
In Silico Genome-Wide Profiling of Conserved miRNAs in AAA, AAB, and ABB Groups of Musa spp.: Unveiling MicroRNA-Mediated Drought Response

 , , , and
, , , and
Abstract
1. Introduction
2. Results
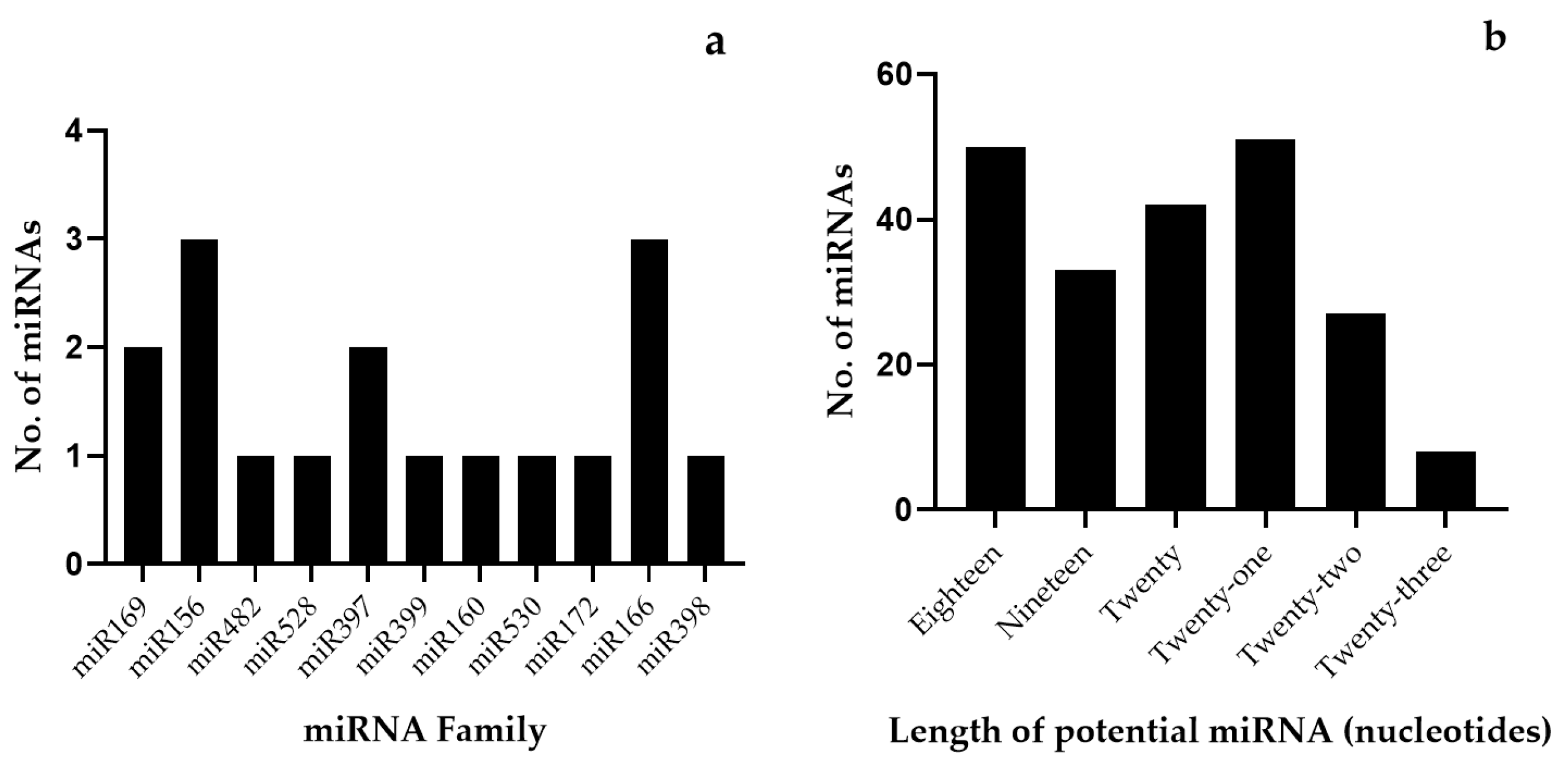
2.1. Identification of Potential miRNAs in Musa spp.
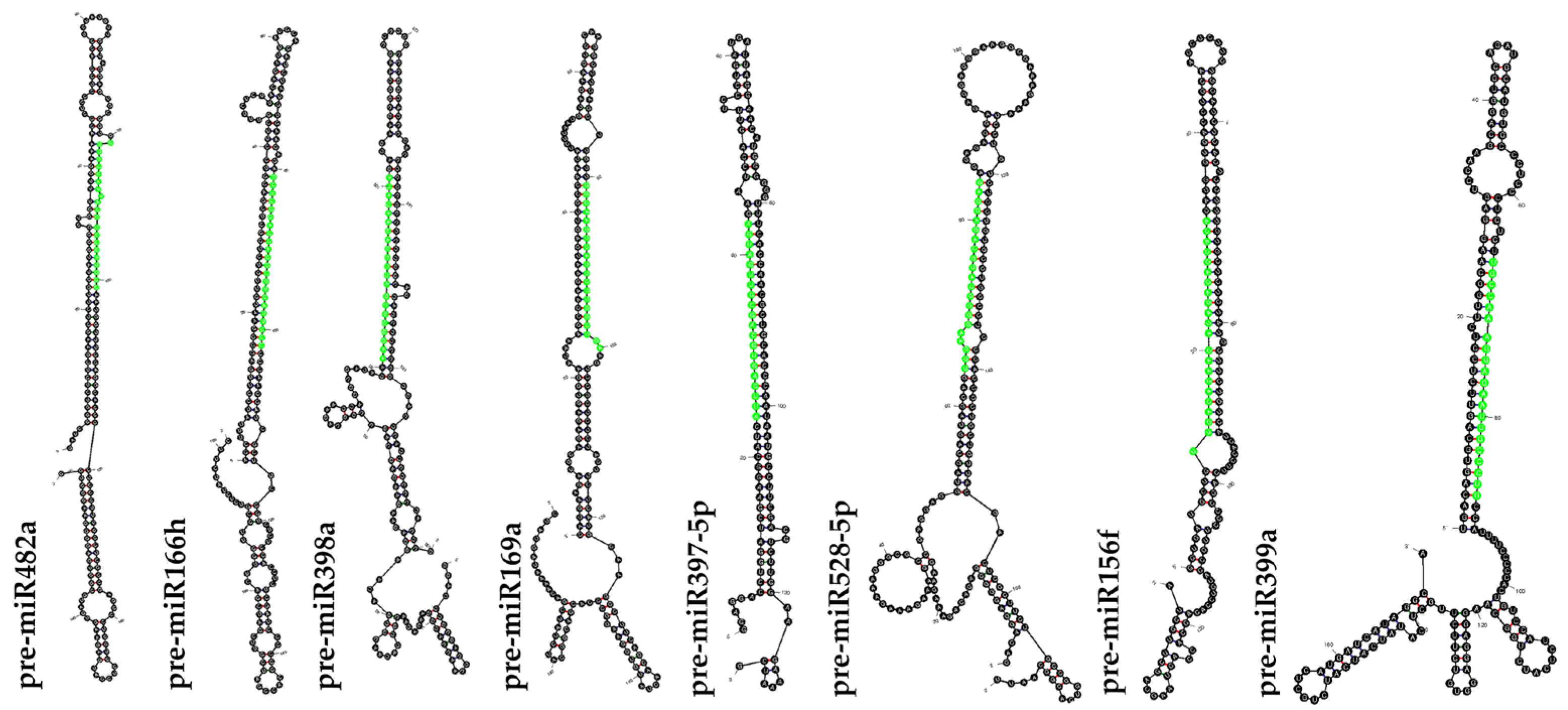
2.2. Prediction and Validation of Secondary Structure of Pre-miRNA
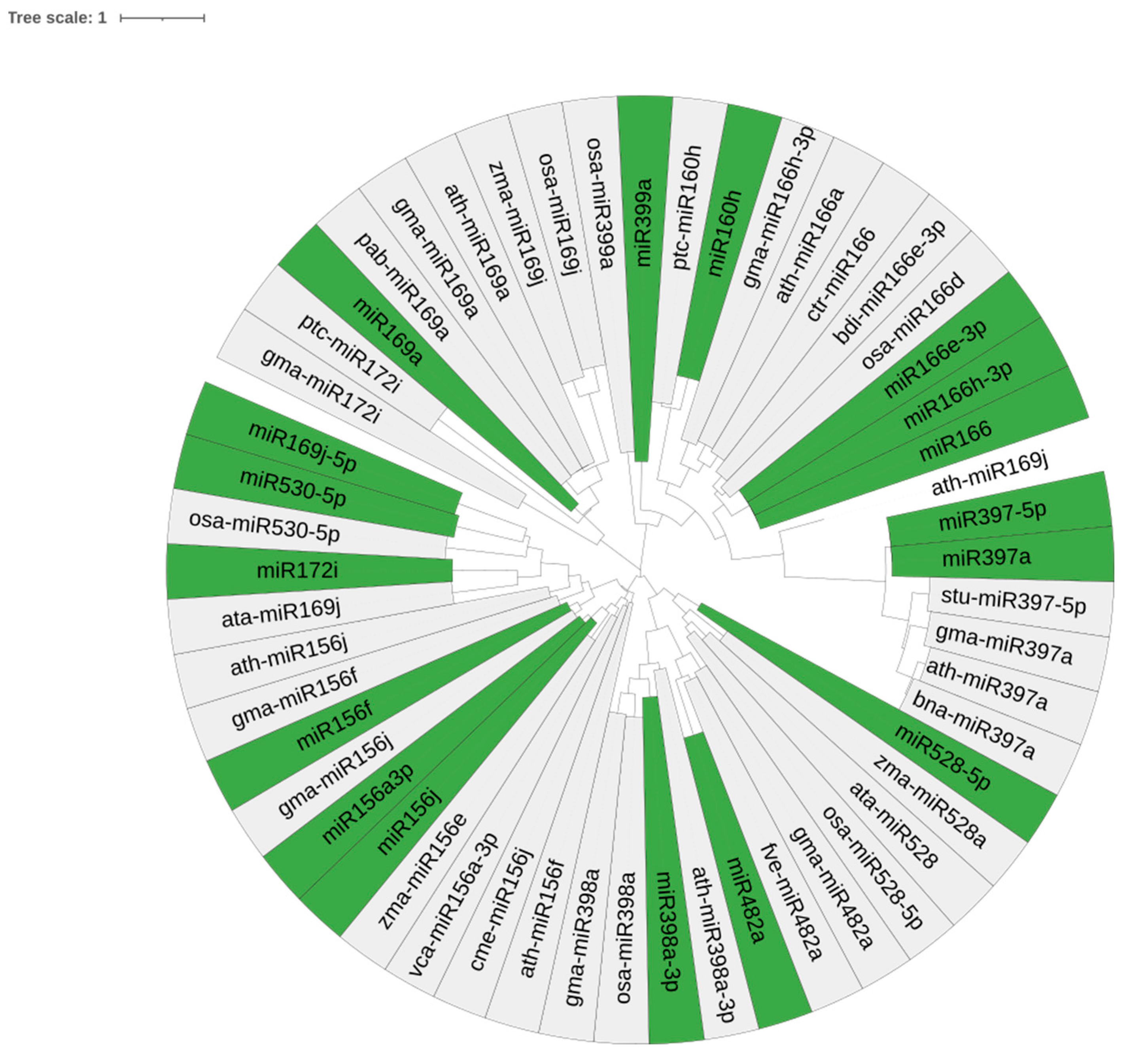
2.3. Conservation and Phylogenetic Analysis
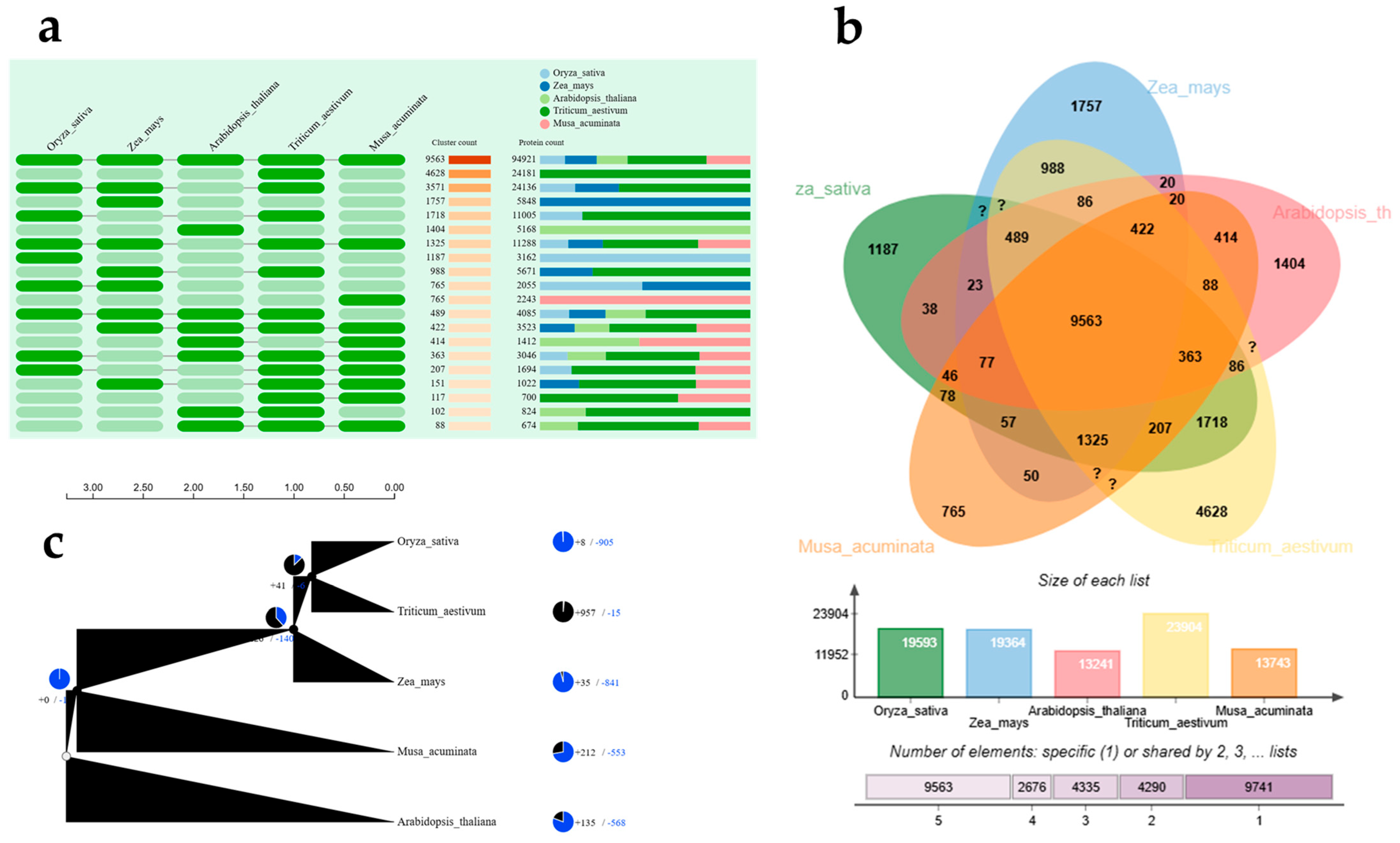
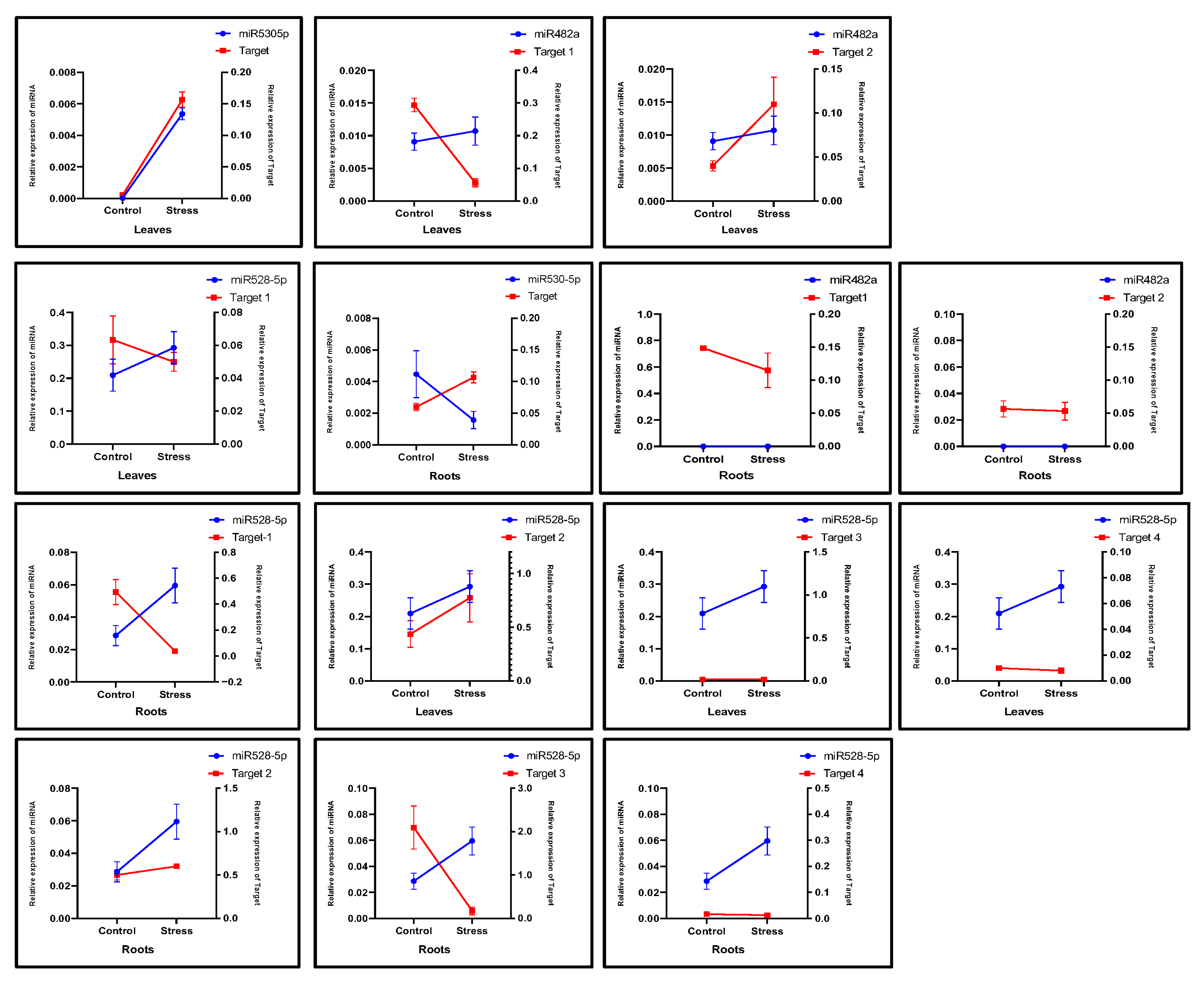
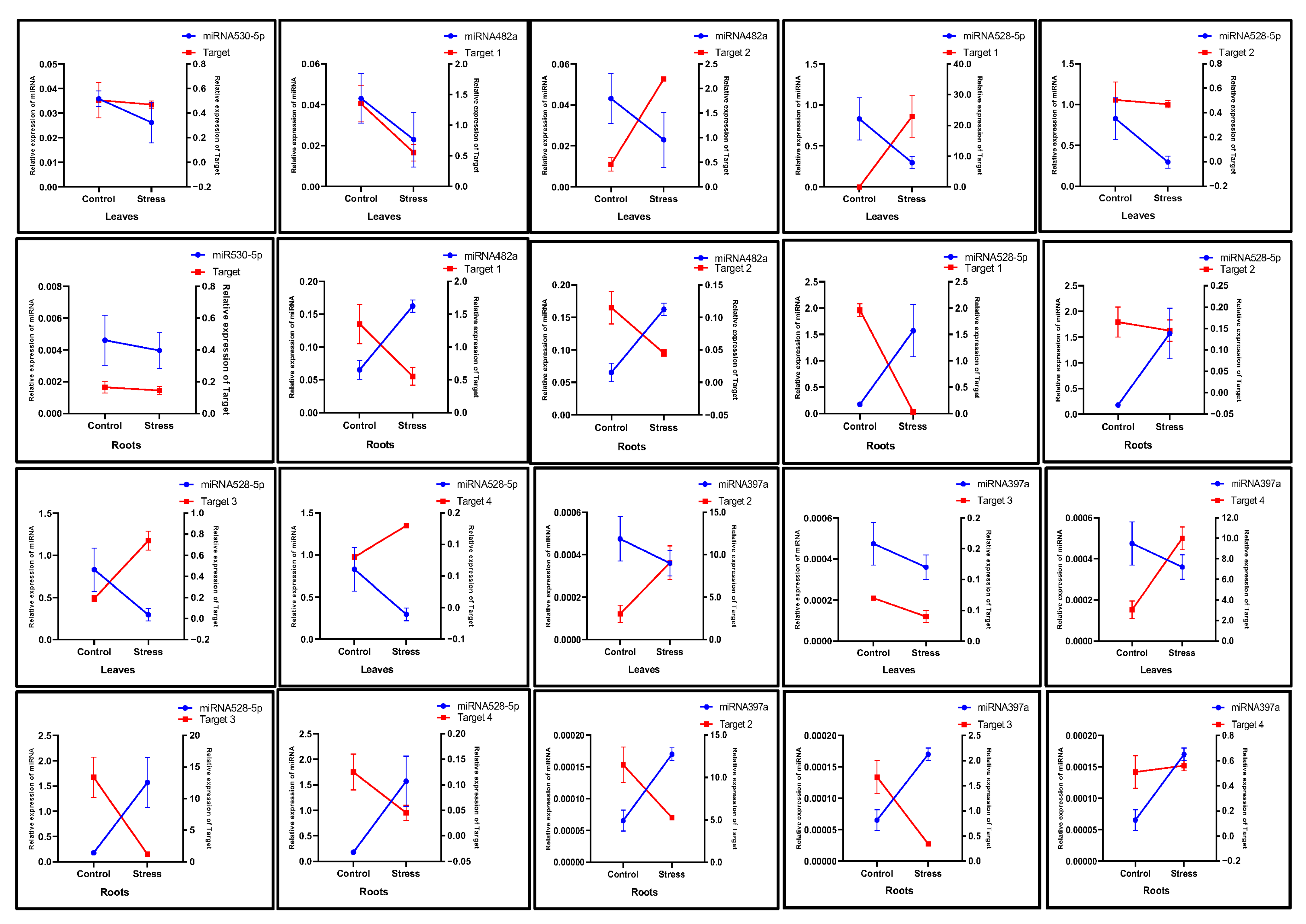
2.4. Prediction of miRNA Target Genes and Functional Analysis
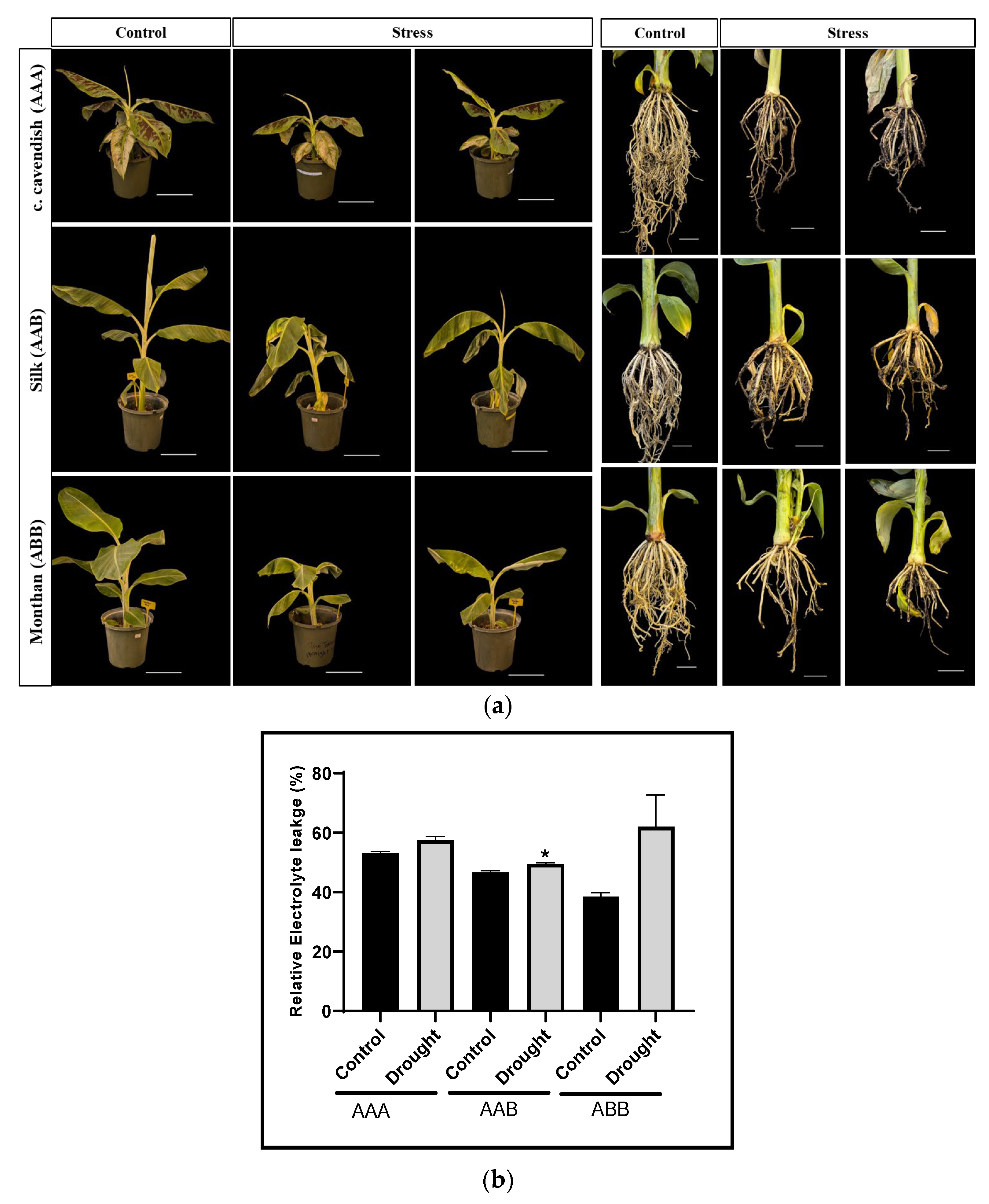
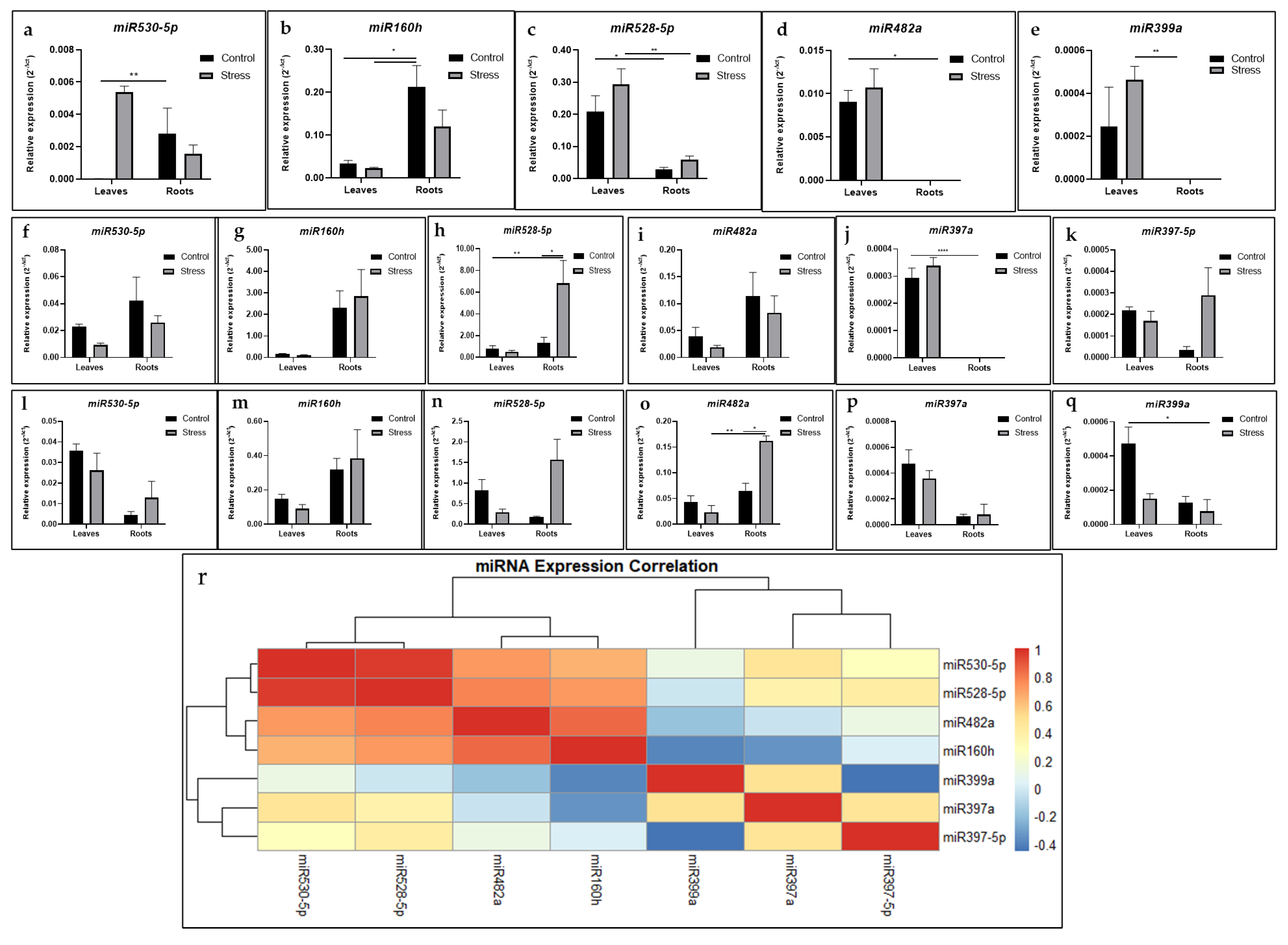
2.5. Expression Analysis of miRNAs and Their Target Genes in Drought-Stressed Plants
3. Discussion
4. Materials and Methods
4.1. Datasets for Query and miRNA Resources
4.2. Bioinformatic Tools Used for Analysis
4.3. Identification of Predicted miRNA Homologs
4.4. Prediction of Secondary miRNA Structure and Validation
4.5. Phylogenetic Analysis
4.6. Prediction and Functional Annotation of miRNA Targets
4.7. Analyses of Orthologous Target Genes in Different Plant spp.
4.8. Plant Materials and Drought Stress
4.9. RNA Isolation and Quantitative Expression Analyses of miRNAs and Their Targets
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, Y.; Jia, T.; Chen, X. The ‘How’ and ‘Where’ of Plant MicroRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef]
- Gao, Z.; Nie, J.; Wang, H. MicroRNA Biogenesis in Plant. Plant Growth Regul. 2021, 93, 1–12. [Google Scholar] [CrossRef]
- Raza, A.; Charagh, S.; Karikari, B.; Sharif, R.; Yadav, V.; Mubarik, M.S.; Habib, M.; Zhuang, Y.; Zhang, C.; Chen, H.; et al. miRNAs for Crop Improvement. Plant Physiol. Biochem. 2023, 201, 107857. [Google Scholar] [CrossRef]
- Gangadhar, B.H.; Venkidasamy, B.; Samynathan, R.; Saranya, B.; Chung, I.-M.; Thiruvengadam, M. Overview of miRNA Biogenesis and Applications in Plants. Biologia 2021, 76, 2309–2327. [Google Scholar] [CrossRef]
- Samynathan, R.; Venkidasamy, B.; Shanmugam, A.; Ramalingam, S.; Thiruvengadam, M. Functional Role of microRNA in the Regulation of Biotic and Abiotic Stress in Agronomic Plants. Front. Genet. 2023, 14, 1272446. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Wang, H.; Xu, H.; Zhou, Y.; Duan, L. MicroRNAs in Plants Development and Stress Resistance. Plant Cell Environ. 2025. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Miura, S.; Nei, M. Origins and Evolution of MicroRNA Genes in Plant Species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef]
- Bologna, N.G.; Schapire, A.L.; Zhai, J.; Chorostecki, U.; Boisbouvier, J.; Meyers, B.C.; Palatnik, J.F. Multiple RNA Recognition Patterns during microRNA Biogenesis in Plants. Genome Res. 2013, 23, 1675–1689. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread Translational Inhibition by Plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Muthusamy, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.S. Computational Prediction, Identification, and Expression Profiling of microRNAs in Banana (Musa spp.) during Soil Moisture Deficit Stress. J. Hortic. Sci. Biotechnol. 2014, 89, 208–214. [Google Scholar] [CrossRef]
- Panda, D.; Dehury, B.; Sahu, J.; Barooah, M.; Sen, P.; Modi, M.K. Computational Identification and Characterization of Conserved miRNAs and Their Target Genes in Garlic (Allium sativum L.) Expressed Sequence Tags. Gene 2014, 537, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Feng, R.; Shi, H.; Ren, M.; Zhang, Y.; Wang, J. Bioinformatic Identification and Expression Analysis of Banana MicroRNAs and Their Targets. PLoS ONE 2015, 10, e0123083. [Google Scholar] [CrossRef]
- Yasin, J.K.; Mishra, B.K.; Pillai, M.A.; Verma, N.; Wani, S.H.; Elansary, H.O.; El-Ansary, D.O.; Pandey, P.S.; Chinnusamy, V. Genome Wide In-Silico miRNA and Target Network Prediction from Stress Responsive Horsegram (Macrotyloma uniflorum) Accessions. Sci. Rep. 2020, 10, 17203. [Google Scholar] [CrossRef]
- Zakeel, M.C.M.; Fernando, S.S.S.T.; Safeena, M.I.S. Computational Discovery of a miRNA and Its Putative Target Genes in Ziziphus Jujuba Using Genome-Wide Expressed Sequence Tags. S. Afr. J. Bot. 2023, 160, 46–54. [Google Scholar] [CrossRef]
- Liu, H.; Qin, C.; Chen, Z.; Zuo, T.; Yang, X.; Zhou, H.; Xu, M.; Cao, S.; Shen, Y.; Lin, H.; et al. Identification of miRNAs and Their Target Genes in Developing Maize Ears by Combined Small RNA and Degradome Sequencing. BMC Genom. 2014, 15, 25. [Google Scholar] [CrossRef]
- Su, C.; Yang, X.; Gao, S.; Tang, Y.; Zhao, C.; Li, L. Identification and Characterization of a Subset of microRNAs in Wheat (Triticum aestivum L.). Genomics 2014, 103, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Archak, S.; Nagaraju, J. Computational Prediction of Rice (Oryza sativa) miRNA Targets. Genom. Proteom. Bioinform. 2007, 5, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, Y.; Liu, Z.; Kong, D.; Duan, M.; Luo, L. Genome-Wide Identification and Analysis of Drought-Responsive microRNAs in Oryza Sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef]
- Kim, H.-J.; Baek, K.-H.; Lee, B.-W.; Choi, D.; Hur, C.-G. In Silico Identification and Characterization of microRNAs and Their Putative Target Genes in Solanaceae Plants. Genome 2011, 54, 91–98. [Google Scholar] [CrossRef]
- Sharma, A.; Bejerano, P.I.A.; Maldonado, I.C.; De Donato Capote, M.; Madariaga-Navarrete, A.; Paul, S. Genome-Wide Computational Prediction and Experimental Validation of Quinoa (Chenopodium quinoa ) microRNAs. Can. J. Plant Sci. 2019, 99, 666–675. [Google Scholar] [CrossRef]
- Ahmed, M.; Ahmed, F.; Ahmed, J.; Akhand, M.R.N.; Azim, K.F.; Imran, M.A.S.; Hoque, S.F.; Hasan, M. In Silico Identification of Conserved miRNAs in the Genome of Fibre Biogenesis Crop Corchorus Capsularis. Heliyon 2021, 7, e06705. [Google Scholar] [CrossRef] [PubMed]
- Wenzhi, W.; Ashraf, M.A.; Ghaffar, H.; Ijaz, Z.; Zaman, W.U.; Mazhar, H.; Zulfqar, M.; Zhang, S. In Silico Identification of Sugarcane Genome-Encoded MicroRNAs Targeting Sugarcane Mosaic Virus. Microbiol. Res. 2024, 15, 273–289. [Google Scholar] [CrossRef]
- Gul, Z.; Barozai, M.Y.K.; Din, M. In-Silico Based Identification and Functional Analyses of miRNAs and Their Targets in Cowpea (Vigna unguiculata L.). AIMS Genet. 2017, 04, 138–165. [Google Scholar] [CrossRef]
- Amah, D.; Alamu, E.; Adesokan, M.; Van Biljon, A.; Maziya-dixon, B.; Swennen, R.; Labuschagne, M. Variability of Carotenoids in a Musa Germplasm Collection and Implications for Provitamin A Biofortification. Food Chem. X 2019, 2, 100024. [Google Scholar] [CrossRef] [PubMed]
- Mbabazi, R.; Harding, R.; Khanna, H.; Namanya, P.; Arinaitwe, G.; Tushemereirwe, W.; Dale, J.; Paul, J. Pro-vitamin A Carotenoids in East African Highland Banana and Other Musa Cultivars Grown in Uganda. Food Sci. Nutr. 2020, 8, 311–321. [Google Scholar] [CrossRef]
- Joshi, R.U.; Singh, A.K.; Singh, V.P.; Rai, R.; Joshi, P. A Review on Adaptation of Banana (Musa spp.) to Cold in Subtropics. Plant Breed. 2023, 142, 269–283. [Google Scholar] [CrossRef]
- Maseko, K.H.; Regnier, T.; Meiring, B.; Wokadala, O.C.; Anyasi, T.A. Musa Species Variation, Production, and the Application of Its Processed Flour: A Review. Sci. Hortic. 2024, 325, 112688. [Google Scholar] [CrossRef]
- Ravi, I. Phenotyping Bananas for Drought Resistance. Front. Physiol. 2013, 4, 9. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Z.; Chen, K.; Yang, M.; Shi, N.; Liao, X. microRNA-mRNA Analysis Reveals Tissue-Specific Regulation of microRNA in Mangrove Clam (Geloina erosa). Biology 2023, 12, 1510. [Google Scholar] [CrossRef]
- Li, Z.; Yang, J.; Cai, X.; Zeng, X.; Zou, J.-J.; Xing, W. A Systematic Review on the Role of miRNAs in Plant Response to Stresses under the Changing Climatic Conditions. Plant Stress 2024, 14, 100674. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, C.; Chen, F.; Ni, S.; Lin, Y.; Lai, Z. High-Throughput Sequencing of Small RNAs Revealed the Diversified Cold-Responsive Pathways during Cold Stress in the Wild Banana (Musa itinerans). BMC Plant Biol. 2018, 18, 308. [Google Scholar] [CrossRef]
- Zhang, F.; Yang, J.; Zhang, N.; Wu, J.; Si, H. Roles of microRNAs in Abiotic Stress Response and Characteristics Regulation of Plant. Front. Plant Sci. 2022, 13, 919243. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Yadav, K.; Srivastava, A.K.; Suprasanna, P.; Ganapathi, T.R. Overexpression of Native Musa-miR397 Enhances Plant Biomass without Compromising Abiotic Stress Tolerance in Banana. Sci. Rep. 2019, 9, 16434. [Google Scholar] [CrossRef]
- Rego, E.C.S.; Pinheiro, T.D.M.; Fonseca, F.C.D.A.; Gomes, T.G.; Costa, E.D.C.; Bastos, L.S.; Alves, G.S.C.; Cotta, M.G.; Amorim, E.P.; Ferreira, C.F.; et al. Characterization of microRNAs and Target Genes in Musa Acuminata Subsp. Burmannicoides, Var. Calcutta 4 during Interaction with Pseudocercospora musae. Plants 2023, 12, 1473. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Chaudhury, S.; Kalita, M.C.; Mondal, T.K. In Silico Identification, Characterization and Expression Analysis of miRNAs in Cannabis sativa L. Plant Gene 2015, 2, 17–24. [Google Scholar] [CrossRef]
- Singh, D.K.; Mehra, S.; Chatterjee, S.; Purty, R.S. In Silico Identification and Validation of miRNA and Their DIR Specific Targets in Oryza Sativa Indica under Abiotic Stress. Non-Coding RNA Res. 2020, 5, 167–177. [Google Scholar] [CrossRef]
- Yin, F.; Qin, C.; Gao, J.; Liu, M.; Luo, X.; Zhang, W.; Liu, H.; Liao, X.; Shen, Y.; Mao, L.; et al. Genome-Wide Identification and Analysis of Drought-Responsive Genes and MicroRNAs in Tobacco. Int. J. Mol. Sci. 2015, 16, 5714–5740. [Google Scholar] [CrossRef]
- Dehury, B.; Panda, D.; Sahu, J.; Sahu, M.; Sarma, K.; Barooah, M.; Sen, P.; Modi, M.K. In Silico Identification and Characterization of Conserved miRNAs and Their Target Genes in Sweet Potato (Ipomoea batatas L.) Expressed Sequence Tags (ESTs). Plant Signal. Behav. 2013, 8, e26543. [Google Scholar] [CrossRef]
- Paul, S.C.; Sharma, A.; Mehta, R.; Paul, S. In Silico Characterization of microRNAs and Their Target Transcripts from Cranberry (Vaccinium macrocarpon). Cytol. Genet. 2020, 54, 82–90. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Novel and Stress-Regulated MicroRNAs and Other Small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef]
- Shriram, V.; Kumar, V.; Devarumath, R.M.; Khare, T.S.; Wani, S.H. MicroRNAs As Potential Targets for Abiotic Stress Tolerance in Plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar] [CrossRef]
- DeYoung, B.J.; Innes, R.W. Plant NBS-LRR Proteins in Pathogen Sensing and Host Defense. Nat. Immunol. 2006, 7, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Mo, W.; Li, Y.; Xiong, Y.; Wang, H.; Zhang, Y.; Lin, M.; Zhang, L.; Li, X. Functional Characterization of NBS-LRR Genes Reveals an NBS-LRR Gene That Mediates Resistance against Fusarium Wilt. BMC Biol. 2024, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, H.; Philippe, F.; Domon, J.-M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell Wall Metabolism in Response to Abiotic Stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Świderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkołazka, A.; Paszczyński, A. Laccase Properties, Physiological Functions, and Evolution. Int. J. Mol. Sci. 2020, 21, 966. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Tang, R.; Qu, H.; Duan, X.; Jiang, Y. Banana sRNAome and Degradome Identify microRNAs Functioning in Differential Responses to Temperature Stress. BMC Genom. 2019, 20, 33. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Zhang, J.; Wu, L.; Qi, Y.; Zhou, J.-M. Identification of microRNAs Involved in Pathogen-Associated Molecular Pattern-Triggered Plant Innate Immunity. Plant Physiol. 2010, 152, 2222–2231. [Google Scholar] [CrossRef]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC Proteins: Regulation and Role in Stress Tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef]
- Begum, Y. Regulatory Role of microRNAs (miRNAs) in the Recent Development of Abiotic Stress Tolerance of Plants. Gene 2022, 821, 146283. [Google Scholar] [CrossRef]
- Xiong, H.; He, H.; Chang, Y.; Miao, B.; Liu, Z.; Wang, Q.; Dong, F.; Xiong, L. Multiple Roles of NAC Transcription Factors in Plant Development and Stress Responses. J. Integr. Plant Biol. 2025, 67, 510–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cheng, C.; Lin, Y.; XuHan, X.; Lai, Z. Genome-Wide Identification and Characterization of mRNAs and lncRNAs Involved in Cold Stress in the Wild Banana (Musa itinerans). PLoS ONE 2018, 13, e0200002. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Sun, B.; Hao, L.; Liu, C.; Zhang, D.; Tang, H.; Li, C.; Li, Y.; Shi, Y.; et al. Genome-Wide Identification and Comparative Analysis of Drought-Related microRNAs in Two Maize Inbred Lines with Contrasting Drought Tolerance by Deep Sequencing. PLoS ONE 2019, 14, e0219176. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, Y.; Teotia, S.; Wang, Z.; Shi, C.; Sun, H.; Gu, Y.; Zhang, Z.; Tang, G. The Interaction between miR160 and miR165/166 in the Control of Leaf Development and Drought Tolerance in Arabidopsis. Sci. Rep. 2019, 9, 2832. [Google Scholar] [CrossRef]
- Hu, Z.; Nie, Z.; Yan, C.; Huang, H.; Ma, X.; Wang, Y.; Ye, N.; Tuskan, G.A.; Yang, X.; Yin, H. Transcriptome and Degradome Profiling Reveals a Role of miR530 in the Circadian Regulation of Gene Expression in Kalanchoë Marnieriana. Cells 2021, 10, 1526. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Lu, Y.; Xie, W.; Zhu, T.; Lian, X. Transcriptome Response to Nitrogen Starvation in Rice. J. Biosci. 2012, 37, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.-F.; Bhutto, S.H.; He, X.-R.; Yang, X.-M.; Zhou, X.-H.; Lin, X.-Y.; Rajput, A.A.; Li, G.-B.; Zhao, J.-H.; et al. Blocking miR530 Improves Rice Resistance, Yield, and Maturity. Front. Plant Sci. 2021, 12, 729560. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Li, K.; Zou, Y.; Chen, L.; Li, X. Identification of Cold-Responsive miRNAs and Their Target Genes in Nitrogen-Fixing Nodules of Soybean. Int. J. Mol. Sci. 2014, 15, 13596–13614. [Google Scholar] [CrossRef]
- Wang, B.; Cheng, D.; Chen, Z.; Zhang, M.; Zhang, G.; Jiang, M.; Tan, M. Bioinformatic Exploration of the Targets of Xylem Sap miRNAs in Maize under Cadmium Stress. Int. J. Mol. Sci. 2019, 20, 1474. [Google Scholar] [CrossRef]
- Li, X.; Chen, P.; Xie, Y.; Yan, Y.; Wang, L.; Dang, H.; Zhang, J.; Xu, L.; Ma, F.; Guan, Q. Apple SERRATE Negatively Mediates Drought Resistance by Regulating MdMYB88 and MdMYB124 and microRNA Biogenesis. Hortic. Res. 2020, 7, 98. [Google Scholar] [CrossRef]
- Zhang, Y.; Waseem, M.; Zeng, Z.; Xu, J.; Chen, C.; Liu, Y.; Zhai, J.; Xia, R. MicroRNA482/2118, a miRNA Superfamily Essential for Both Disease Resistance and Plant Development. New Phytol. 2022, 233, 2047–2057. [Google Scholar] [CrossRef]
- Liao, L.; Xie, B.; Guan, P.; Jiang, N.; Cui, J. New Insight into the Molecular Mechanism of miR482/2118 during Plant Resistance to Pathogens. Front. Plant Sci. 2022, 13, 1026762. [Google Scholar] [CrossRef]
- Yang, R.; Li, P.; Mei, H.; Wang, D.; Sun, J.; Yang, C.; Hao, L.; Cao, S.; Chu, C.; Hu, S.; et al. Fine-Tuning of MiR528 Accumulation Modulates Flowering Time in Rice. Mol. Plant 2019, 12, 1103–1113. [Google Scholar] [CrossRef]
- Liu, A.; Zhou, Z.; Yi, Y.; Chen, G. Transcriptome Analysis Reveals the Roles of Stem Nodes in Cadmium Transport to Rice Grain. BMC Genom. 2020, 21, 127. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kumari, S.; Zhang, L.; Zheng, Y.; Ware, D. Characterization of miRNAs in Response to Short-Term Waterlogging in Three Inbred Lines of Zea mays. PLoS ONE 2012, 7, e39786. [Google Scholar] [CrossRef]
- Song, S.; Xu, Y.; Huang, D.; Ashraf, M.A.; Li, J.; Hu, W.; Jin, Z.; Zeng, C.; Tang, F.; Xu, B.; et al. Identification and Characterization of miRNA169 Family Members in Banana (Musa acuminata L.) That Respond to Fusarium oxysporum f. sp. cubense Infection in Banana Cultivars. PeerJ 2018, 6, e6209. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, M.; Uma, S.; Backiyarani, S.; Saraswathi, M.S.; Chandrasekar, A. Transcriptomic Changes of Drought-Tolerant and Sensitive Banana Cultivars Exposed to Drought Stress. Front. Plant Sci. 2016, 7, 1609. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ Integrated into Galaxy. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A Plant Small RNA Target Analysis Server (2017 Release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An Integrated Platform for Exploring and Visualizing Orthologous Data across Genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-Throughput Functional Annotation and Data Mining with the Blast2GO Suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Nagaraju, J. In Silico Prediction and Characterization of microRNAs from Red Flour Beetle (Tribolium castaneum). Insect Mol. Biol. 2008, 17, 427–436. [Google Scholar] [CrossRef]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A Uniform System for microRNA Annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 Models Variation in Evolutionary Rates among Gene Families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef]
- Meer, L.; Mumtaz, S.; Labbo, A.M.; Khan, M.J.; Sadiq, I. Genome-Wide Identification and Expression Analysis of Calmodulin-Binding Transcription Activator Genes in Banana under Drought Stress. Sci. Hortic. 2019, 244, 10–14. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, W.; Song, S.; Ye, X.; Ding, Z.; Liu, J.; Wang, Z.; Li, J.; Hou, X.; Xu, B.; et al. MaDREB1F Confers Cold and Drought Stress Resistance through Common Regulation of Hormone Synthesis and Protectant Metabolite Contents in Banana. Hortic. Res. 2023, 10, uhac275. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, J.; Xie, X.; Zhang, F.; Dong, H.; Li, Y.; Bi, F.; Huang, X.; Dou, T. Transcriptome Sequence Reveal the Roles of MaGME777 and MabHLH770 in Drought Tolerance in Musa Acuminata. Plant Sci. 2025, 356, 112495. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Sánchez, M.; Huber, D.J.; Vallejos, C.E.; Kelley, K. Physiological, Molecular and Ultrastructural Analyses during Ripening and Over-ripening of Banana (Musa spp., AAA Group, Cavendish Sub-group) Fruit Suggest Characteristics of Programmed Cell Death. J. Sci. Food Agric. 2018, 98, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F. Stem-Loop RT-qPCR for miRNAs. CP Mol. Biol. 2011, 95, 15.10.1–15.10.15. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A Highly Sensitive RT-PCR Method for Detection and Quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EST/TSA ID. | miRNA Families | LE/T | LP | AU% | GC% | A | C | G | U/T | MFE | AMFE | MFEI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FL647992 | miR169j-5p | 571 | 110 | 44.91 | 55.09 | 40 | 58 | 34 | 35 | −55.70 | −50.630 | 0.92 |
| JK538379 | miR156f | 229 | 127 | 49.28 | 50.72 | 30 | 41 | 30 | 39 | −58.40 | −45.980 | 0.91 |
| ES434836 | miR156a-3p | 789 | 91 | 41.21 | 58.79 | 27 | 49 | 51 | 55 | −76.80 | −84.400 | 1.44 |
| DN238517 | miR482a | 236 | 65 | 38.12 | 61.88 | 29 | 55 | 51 | 46 | −88.40 | −136.000 | 2.20 |
| FL667486 | miR528-5p | 674 | 62 | 45.56 | 54.44 | 52 | 38 | 67 | 23 | −66.00 | −106.000 | 1.95 |
| FL666615 | miR397a | 586 | 96 | 56.39 | 43.61 | 38 | 28 | 37 | 30 | −58.50 | −60.930 | 1.40 |
| FL666459 | miR399a | 561 | 136 | 44.12 | 55.88 | 35 | 50 | 38 | 47 | −55.80 | −41.029 | 0.73 |
| FL666054 | miR160h | 392 | 96 | 37.57 | 62.43 | 44 | 49 | 53 | 35 | −71.30 | −74.270 | 1.19 |
| FL659295 | miR530-5p | 807 | 65 | 68.33 | 31.67 | 29 | 29 | 33 | 29 | −46.40 | −71.380 | 2.25 |
| FL666615 | miR397-5p | 586 | 78 | 60.77 | 39.23 | 35 | 28 | 37 | 30 | −56.60 | −72.560 | 1.85 |
| FL647629 | miR169a | 721 | 116 | 32.22 | 67.78 | 54 | 39 | 42 | 45 | −67.00 | −57.760 | 0.85 |
| GABH01012340 | miR166 | 4052 | 153 | 47.25 | 52.75 | 29 | 56 | 40 | 57 | −59.8 | −39.085 | 0.71 |
| GABH01015288 | miR156j | 4850 | 81 | 44.79 | 55.21 | 42 | 34 | 72 | 44 | −87.7 | −108.272 | 2.13 |
| GABH01000646 | miR398a-3p | 1536 | 113 | 50.83 | 49.17 | 50 | 40 | 49 | 42 | −63.5 | −56.195 | 0.96 |
| GABH01012340 | miR166e-3p | 4052 | 141 | 47.51 | 52.49 | 29 | 55 | 40 | 57 | −59.9 | −42.482 | 0.69 |
| GABH01012340 | miR166h-3p | 4052 | 142 | 48.07 | 51.93 | 30 | 55 | 39 | 57 | −60.4 | −42.535 | 0.78 |
| GABH01002598 | miR172i | 2269 | 160 | 51.11 | 48.89 | 39 | 35 | 53 | 53 | −51.3 | −32.063 | 0.74 |
| Musa miRNA | miRNA Homolog | Mature miRNA Sequences | LM | Loc | Strand |
|---|---|---|---|---|---|
| miR169j-5p | ata-miR169j-5p | UAGCCAAGGAUGAUUUGCCUGUG | 23 | 5′ | − |
| miR156f | gma-miR156f | UUGACAGAAGAGAGAGAGCACA | 22 | 5′ | − |
| miR156a-3p | vca-miR156a-3p | UGCUCACUUCUCUUUCUGUCAG | 21 | 3′ | + |
| miR482a | fve-miR482a | UCUUUCCAAUUCCUCCCAUGCC | 22 | 3′ | + |
| miR528-5p | osa-miR528-5p | UGGAAGGGGCAUGCAGAGGAG | 21 | 5′ | + |
| miR397a | bna-miR397a | UCAUUGAGUGCAGCGUUGAUGU | 21 | 5′ | + |
| miR399a | osa-miR399a | UGCCAAAGGAGAAUUGCCCUG | 21 | 3′ | + |
| miR160h | ptc-miR160h | UGCCUGGCUCCCUGCAUGCCA | 21 | 5′ | − |
| miR530-5p | osa-miR530-5p | UGCAUUUGCACCUGCACCUA | 20 | 5′ | + |
| miR397-5p | stu-miR397-5p | AUUGAGUGCAGCGUUGAUGAC | 20 | 5′ | + |
| miR169a | pab-miR169a | UCAGCCAAGAAUGACUUGCCC | 20 | 5′ | − |
| miR160 | ctr-miR166 | UCGGACCAGGCUUCAUUCCCCC | 22 | 5′ | + |
| miR156j | cme-miR156j | GUUGACAGAAGAGAGUGAGCAC | 22 | 5′ | + |
| miR398a-3p | ath-miR398a-3p | UGUGUUCUCAGGUCACCCCUU | 21 | 3′ | − |
| miR166e-3p | bdi-miR166e-3p | CUCGGACCAGGCUUCAUUCCC | 21 | 3′ | + |
| miR166h-3p | gma-miR166h-3p | UCUCGGACCAGGCUUCAUUCC | 21 | 3′ | + |
| miR172i | ptc-miR172i | AGAAUCCUGAUGAUGCUGCAA | 20 | 3′ | + |
| miRNA Family | Name of Targets | Target Description | E-Value | Inhibition | Functions |
|---|---|---|---|---|---|
| miR482a | Target-1 | NBS-LRR class resistance protein (Fragment) | 2 | Cleavage | Defense Response |
| Target-2 | Disease resistance protein RGA2, putative, expressed | 3 | Translation | Defense Response | |
| miR528-5p | Target-1 | Polyphenol oxidase, chloroplastic | 1 | Cleavage | Stress Responses |
| Target-2 | Putative Leucyl-tRNA synthetase, cytoplasmic | 2.5 | Cleavage | Metabolism | |
| Target-3 | Mavicyanin | 3 | Cleavage | Transport | |
| Target-4 | Putative serine/threonine protein kinase fray2 | 3 | Cleavage | Signal Transduction | |
| miR397a | Target-1 | Laccase-4 | 1.5 | Cleavage | Stress Responses |
| Target-2 | Putative S-(hydroxymethyl)glutathione dehydrogenase | 2 | Cleavage | Stress Responses | |
| Target-3 | Serine carboxypeptidase-like 35 | 3 | Cleavage | Stress Responses | |
| Target-4 | Putative NAC domain-containing protein 74 | 3 | Cleavage | Transcription Factor | |
| miR530-5p | Target | Tetratricopeptide repeat domain-containing protein, expressed | 2.5 | Cleavage | Cellular Process |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, K.; Ihearahu, O.C.; Agbor, V.E.J.; Evans, T.; Naitchede, L.H.S.; Ray, S.; Ude, G. In Silico Genome-Wide Profiling of Conserved miRNAs in AAA, AAB, and ABB Groups of Musa spp.: Unveiling MicroRNA-Mediated Drought Response. Int. J. Mol. Sci. 2025, 26, 6385. https://doi.org/10.3390/ijms26136385
Saha K, Ihearahu OC, Agbor VEJ, Evans T, Naitchede LHS, Ray S, Ude G. In Silico Genome-Wide Profiling of Conserved miRNAs in AAA, AAB, and ABB Groups of Musa spp.: Unveiling MicroRNA-Mediated Drought Response. International Journal of Molecular Sciences. 2025; 26(13):6385. https://doi.org/10.3390/ijms26136385
Chicago/Turabian StyleSaha, Kishan, Onyinye C. Ihearahu, Vanessa E. J. Agbor, Teon Evans, Labode Hospice Stevenson Naitchede, Supriyo Ray, and George Ude. 2025. "In Silico Genome-Wide Profiling of Conserved miRNAs in AAA, AAB, and ABB Groups of Musa spp.: Unveiling MicroRNA-Mediated Drought Response" International Journal of Molecular Sciences 26, no. 13: 6385. https://doi.org/10.3390/ijms26136385
APA StyleSaha, K., Ihearahu, O. C., Agbor, V. E. J., Evans, T., Naitchede, L. H. S., Ray, S., & Ude, G. (2025). In Silico Genome-Wide Profiling of Conserved miRNAs in AAA, AAB, and ABB Groups of Musa spp.: Unveiling MicroRNA-Mediated Drought Response. International Journal of Molecular Sciences, 26(13), 6385. https://doi.org/10.3390/ijms26136385