.; Weigert, J.; Wiest, R.; Bauer, S.; Karrasch, T.; Farkas, S.; Scherer, M.N.; Walter, R.; Weiss, T.S.; Hellerbrand, C.; et al. Systemic and hepatic vein galectin-3 are increased in patients with alcoholic liver cirrhosis and negatively correlate with liver function. Cytokine 2011, 55, 435–440. [Google Scholar] [CrossRef]
- Los, M.; Mozoluk, M.; Ferrari, D.; Stepczynska, A.; Stroh, C.; Renz, A.; Herceg, Z.; Wang, Z.Q.; Schulze-Osthoff, K. Activation and caspase-mediated inhibition of PARP: A molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 2002, 13, 978–988. [Google Scholar] [CrossRef]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef]
- Charununtakorn, S.T.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Potential Roles of Humanin on Apoptosis in the Heart. Cardiovasc. Ther. 2016, 34, 107–114. [Google Scholar] [CrossRef]
- Liu, H.; Nguyen, H.H.; Yoon, K.T.; Lee, S.S. Pathogenic Mechanisms Underlying Cirrhotic Cardiomyopathy. Front. Netw. Physiol. 2022, 2, 849253. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, C.; Zhang, J.; Jiao, Z.; Dong, N.; Wang, G.; Wang, Z.; Wang, L. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics 2019, 9, 2346–2360. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Mai, Z.; Zhu, X.; Wu, T.; Chen, Y.; Geng, D.; Wang, J. Mesenchymal stem cell-derived exosomes ameliorate cardiomyocyte apoptosis in hypoxic conditions through microRNA144 by targeting the PTEN/AKT pathway. Stem Cell Res. Ther. 2020, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef] [PubMed]
- Roslan, J.; Giribabu, N.; Karim, K.; Salleh, N. Quercetin ameliorates oxidative stress, inflammation and apoptosis in the heart of streptozotocin-nicotinamide-induced adult male diabetic rats. Biomed. Pharmacother. 2017, 86, 570–582. [Google Scholar] [CrossRef]
- Okafor, C.C.; Perreault-Micale, C.; Hajjar, R.J.; Lebeche, D.; Skiroman, K.; Jabbour, G.; Doye, A.A.; Lee, M.X.; Laste, N.; Gwathmey, J.K. Chronic treatment with carvedilol improves ventricular function and reduces myocyte apoptosis in an animal model of heart failure. BMC Physiol. 2003, 3, 6. [Google Scholar] [CrossRef]
- Liu, H.; Ryu, D.; Hwang, S.; Lee, S.S. Therapies for Cirrhotic Cardiomyopathy: Current Perspectives and Future Possibilities. Int. J. Mol. Sci. 2024, 25, 5849. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Criteria | Systolic Dysfunction | Diastolic Dysfunction |
|---|---|---|
| CCC criteria (2019) | LVEF ≤ 50% Or GLS < 18% | ≥3 of the following: E/e’ ratio ≥ 15 e’ septal < 7 cm/s TR velocity > 2.8 m/s LAVI > 34 mL/m2 |
| First Author (Ref.) | Subject | Disease/Model | Effects |
|---|---|---|---|
| Sun [20] | Mouse | Isoprenaline-induced heart failure | Ivabradine decreases apoptosis and improves cardiac function |
| Qin [21] | Rabbit | Pacing-induced heart failure | Selegiline decreases apoptosis and improves cardiac function |
| Medali [22] | Mouse | Coronary artery ligation-induced MI | Thioredoxin decreases apoptosis and improves cardiac function |
| Ibarrola [23] | Rat | coronary artery ligation-induced ischemia–reperfusion | MCP decreases galectin-3 and apoptosis, and improves cardiac function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.; Ryu, D.G.; Yoon, K.T.; Liu, H.; Lee, S.S. Apoptosis in Cardiac Conditions Including Cirrhotic Cardiomyopathy. Int. J. Mol. Sci. 2025, 26, 6423. https://doi.org/10.3390/ijms26136423
Yu F, Ryu DG, Yoon KT, Liu H, Lee SS. Apoptosis in Cardiac Conditions Including Cirrhotic Cardiomyopathy. International Journal of Molecular Sciences. 2025; 26(13):6423. https://doi.org/10.3390/ijms26136423
Chicago/Turabian StyleYu, Fengxue, Dae Gon Ryu, Ki Tae Yoon, Hongqun Liu, and Samuel S. Lee. 2025. "Apoptosis in Cardiac Conditions Including Cirrhotic Cardiomyopathy" International Journal of Molecular Sciences 26, no. 13: 6423. https://doi.org/10.3390/ijms26136423
APA StyleYu, F., Ryu, D. G., Yoon, K. T., Liu, H., & Lee, S. S. (2025). Apoptosis in Cardiac Conditions Including Cirrhotic Cardiomyopathy. International Journal of Molecular Sciences, 26(13), 6423. https://doi.org/10.3390/ijms26136423