
Biomimetic Tumour Model Systems for Pancreatic Ductal Adenocarcinoma in Relation to Photodynamic Therapy

 , , , , ,
, , , , ,  ,
on behalf of the Photodynamic Therapy Study Group
,
on behalf of the Photodynamic Therapy Study Group
Abstract
1. Introduction
2. Two-Dimensional and Three-Dimensional PDAC Models
2.1. Spheroid Models
Limitations of Spheroid Models
2.2. Co-Culture Models
Limitations of Co-Culture Models
2.3. Organoid Models
Limitations of Organoid Models
2.4. Hydrogel Scaffold-Based Models
Limitations of Hydrogel Scaffold-Based Models
2.5. Assembloid Models
Limitations of Assembloid Models
2.6. Microfluidic Models
Limitations of Microfluidics Models
2.7. Xenograft Models
2.7.1. Cell Line-Derived Tumour Xenograft Models
Limitations of Xenograft Models
2.7.2. Organoid-Based Xenograft Models
Limitations of Organoid-Based Xenograft Models
2.7.3. Patient-Derived Xenograft Model
Limitations of Patient-Derived Xenograft Models
2.8. Evolving Technologies for PDAC Mimicry
3. Application of Biomimetic PDAC Models in PDT Research
3.1. In Vivo PDAC Models Used in PDT Research
Mouse Models
3.2. Ex Vivo PDAC Models Used in PDT Research
PDX Models
3.3. In Vitro PDAC Models Used in PDT Research
3.3.1. Cell Culture Monolayers (2-D)
3.3.2. Heterotypic Spheroid Cultures
3.3.3. Hydrogel Scaffold-Supported Spheroids
Matrigel
Collagen
Alginate and Gelatin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Disease | Source | Models | Methods | Tested in PDT | Ref. |
|---|---|---|---|---|---|---|
| A818-1 | PDAC | Metastatic | Spheroids | plates coated with agarose in non-supplemented medium at a ratio of 1:3 | No | [237] |
| A818-4 | PDAC | Metastatic | Spheroids | nonadherent round-bottom plates with medium containing 20% methyl cellulose | No | [238] |
| A818-6 | PDAC | Metastatic | Spheroids | plates coated with agarose or cultured in rotating culture vessels | No | [239] |
| ASPC-1 | PDAC | Metastatic | Spheroids | ultra-low attachment round-bottom plates; PDT regimen: BPD, 690 nm, 150 mW/cm2, 0–80 J/cm2 | Yes | [219,220] |
| Co-cultured spheroids | MRC-5, patient-derived CAFs; PDT regimen: BPD, 690 nm, 150 mW/cm2, 0–80 J/cm2 | Yes | [219,220] | |||
| Microfluidic spheroids | spheroids were generated by liquid overlay method and then transferred to a microfluidic chip | No | [240] | |||
| Hydrogel-based spheroids | PEG hydrogel | No | [241] | |||
| Organoids † | Matrigel; Cultrex Reduced Growth Factor BME, low attachment plates | No | [242,243] | |||
| Cell line-derived xenografts | male nude mice (16 wk), subcutaneous; PDT regimen: zinc phthalocyanine-loaded mesoporous silica nanoparticles, 685 nm, 50 mW/cm2, 100 J/cm2, 1980 s | Yes | [244] | |||
| Cell line-derived xenografts | male SCID nude mice (6 wk), orthotopic; PDT regimen: verteporfin, 690 nm, 74 mW/cm2, 10–40 J/cm2, 135–540 s | Yes | [245] | |||
| BXPC-3 | PDAC | Primary | Spheroids | medium containing 0.24% methylcellulose | No | [40] |
| Co-cultured spheroids | MRC-5, suspended in polyacrylamide hydrogel coated with collagen type I | No | [246] | |||
| Microfluidic spheroids | HepaChip device | No | [117] | |||
| Hydrogel-based spheroids | Matrigel, collagen I; PDT regimen: BPD, 690 nm, 100 mW/cm2, 0.5–25 J/cm2 | Yes | [232] | |||
| Organoids † | Matrigel, collagen I, tumour-associated PSCs and M2 macrophages in suspension | No | [247] | |||
| Cell line-derived xenografts | female athymic NCR-Nu-F nude mice (5–8 wk), subcutaneous | No | [248] | |||
| Cell line-derived xenografts | BALB/c nude mice (6 wk), orthotopic; PDT regimen: Ce6, 660 nm, 200 mW/cm2, 200 J/cm2, 1000 s | Yes | [249] | |||
| Capan-1 | PDAC | Metastatic | Spheroids | medium containing 0.24% methylcellulose | No | [40] |
| Co-cultured spheroids | PSCs | No | [250] | |||
| Hydrogel-based spheroids | Matrigel and medium mixture (1:2) | No | [250] | |||
| Cell line-derived xenografts | female BALB/c nude mice (6 wk), subcutaneous; PDT regimen: rBC2-IR700, NIR light 670–710 nm, 100 J/cm2 | Yes | [251] | |||
| Cell line-derived xenografts | female BALB/c nude mice (6 wk), orthotopic; PDT regimen: rBC2-IR700, NIR light 670–710 nm, 100 J/cm2 | Yes | [251] | |||
| Capan-2 | PDAC | Primary | Spheroids | ultra-low attachment round-bottom plates; PDT regimen: BPD, 690 nm, 150 mW/cm2, 0.5–40 J/cm2 | Yes | [219,252] |
| Co-cultured spheroids | patient-derived CAFs; PDT regimen: BPD, 690 nm, 150 mW/cm2, 0.5–40 J/cm2 | Yes | [219] | |||
| Cell line-derived xenografts | athymic nude mice, subcutaneous; PDT regimen: temoporfin, 980 nm, 500 mW/cm2, 90 J/cm2, 180 s | Yes | [253] | |||
| CFPAC-1 | PDAC | Metastatic | Spheroids | cancer stem cell medium | No | [254] |
| Co-cultured spheroids | MRC-5 or PSCs; PDT regimen: BPD, 690 nm, 100 mW/cm2, 0.5–25 J/cm2 | Yes | [204] | |||
| Organoids † | collagen I, CFPAC-1 cells expressing GRHL2 | No | [255] | |||
| Cell line-derived xenografts | athymic CD1 nude mice (6–8 wk), subcutaneous | No | [256] | |||
| Cell line-derived xenografts | female BALB/c nude mice (5 wk), orthotopic | No | [257] | |||
| COLO 357 | PASC | Metastatic | Spheroids | nonadherent round-bottom plates with medium containing 20% methyl cellulose | No | [238] |
| Co-cultured spheroids | patient-derived CAFs | No | [258] | |||
| Hydrogel-based spheroids | gelatin-norbornene (GelNB)-based hydrogels | No | [258] | |||
| Cell line-derived xenografts | female SCID/bg mice (4 wk), subcutaneous | No | [259] | |||
| Cell line-derived xenografts | female SCID/bg mice (4 wk), orthotopic | No | [259] | |||
| DAN-G | PAC | Primary | Spheroids | polystyrene-coated ultra-low attachment plates | No | [260] |
| Co-cultured spheroids | fibroblast-conditioned medium | No | [260] | |||
| Cell line-derived xenografts | male NMRI nude mice (4–6 wk), subcutaneous | No | [261] | |||
| HPAC | PAC | Primary | Spheroids | round-bottom plates pretreated with 0.5% polyHEMA, plates coated with 1% agarose in DMEM; PDT regimen: Ru-bqp-ester, 470 nm, 2.4 ± 0.2 mW/cm2, 4.3 ± 0.4 J/cm2, 1800 s | Yes | [262,263] |
| Cell line-derived xenografts | female athymic BALB/c nude mice (7 wk), subcutaneous | No | [264] | |||
| Cell line-derived xenografts | female SCID nude mice (5 wk), orthotopic | No | [265] | |||
| HPAF-II | PDAC | Metastatic | Spheroids | plates coated with 1% agarose in DMEM | No | [263] |
| Co-cultured spheroids | fibroblast (DF-1) cells | No | [266] | |||
| Cell line-derived xenografts | female athymic NCR-Nu-F nude mice (5–8 wk), subcutaneous | No | [248] | |||
| Cell line-derived xenografts | female NOD/SCID nude mice (8 wk), orthotopic | No | [267] | |||
| Hs 766T | PAC | Metastatic | Hydrogel-based spheroids | Matrigel | No | [268] |
| Cell line-derived xenografts | female athymic nude mice (6 wk), subcutaneous | No | [268] | |||
| JoPaCa-1 | PDAC | Primary | Cell line-derived xenografts | NOD.Cg-Prkdcscid Il2rgtm1Wjl (NOD/SCID/c or NSG) mice, orthotopic | No | [269] |
| KCI-MOH1 | PAC | Primary | Cell line-derived xenografts | female SCID mice (4 wk), subcutaneous | No | [270] |
| KLM-1 | PDAC | Metastatic | Spheroids | NanoCulture plates | No | [271] |
| Hydrogel-based spheroids | 2-methoxyethyl methacrylate and 2-(diethylamino)ethyl methacrylate heteropolymer | No | [272] | |||
| Cell line-derived xenografts | female athymic nude mice (5 wk), subcutaneous | No | [273] | |||
| Cell line-derived xenografts | female NGS mice (5–6 wk), orthotopic | No | [274] | |||
| KP-1N | PASC | Metastatic | Cell line-derived xenografts | nude mice (6–8 wk), subcutaneous | No | [275] |
| KP-2 | PA | Primary | Cell line-derived xenografts | nude mice (6–8 wk), subcutaneous | No | [275] |
| KP-3 | PDAC | Metastatic | Cell line-derived xenografts | nude mice (6–8 wk), subcutaneous | No | [275] |
| KP-4 | PA | Metastatic | Spheroids | ultra-low attachment round-bottom plates | No | [276] |
| Cell line-derived xenografts | BALB/c nude mice (6–12 wk), subcutaneous | No | [277] | |||
| MIA PaCa-2 | PDAC | Primary | Spheroids | ultra-low attachment round-bottom plates; PDT regimen: BPD (including antibody-targeted BPD and liposomal BPD), 690 nm, 150 mW/cm2, 0–80 J/cm2 | Yes | [200,218,219,220] |
| Co-cultured spheroids | MRC-5, patient-derived CAFs; PDT regimen: BPD (including antibody-targeted BPD and liposomal BPD), 690 nm, 150 mW/cm2, 0–80 J/cm2 | Yes | [218,219,220] | |||
| Microfluidic spheroids | HepaChip device | No | [117] | |||
| Hydrogel-based spheroids | Matrigel; PDT regimen: BPD, 690 nm, 150 mW/cm2, 1–50 J/cm2 | Yes | [200,230] | |||
| Organoids † | Matrigel, collagen I, tumour-associated PSCs and M2 macrophages in suspension | [247] | ||||
| Cell line-derived xenografts | female nude mice (6 wk), subcutaneous; PDT regimen: LC-Dox-PoP, 665 nm, 150 mW/cm2, 50 J/cm2 | Yes | [278] | |||
| Cell line-derived xenografts | male Swiss nude mice (4 wk), orthotopic; male Swiss nude mice (4–6 wk) co-implanted with pCAFs, orthotopic; PDT regimen: BPD, 690 nm, 100 mW/cm2, 50 J/cm2; verteporfin or liposomal irinotecan, 690 nm, 100 mW/cm2, 75 J/cm2 | Yes | [199,230] | |||
| MZ-PC-1 | PDAC | Metastatic | Cell line-derived xenografts | NMRI nude mice (4–6 wk), subcutaneous | No | [279] |
| PaCa-44 | PDAC | Primary | Cell line-derived xenografts | C.B-17/IcrHsd-Prkcdscid Lystbg mice (8–10 wk), subcutaneous | No | [280] |
| Cell line-derived xenografts | C.B-17/IcrHsd-Prkcdscid Lystbg mice (8–10 wk), orthotopic | No | [280] | |||
| PaCa 5061 | PDAC | Primary | Cell line-derived xenografts | male and female C57BL/6 mice (14–16 wk), subcutaneous | No | [281] |
| Pan2M | PDAC | Metastatic | Cell line-derived xenografts | female BALB/c nude mice (4 wk), orthotopic | No | [282] |
| PANC03.27 | PAC | Primary | Cell line-derived xenografts | athymic C57BL/6 nude mice, subcutaneous | No | [283] |
| PANC04.03 | PDAC | Primary | Co-cultured spheroids | PSCs | No | [284] |
| Hydrogel-based spheroids | Matrigel and collagen I mixture (3:1) | No | [284] | |||
| PANC 04.14 | PAC | Unknown | Cell line-derived xenografts | nude mice, orthotopic | No | [285] |
| PANC 10.05 | PDAC | Primary | Cell line-derived xenografts | male nude mice (8 wk), subcutaneous | No | [286] |
| PANC-1 | PDAC | Primary | Spheroids | Nunclon Sphera plates, NanoCulture plates; PDT regimen: 6-amine-2,5-bromophenalenone (OE19), 525 nm, 18.6 mW/cm2, 16.6 J/cm2, 900 s | Yes | [271,287] |
| Co-cultured spheroids | MRC-5, PSCs; PDT regimen: BPD, 690 nm, 100 mW/cm2, 0.5–25 J/cm2 | Yes | [204] | |||
| Microfluidic spheroids | HepaChip device | No | [117] | |||
| Hydrogel-based spheroids | Matrigel, collagen I, riboflavin-mediated collagen photocrosslinking hydrogel, alginate-gelatin hydrogel; PDT regimen: BPD, 690 nm, 100 mW/cm2, 0.5–25 J/cm2; BPD, 690 nm, 150 mW/cm2 | Yes | [204,231,232,233] | |||
| Organoids † | Matrigel, collagen I, tumour-associated PSCs and M2-like differentiated macrophages in suspension | No | [247] | |||
| Cell line-derived xenografts | female BALB/c nude mice (6 wk), subcutaneous; female athymic CD1 mice (4 wk), subcutaneous (both 2D and spheroids-based); PDT regimen: YLG-1, 650 nm, 100 J/cm2 | Yes | [254,288] | |||
| Cell line-derived xenografts | male SCID nude mice (6 wk), orthotopic; PDT regimen: verteporfin, 690 nm, 74 mW/cm2, 10–40 J/cm2, 135–540 s | Yes | [245] | |||
| PancTU-I | PDAC | Unknown | Spheroids | nonadherent U-form plates with medium containing 20% methyl cellulose | No | [238] |
| Cell line-derived xenografts | male or female SCID mice (13–20 wk), subcutaneous | No | [289] | |||
| Cell line-derived xenografts | male or female SCID mice (13–20 wk), orthotopic | No | [289] | |||
| PaTu 8902 | PAC | Primary | Spheroids | ultra-low attachment round-bottom plates | No | [252] |
| Co-cultured spheroids | undifferentiated monocyte-like (THP-1) cells or THP-1 conditioned medium | No | [252] | |||
| Cell line-derived xenografts | athymic nude mice, subcutaneous | No | [290] | |||
| PaTu 8988 | PAC | Metastatic | Cell line-derived xenografts | male BALB/c nude mice (5–6 wk), subcutaneous | No | [291] |
| Cell line-derived xenografts | BALB/c nude mice (5 wk), orthotopic | No | [291] | |||
| PC-1 | PDAC | Metastatic | Cell line-derived xenografts | male or female NIH athymic nude mice (4–6 wk), subcutaneous | No | [292] |
| PC-2 | PDAC | Metastatic | Spheroids | serum-free medium DMEM/F12 supplemented with bFGF, EGF, insulin, transferrin, sodium selenite, and bovine serum albumin | No | [293] |
| Cell line-derived xenografts | male or female NIH athymic nude mice (4–6 wk), subcutaneous | No | [292] | |||
| PC-3 | PDAC | Unknown | Cell line-derived xenografts | male BALB/c athymic nude mice (5 wk), subcutaneous | No | [294] |
| PC-7 | PDAC | Unknown | Cell line-derived xenografts | female specific pathogen-free athymic nude mice (4 wk), subcutaneous | No | [295] |
| Cell line-derived xenografts | BALB/c nude mice (5 wk), orthotopic | No | [291] | |||
| PCI-24 | PAC | Primary | Cell line-derived xenografts | female BALB/c nude mice (4–6 wk), subcutaneous | No | [296] |
| PCI-35 | PDAC | Primary | Cell line-derived xenografts | KSN Slc nude mice, subcutaneous | No | [297] |
| PCI-43 | PAC | Primary | Cell line-derived xenografts | female BALB/c nude mice (4–6 wk), subcutaneous | No | [296] |
| PDXPC1 | PDAC | Primary | Spheroids | serum-free medium DMEM/F12 supplemented with basic bFGF, EGF, and insulin | No | [298] |
| Cell line-derived xenografts | female BALB/c nude mice (4–6 wk), subcutaneous | No | [298] | |||
| PK-1 | PDAC | Metastatic | Cell line-derived xenografts | male BALB/c nude mice (5 wk), subcutaneous | No | [299] |
| PK-45P | PA | Unknown | Spheroids | ultra-low attachment round-bottom plates | No | [276] |
| PK-8 | PDAC | Metastatic | Spheroids | ultra-low attachment plates | No | [300] |
| Cell line-derived xenografts | SCID mice, subcutaneous | No | [301] | |||
| PL45 | PAC | Primary | Spheroids | plates coated with 1% agarose in DMEM | No | [263] |
| Cell line-derived xenografts | NOD/SCID nude mice (7–9 wk), subcutaneous | No | [302] | |||
| Cell line-derived xenografts | athymic nude mice, orthotopic | No | [303] | |||
| PSN1 | PAC | Primary | Spheroids | cancer stem cell medium | No | [254] |
| Cell line-derived xenografts | male BALB/c nude mice (12–14 wk), subcutaneous | No | [304] | |||
| PT45 | PDAC | Primary | Spheroids | gelatin porous microbeads | No | [305] |
| Co-cultured spheroids | human normal fibroblasts or CAF | No | [305] | |||
| Cell line-derived xenografts | C57BL athymic ICRF nude mice, subcutaneous | No | [306] | |||
| PT45-P1 | PDAC | Primary | Spheroids | nonadherent round-bottom plates with medium containing 20% methyl cellulose | No | [238] |
| S2-007 | PDAC | Metastatic | Spheroids | not listed | No | [307] |
| Hydrogel-based spheroids | polypeptide network hydrogel | No | [308] | |||
| Cell line-derived xenografts | BALB/c nude mice (8–16 wk), subcutaneous | No | [309] | |||
| Cell line-derived xenografts | male athymic nude mice (5 wk), orthotopic | No | [307] | |||
| S2-013 | PDAC | Metastatic | Co-cultured spheroids | HUVECs and human MSCs | No | [135] |
| Organoids † | HUVECs and MSCs | No | [135] | |||
| Cell line-derived xenografts | female athymic BALB/cSlc-nu/nu mice (7 wk), subcutaneous | No | [135] | |||
| Cell line-derived xenografts | female athymic nude mice (6–8 wk), orthotopic | No | [310] | |||
| Organoid-based xenografts | S2-013 organoids, female athymic BALB/cSlc-nu/nu (7 wk), subcutaneous | No | [135] | |||
| S2-020 | PDAC | Metastatic | Cell line-derived xenografts | BALB/c nude mice (8–16 wk), subcutaneous | No | [309] |
| S2-028 | PDAC | Metastatic | Microfluidic spheroids | three-lane OrganoPlate channels | No | [311] |
| Cell line-derived xenografts | BALB/c nude mice (8–16 wk), subcutaneous | No | [309] | |||
| Cell line-derived xenografts | athymic mice, intrasplenic injection | No | [312] | |||
| S2-CP8 | PDAC | Metastatic | Cell line-derived xenografts | male BALB/cAJcl nude mice (6 wk), orthotopic | No | [313] |
| SK-PC-1 | PDAC | Unknown | Cell line-derived xenografts | female athymic BALB/c nude mice (5 wk), subcutaneous | No | [314] |
| SU8686 | PAC | Metastatic | Cell line-derived xenografts | male BALB/cAJcl nude mice (6–8 wk), orthotopic | No | [315] |
| Sui66-Sui70, Sui72-Sui74 | PDAC | Primary | Cell line-derived xenografts | female C.B.17/Icr Jcl-scid SCID mice (6–8 wk), subcutaneous | No | [316] |
| Sui65, Sui71 | PDAC | Metastatic | Cell line-derived xenografts | female C.B.17/Icr Jcl-scid SCID mice (6–8 wk), subcutaneous | No | [316] |
| SUIT-2 | PDAC | Metastatic | Co-cultured spheroids | PSCs | No | [284] |
| Hydrogel-based spheroids | Matrigel and collagen I mixture (3:1) | No | [284] | |||
| Cell line-derived xenografts | female BALB/c nude mice (6 wk), subcutaneous; PDT regimen: rBC2-IR700, NIR light 670–710 nm, 100 J/cm2 | Yes | [251] | |||
| Cell line-derived xenografts | female nude mice (6 wk), co-implanted with PSCs, orthotopic | No | [317] | |||
| SUIT-4 | PDAC | Metastatic | Cell line-derived xenografts | BALB/c athymic nude mice (6 wk), subcutaneous | No | [318] |
| SUIT-58 | PDAC | Metastatic | Hydrogel-based spheroids | Collagen I and cell culture insert | No | [319] |
| SW1990 | PAC | Primary | Spheroids | serum-free sphere medium DMEM/F12 supplemented with B27, bFGF, and EGF | No | [320] |
| Cell line-derived xenografts | female BALB/c nude mice (5 wk), subcutaneous; PDT regimen: quantum dots conjugated with integrin antagonist arginine-glycine-aspartic acid peptides, 630 nm, 100 mW/cm2, 1200 s | Yes | [321] | |||
| Cell line-derived xenografts | male and female athymic N:NIH (S) nude mice (5–6 wk), orthotopic | No | [322] | |||
| T3M-4 | PDAC | Primary | Spheroids | ultra-low attachment round-bottom plates | No | [276] |
| Cell line-derived xenografts | BALB/c athymic nude mice (7 wk), subcutaneous | No | [323] | |||
| Cell line-derived xenografts | female BALB/c athymic nude mice (6–8 wk), orthotopic | No | [324] | |||
| TCC-Pan2 | PDAC | Metastatic | Cell line-derived xenografts | female BALB/c nude mice (4 wk), orthotopic | No | [282] |
| YAPC | PA | Metastatic | Spheroids | hanging drop method | No | [325] |
| Cell line-derived xenografts | male NMRI mice (4–6 wk), subcutaneous | No | [326] |
3.4. Challenges and Caveats of Biomimetic PDAC Models in the Context of PDT
3.5. Future Directions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jager, D.; Buchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zulke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Hank, T.; Hinz, U.; Bergmann, F.; Schneider, L.; Springfeld, C.; Jager, D.; Schirmacher, P.; Hackert, T.; Buchler, M.W. Pancreatic Cancer Surgery: The New R-status Counts. Ann. Surg. 2017, 265, 565–573. [Google Scholar] [CrossRef]
- Hank, T.; Hinz, U.; Tarantino, I.; Kaiser, J.; Niesen, W.; Bergmann, F.; Hackert, T.; Buchler, M.W.; Strobel, O. Validation of at least 1 mm as cut-off for resection margins for pancreatic adenocarcinoma of the body and tail. Br. J. Surg. 2018, 105, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Vitali, F.; Pfeifer, L.; Janson, C.; Goertz, R.S.; Neurath, M.F.; Strobel, D.; Wildner, D. Quantitative perfusion analysis in pancreatic contrast enhanced ultrasound (DCE-US): A promising tool for the differentiation between autoimmune pancreatitis and pancreatic cancer. Z. Gastroenterol. 2015, 53, 1175–1181. [Google Scholar] [CrossRef]
- Liu, X.; Fu, Y.; Chen, Q.; Wu, J.; Gao, W.; Jiang, K.; Miao, Y.; Wei, J. Predictors of distant metastasis on exploration in patients with potentially resectable pancreatic cancer. BMC Gastroenterol. 2018, 18, 168. [Google Scholar] [CrossRef]
- Versteijne, E.; Suker, M.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; Creemers, G.M.; van Dam, R.M.; et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J. Clin. Oncol. 2020, 38, 1763–1773. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Dangi-Garimella, S.; Sahai, V.; Ebine, K.; Kumar, K.; Munshi, H.G. Three-dimensional collagen I promotes gemcitabine resistance in vitro in pancreatic cancer cells through HMGA2-dependent histone acetyltransferase expression. PLoS ONE 2013, 8, e64566. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Hsiao, C.H.; Chow, D.S.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Sensi, F.; D’Angelo, E.; Biccari, A.; Marangio, A.; Battisti, G.; Crotti, S.; Fassan, M.; Laterza, C.; Giomo, M.; Elvassore, N.; et al. Establishment of a human 3D pancreatic adenocarcinoma model based on a patient-derived extracellular matrix scaffold. Transl. Res. 2023, 253, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhou, S.; Lv, T.; Xia, F.; Shen, L.; Wan, J.; Wang, Y.; Zhang, H.; Cai, S.; Peng, J.; et al. Newly developed 3D in vitro models to study tumor-immune interaction. J. Exp. Clin. Cancer Res. 2023, 42, 81. [Google Scholar] [CrossRef]
- Karimnia, V.; Stanley, M.E.; Fitzgerald, C.T.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodynamic Stromal Depletion Enhances Therapeutic Nanoparticle Delivery in 3D Pancreatic Ductal Adenocarcinoma Tumor Models. Photochem. Photobiol. 2023, 99, 120–131. [Google Scholar] [CrossRef]
- Lintern, N.; Smith, A.M.; Jayne, D.G.; Khaled, Y.S. Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 4135. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Du, W.; Ma, W.W. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: Controversies and promises. J. Gastrointest. Oncol. 2016, 7, 487–494. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Mardhian, D.F.; van Baarlen, J.; Ostman, A.; Prakash, J. Integrin alpha11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J. 2019, 33, 6609–6621. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Du, W.; Pasca di Magliano, M.; Zhang, Y. Therapeutic Potential of Targeting Stromal Crosstalk-Mediated Immune Suppression in Pancreatic Cancer. Front. Oncol. 2021, 11, 682217. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Kühnemuth, B.; Duewell, P.; Ormanns, S.; Gress, T.; Schnurr, M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett. 2016, 381, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Brunetti, O.; Gnoni, A.; Cascinu, S.; Gasparini, G.; Lorusso, V.; Ribatti, D.; Silvestris, N. Angiogenesis in pancreatic ductal adenocarcinoma: A controversial issue. Oncotarget 2016, 7, 58649–58658. [Google Scholar] [CrossRef]
- Le Large, T.Y.; Mantini, G.; Meijer, L.L.; Pham, T.V.; Funel, N.; van Grieken, N.C.; Kok, B.; Knol, J.; van Laarhoven, H.W.; Piersma, S.R.; et al. Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets. JCI Insight 2020, 5, e138290. [Google Scholar] [CrossRef]
- Pan, Z.; Li, L.; Fang, Q.; Zhang, Y.; Hu, X.; Qian, Y.; Huang, P. Analysis of dynamic molecular networks for pancreatic ductal adenocarcinoma progression. Cancer Cell Int. 2018, 18, 214. [Google Scholar] [CrossRef]
- Shen, Y.; Pu, K.; Zheng, K.; Ma, X.; Qin, J.; Jiang, L.; Li, J. Differentially Expressed microRNAs in MIA PaCa-2 and PANC-1 Pancreas Ductal Adenocarcinoma Cell Lines are Involved in Cancer Stem Cell Regulation. Int. J. Mol. Sci. 2019, 20, 4473. [Google Scholar] [CrossRef]
- Khosravani, F.; Mir, H.; Mirzaei, A.; Kobarfard, F.; Bardania, H.; Hosseini, E. Arsenic trioxide and Erlotinib loaded in RGD-modified nanoliposomes for targeted combination delivery to PC3 and PANC-1 cell lines. Biotechnol. Appl. Biochem. 2023, 70, 811–823. [Google Scholar] [CrossRef]
- Malinda, R.R.; Zeeberg, K.; Sharku, P.C.; Ludwig, M.Q.; Pedersen, L.B.; Christensen, S.T.; Pedersen, S.F. TGFβ Signaling Increases Net Acid Extrusion, Proliferation and Invasion in Panc-1 Pancreatic Cancer Cells: SMAD4 Dependence and Link to Merlin/NF2 Signaling. Front. Oncol. 2020, 10, 687. [Google Scholar] [CrossRef]
- Schnittert, J.; Heinrich, M.A.; Kuninty, P.R.; Storm, G.; Prakash, J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett. 2018, 420, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Bojmar, L.; Tjomsland, V.; Larsson, M.; Storm, G.; Ostman, A.; Sandstrom, P.; Prakash, J. MicroRNA-199a and -214 as potential therapeutic targets in pancreatic stellate cells in pancreatic tumor. Oncotarget 2016, 7, 16396–16408. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Mei, W.; Zheng, Z.; Cao, F.; Liang, K.; Jia, Y.; Wang, Y.; Liu, D.; Li, J.; Li, F. Exosomes secreted from human umbilical cord mesenchymal stem cells promote pancreatic ductal adenocarcinoma growth by transferring miR-100-5p. Tissue Cell 2021, 73, 101623. [Google Scholar] [CrossRef] [PubMed]
- Suri, R.; Zimmerman, J.W.; Burkhart, R.A. Modeling human pancreatic ductal adenocarcinoma for translational research: Current options, challenges, and prospective directions. Ann. Pancreat. Cancer 2020, 3, 17. [Google Scholar] [CrossRef]
- Hwang, C.I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef]
- Audero, M.M.; Carvalho, T.M.A.; Ruffinatti, F.A.; Loeck, T.; Yassine, M.; Chinigo, G.; Folcher, A.; Farfariello, V.; Amadori, S.; Vaghi, C.; et al. Acidic Growth Conditions Promote Epithelial-to-Mesenchymal Transition to Select More Aggressive PDAC Cell Phenotypes In Vitro. Cancers 2023, 15, 2572. [Google Scholar] [CrossRef]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)evolution: Unveiling Tumor-Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef]
- Rescigno, F.; Ceriotti, L.; Meloni, M. Extra Cellular Matrix Deposition and Assembly in Dermis Spheroids. Clin. Cosmet. Investig. Dermatol. 2021, 14, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Ncube, K.N.; Jurgens, T.; Steenkamp, V.; Cromarty, A.D.; van den Bout, I.; Cordier, W. Comparative Evaluation of the Cytotoxicity of Doxorubicin in BT-20 Triple-Negative Breast Carcinoma Monolayer and Spheroid Cultures. Biomedicines 2023, 11, 1484. [Google Scholar] [CrossRef]
- Nkune, N.W.; Simelane, N.W.N.; Montaseri, H.; Abrahamse, H. Photodynamic Therapy-Mediated Immune Responses in Three-Dimensional Tumor Models. Int. J. Mol. Sci. 2021, 22, 12618. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Alix, C.; Bouakaz, A.; Serriere, S.; Escoffre, J.M. Tumor Spheroids as Model to Design Acoustically Mediated Drug Therapies: A Review. Pharmaceutics 2023, 15, 806. [Google Scholar] [CrossRef] [PubMed]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]
- Kuntze, A.; Goetsch, O.; Fels, B.; Najder, K.; Unger, A.; Wilhelmi, M.; Sargin, S.; Schimmelpfennig, S.; Neumann, I.; Schwab, A.; et al. Protonation of Piezo1 Impairs Cell-Matrix Interactions of Pancreatic Stellate Cells. Front. Physiol. 2020, 11, 89. [Google Scholar] [CrossRef]
- Ware, M.J.; Keshishian, V.; Law, J.J.; Ho, J.C.; Favela, C.A.; Rees, P.; Smith, B.; Mohammad, S.; Hwang, R.F.; Rajapakshe, K.; et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 2016, 108, 129–142. [Google Scholar] [CrossRef]
- Lee, K.-H.; Kim, T.-H. Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors 2021, 11, 445. [Google Scholar] [CrossRef]
- Dufau, I.; Frongia, C.; Sicard, F.; Dedieu, L.; Cordelier, P.; Ausseil, F.; Ducommun, B.; Valette, A. Multicellular tumor spheroid model to evaluate spatio-temporal dynamics effect of chemotherapeutics: Application to the gemcitabine/CHK1 inhibitor combination in pancreatic cancer. BMC Cancer 2012, 12, 15. [Google Scholar] [CrossRef]
- Maietta, I.; Martínez-Pérez, A.; Álvarez, R.; De Lera, Á.R.; González-Fernández, Á.; Simón-Vázquez, R. Synergistic Antitumoral Effect of Epigenetic Inhibitors and Gemcitabine in Pancreatic Cancer Cells. Pharmaceuticals 2022, 15, 824. [Google Scholar] [CrossRef]
- Wang, Z.; He, R.; Dong, S.; Zhou, W. Pancreatic stellate cells in pancreatic cancer: As potential targets for future therapy. Front. Oncol. 2023, 13, 1185093. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.P.; Gaspar, V.M.; Mendes, L.; Duarte, I.F.; Mano, J.F. Organotypic 3D decellularized matrix tumor spheroids for high-throughput drug screening. Biomaterials 2021, 275, 120983. [Google Scholar] [CrossRef]
- Scalise, M.; Marino, F.; Salerno, L.; Cianflone, E.; Molinaro, C.; Salerno, N.; De Angelis, A.; Viglietto, G.; Urbanek, K.; Torella, D. From Spheroids to Organoids: The Next Generation of Model Systems of Human Cardiac Regeneration in a Dish. Int. J. Mol. Sci. 2021, 22, 13180. [Google Scholar] [CrossRef]
- Khursheed, M.; Bashyam, M.D. Apico-basal polarity complex and cancer. J. Biosci. 2014, 39, 145–155. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Shinkawa, T.; Ohuchida, K.; Nakamura, M. Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer. Cancers 2022, 14, 3994. [Google Scholar] [CrossRef]
- Luo, Y.; Li, Z.; Kong, Y.; He, W.; Zheng, H.; An, M.; Lin, Y.; Zhang, D.; Yang, J.; Zhao, Y.; et al. KRAS mutant-driven SUMOylation controls extracellular vesicle transmission to trigger lymphangiogenesis in pancreatic cancer. J. Clin. Investig. 2022, 132, e157644. [Google Scholar] [CrossRef]
- McGuigan, A.J.; Coleman, H.G.; McCain, R.S.; Kelly, P.J.; Johnston, D.I.; Taylor, M.A.; Turkington, R.C. Immune cell infiltrates as prognostic biomarkers in pancreatic ductal adenocarcinoma: A systematic review and meta-analysis. J. Pathol. Clin. Res. 2021, 7, 99–112. [Google Scholar] [CrossRef]
- Liu, X.; Iovanna, J.; Santofimia-Castaño, P. Stroma-targeting strategies in pancreatic cancer: A double-edged sword. J. Physiol. Biochem. 2023, 79, 213–222. [Google Scholar] [CrossRef]
- Stouten, I.; van Montfoort, N.; Hawinkels, L.J.A.C. The Tango between Cancer-Associated Fibroblasts (CAFs) and Immune Cells in Affecting Immunotherapy Efficacy in Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 8707. [Google Scholar] [CrossRef] [PubMed]
- Maneshi, P.; Mason, J.; Dongre, M.; Oehlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oda, T.; Inagaki, Y.; Kushige, H.; Saito, Y.; Mori, N.; Takayama, Y.; Kumagai, Y.; Mitsuyama, T.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into heterogeneous cancer-associated fibroblasts in a stroma-rich xenograft model. Sci. Rep. 2021, 11, 4690. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef]
- Knight, E.; Przyborski, S. Advances in 3D cell culture technologies enabling tissue-like structures to be created in vitro. J. Anat. 2015, 227, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, S.K.; Khawar, I.A.; Jeong, S.Y.; Chung, S.; Kuh, H.J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef]
- Jang, S.D.; Song, J.; Kim, H.A.; Im, C.N.; Khawar, I.A.; Park, J.K.; Kuh, H.J. Anti-Cancer Activity Profiling of Chemotherapeutic Agents in 3D Co-Cultures of Pancreatic Tumor Spheroids with Cancer-Associated Fibroblasts and Macrophages. Cancers 2021, 13, 5955. [Google Scholar] [CrossRef]
- Kpeglo, D.; Hughes, M.D.G.; Dougan, L.; Haddrick, M.; Knowles, M.A.; Evans, S.D.; Peyman, S.A. Modeling the mechanical stiffness of pancreatic ductal adenocarcinoma. Matrix Biol. Plus 2022, 14, 100109. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, Z.; Shang, D.; Cheng, J.; Yuan, H.; Wu, Y.; Song, X.; Jiang, H. α-Smooth muscle actin-positive myofibroblasts, in association with epithelial-mesenchymal transition and lymphogenesis, is a critical prognostic parameter in patients with oral tongue squamous cell carcinoma. J. Oral Pathol. Med. 2014, 43, 335–343. [Google Scholar] [CrossRef]
- Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of Cadherins in Cancer-A Review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef]
- Kim, S.; You, D.; Jeong, Y.; Yu, J.; Kim, S.W.; Nam, S.J.; Lee, J.E. TP53 upregulates α-smooth muscle actin expression in tamoxifen-resistant breast cancer cells. Oncol. Rep. 2019, 41, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Priwitaningrum, D.L.; Blonde, J.G.; Sridhar, A.; van Baarlen, J.; Hennink, W.E.; Storm, G.; Le Gac, S.; Prakash, J. Tumor stroma-containing 3D spheroid arrays: A tool to study nanoparticle penetration. J. Control. Release 2016, 244, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Bansal, R.; De Geus, S.W.L.; Mardhian, D.F.; Schnittert, J.; van Baarlen, J.; Storm, G.; Bijlsma, M.F.; van Laarhoven, H.W.; Metselaar, J.M.; et al. ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci. Adv. 2019, 5, eaax2770. [Google Scholar] [CrossRef]
- Anane-Adjei, A.B.; Fletcher, N.L.; Cavanagh, R.J.; Houston, Z.H.; Crawford, T.; Pearce, A.K.; Taresco, V.; Ritchie, A.A.; Clarke, P.; Grabowska, A.M.; et al. Synthesis, characterisation and evaluation of hyperbranched N-(2-hydroxypropyl) methacrylamides for transport and delivery in pancreatic cell lines in vitro and in vivo. Biomater. Sci. 2022, 10, 2328–2344. [Google Scholar] [CrossRef]
- Saito, K.; Sakaguchi, M.; Maruyama, S.; Iioka, H.; Putranto, E.W.; Sumardika, I.W.; Tomonobu, N.; Kawasaki, T.; Homma, K.; Kondo, E. Stromal mesenchymal stem cells facilitate pancreatic cancer progression by regulating specific secretory molecules through mutual cellular interaction. J. Cancer 2018, 9, 2916–2929. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef] [PubMed]
- Pednekar, K.P.; Heinrich, M.A.; van Baarlen, J.; Prakash, J. Novel 3D microtissues Mimicking the Fibrotic Stroma in Pancreatic Cancer to Study Cellular Interactions and Stroma-Modulating Therapeutics. Cancers 2021, 13, 5006. [Google Scholar] [CrossRef]
- Ammar, N.; Hildebrandt, M.; Geismann, C.; Roder, C.; Gemoll, T.; Sebens, S.; Trauzold, A.; Schafer, H. Monocarboxylate Transporter-1 (MCT1)-Mediated Lactate Uptake Protects Pancreatic Adenocarcinoma Cells from Oxidative Stress during Glutamine Scarcity Thereby Promoting Resistance against Inhibitors of Glutamine Metabolism. Antioxidants 2023, 12, 1818. [Google Scholar] [CrossRef]
- Kitamura, F.; Semba, T.; Yasuda-Yoshihara, N.; Yamada, K.; Nishimura, A.; Yamasaki, J.; Nagano, O.; Yasuda, T.; Yonemura, A.; Tong, Y.; et al. Cancer-associated fibroblasts reuse cancer-derived lactate to maintain a fibrotic and immunosuppressive microenvironment in pancreatic cancer. JCI Insight 2023, 8, e163022. [Google Scholar] [CrossRef]
- Xu, R.; Yang, J.; Ren, B.; Wang, H.; Yang, G.; Chen, Y.; You, L.; Zhao, Y. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front. Oncol. 2020, 10, 572722. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Han, R.-R.; Qiu, G.-Z.; Ju, X.-C.; Lou, G.; Jin, W.-L. Organoids: An intermediate modeling platform in precision oncology. Cancer Lett. 2018, 414, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Swingen, C.; Zhang, J. Induced pluripotent stem cells and their potential for basic and clinical sciences. Curr. Cardiol. Rev. 2013, 9, 63–72. [Google Scholar] [CrossRef]
- Zhang, Y.; Houchen, C.W.; Li, M. Patient-Derived Organoid Pharmacotyping Guides Precision Medicine for Pancreatic Cancer. Clin. Cancer Res. 2022, 28, 3176–3178. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Wang, P.; Zhang, N.; Wang, P. Tumor organoids for cancer research and personalized medicine. Cancer Biol. Med. 2021, 18, 319–332. [Google Scholar] [CrossRef]
- Broguiere, N.; Isenmann, L.; Hirt, C.; Ringel, T.; Placzek, S.; Cavalli, E.; Ringnalda, F.; Villiger, L.; Züllig, R.; Lehmann, R.; et al. Growth of Epithelial Organoids in a Defined Hydrogel. Adv. Mater. 2018, 30, e1801621. [Google Scholar] [CrossRef]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 5271. [Google Scholar] [CrossRef]
- Aberle, M.R.; Burkhart, R.A.; Tiriac, H.; Damink, S.W.M.O.; Dejong, C.H.C.; Tuveson, D.A.; van Dam, R.M. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 2018, 105, E48–E60. [Google Scholar] [CrossRef]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11, 603640. [Google Scholar] [CrossRef]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Zeöld, A.; Sándor, G.O.; Kiss, A.; Soós, A.Á.; Tölgyes, T.; Bursics, A.; Szűcs, Á.; Harsányi, L.; Kittel, Á.; Gézsi, A.; et al. Shared extracellular vesicle miRNA profiles of matched ductal pancreatic adenocarcinoma organoids and blood plasma samples show the power of organoid technology. Cell. Mol. Life Sci. 2021, 78, 3005–3020. [Google Scholar] [CrossRef]
- Sereti, E.; Papapostolou, I.; Dimas, K. Pancreatic Cancer Organoids: An Emerging Platform for Precision Medicine? Biomedicines 2023, 11, 890. [Google Scholar] [CrossRef]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.; Baenke, F.; Klimova, A.; Drukewitz, S.; Jahnke, B.; Brückmann, S.; Secci, R.; Winter, C.; Schmäche, T.; Seidlitz, T.; et al. Detecting drug resistance in pancreatic cancer organoids guides optimized chemotherapy treatment. J. Pathol. 2022, 257, 607–619. [Google Scholar] [CrossRef]
- Krieger, T.G.; Le Blanc, S.; Jabs, J.; Ten, F.W.; Ishaque, N.; Jechow, K.; Debnath, O.; Leonhardt, C.-S.; Giri, A.; Eils, R.; et al. Single-cell analysis of patient-derived PDAC organoids reveals cell state heterogeneity and a conserved developmental hierarchy. Nat. Commun. 2021, 12, 5826. [Google Scholar] [CrossRef]
- Lee, S.; Shanti, A. Effect of Exogenous pH on Cell Growth of Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 9910. [Google Scholar] [CrossRef]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef]
- Givant-Horwitz, V.; Davidson, B.; Reich, R. Laminin-induced signaling in tumor cells: The role of the M(r) 67,000 laminin receptor. Cancer Res. 2004, 64, 3572–3579. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef]
- Athukorala, S.S.; Tran, T.S.; Balu, R.; Truong, V.K.; Chapman, J.; Dutta, N.K.; Roy Choudhury, N. 3D Printable Electrically Conductive Hydrogel Scaffolds for Biomedical Applications: A Review. Polymers 2021, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, K.; Thomas, L.V.; Ram Kumar, R.M. Advancement of Scaffold-Based 3D Cellular Models in Cancer Tissue Engineering: An Update. Front. Oncol. 2021, 11, 733652. [Google Scholar] [CrossRef]
- Ermis, M.; Falcone, N.; Roberto de Barros, N.; Mecwan, M.; Haghniaz, R.; Choroomi, A.; Monirizad, M.; Lee, Y.; Song, J.; Cho, H.J.; et al. Tunable hybrid hydrogels with multicellular spheroids for modeling desmoplastic pancreatic cancer. Bioact. Mater. 2023, 25, 360–373. [Google Scholar] [CrossRef]
- Ma, B.; Wang, X.; Bove, A.M.; Simone, G. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Yan, M.; Wang, L.; Wu, Y.; Lu, Y. Three-dimensional highly porous hydrogel scaffold for neural circuit dissection and modulation. Acta Biomater. 2023, 157, 252–262. [Google Scholar] [CrossRef]
- Curvello, R.; Kast, V.; Abuwarwar, M.H.; Fletcher, A.L.; Garnier, G.; Loessner, D. 3D Collagen-Nanocellulose Matrices Model the Tumour Microenvironment of Pancreatic Cancer. Front. Digit. Health 2021, 3, 704584. [Google Scholar] [CrossRef]
- Khan, A.H.; Zhou, S.P.; Moe, M.; Ortega Quesada, B.A.; Bajgiran, K.R.; Lassiter, H.R.; Dorman, J.A.; Martin, E.C.; Pojman, J.A.; Melvin, A.T. Generation of 3D Spheroids Using a Thiol–Acrylate Hydrogel Scaffold to Study Endocrine Response in ER+ Breast Cancer. ACS Biomater. Sci. Eng. 2022, 8, 3977–3985. [Google Scholar] [CrossRef]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef]
- Geckil, H.; Xu, F.; Zhang, X.; Moon, S.; Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine 2010, 5, 469–484. [Google Scholar] [CrossRef]
- Kanton, S.; Paşca, S.P. Human assembloids. Development 2022, 149, dev201120. [Google Scholar] [CrossRef]
- Choi, J.I.; Rim, J.H.; Jang, S.I.; Park, J.S.; Park, H.; Cho, J.H.; Lim, J.B. The role of Jagged1 as a dynamic switch of cancer cell plasticity in PDAC assembloids. Theranostics 2022, 12, 4431–4445. [Google Scholar] [CrossRef] [PubMed]
- Mondadori, C.; Crippa, M.; Moretti, M.; Candrian, C.; Lopa, S.; Arrigoni, C. Advanced Microfluidic Models of Cancer and Immune Cell Extravasation: A Systematic Review of the Literature. Front. Bioeng. Biotechnol. 2020, 8, 907. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, N.; Gonzalez-Suarez, A.M.; Fattahi, P.; Hou, X.; Weroha, J.S.; Gaspar-Maia, A.; Stybayeva, G.; Revzin, A. A microfluidic platform for cultivating ovarian cancer spheroids and testing their responses to chemotherapies. Microsyst. Nanoeng. 2020, 6, 93. [Google Scholar] [CrossRef]
- Lim, W.; Park, S. A Microfluidic Spheroid Culture Device with a Concentration Gradient Generator for High-Throughput Screening of Drug Efficacy. Molecules 2018, 23, 3355. [Google Scholar] [CrossRef]
- Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small 2020, 16, e1905500. [Google Scholar] [CrossRef]
- Sonmez, U.M.; Cheng, Y.-W.; Watkins, S.C.; Roman, B.L.; Davidson, L.A. Endothelial cell polarization and orientation to flow in a novel microfluidic multimodal shear stress generator. Lab Chip 2020, 2, 4373–4439. [Google Scholar] [CrossRef]
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts. Sci. Rep. 2017, 7, 1325. [Google Scholar] [CrossRef]
- Sato, O.; Tsuchikawa, T.; Kato, T.; Amaishi, Y.; Okamoto, S.; Mineno, J.; Takeuchi, Y.; Sasaki, K.; Nakamura, T.; Umemoto, K.; et al. Tumor Growth Suppression of Pancreatic Cancer Orthotopic Xenograft Model by CEA-Targeting CAR-T Cells. Cancers 2023, 15, 601. [Google Scholar] [CrossRef]
- Wu, C.; Hu, B.; Wang, L.; Wu, X.; Gu, H.; Dong, H.; Yan, J.; Qi, Z.; Zhang, Q.; Chen, H.; et al. Assessment of stromal SCD-induced drug resistance of PDAC using 3D-printed zPDX model chips. iScience 2023, 26, 105723. [Google Scholar] [CrossRef]
- Mallya, K.; Gautam, S.K.; Aithal, A.; Batra, S.K.; Jain, M. Modeling pancreatic cancer in mice for experimental therapeutics. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188554. [Google Scholar] [CrossRef]
- Zeng, Z.; Wong, C.J.; Yang, L.; Ouardaoui, N.; Li, D.; Zhang, W.; Gu, S.; Zhang, Y.; Liu, Y.; Wang, X.; et al. TISMO: Syngeneic mouse tumor database to model tumor immunity and immunotherapy response. Nucleic Acids Res. 2022, 50, D1391–D1397. [Google Scholar] [CrossRef] [PubMed]
- Rovithi, M.; Avan, A.; Funel, N.; Leon, L.G.; Gomez, V.E.; Wurdinger, T.; Griffioen, A.W.; Verheul, H.M.W.; Giovannetti, E. Development of bioluminescent chick chorioallantoic membrane (CAM) models for primary pancreatic cancer cells: A platform for drug testing. Sci. Rep. 2017, 7, 44686. [Google Scholar] [CrossRef]
- Johnson, J.I.; Decker, S.; Sausville, E.A.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical Predictive Value of the in Vitro Cell Line, Human Xenograft, and Mouse Allograft Preclinical Cancer Models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar] [PubMed]
- Garber, K. From Human to Mouse and Back: “Tumorgraft” Models Surge in Popularity. JNCI J. Natl. Cancer Inst. 2009, 101, 6–8. [Google Scholar] [CrossRef]
- Bruns, C.J.; Harbison, M.T.; Davis, D.W.; Portera, C.A.; Tsan, R.; McConkey, D.J.; Evans, D.B.; Abbruzzese, J.L.; Hicklin, D.J.; Radinsky, R. Epidermal Growth Factor Receptor Blockade with C225 Plus Gemcitabine Results in Regression of Human Pancreatic Carcinoma Growing Orthotopically in Nude Mice by Antiangiogenic Mechanisms. Clin. Cancer Res. 2000, 6, 1936–1948. [Google Scholar] [PubMed]
- Philip, P.A.; Benedetti, J.; Khorana, A.A.; Goldman, B.; Fenoglio-Preiser, C.M.; Abbruzzese, J.L.; Blanke, C.D.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; et al. Phase III Study Comparing Gemcitabine Plus Cetuximab Versus Gemcitabine in Patients with Advanced Pancreatic Adenocarcinoma: Southwest Oncology Group–Directed Intergroup Trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef]
- Koutsounas, I.; Giaginis, C.; Theocharis, S. Histone deacetylase inhibitors and pancreatic cancer: Are there any promising clinical trials? World J. Gastroenterol. 2013, 19, 1173–1181. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Van Hemelryk, A.; Tomljanovic, I.; Stuurman, D.; de Ridder, C.M.A.; Teubel, W.J.; Erkens-Schulze, S.; van de Werken, H.J.G.; van Royen, M.; Grudniewska, M.; Jenster, G.W.; et al. Patient-derived xenografts and organoids recapitulate castration-resistant prostate cancer with sustained androgen receptor signaling. Eur. J. Cancer 2022, 174, S43. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Uboldi, I.; Kuninty, P.R.; Ankone, M.J.K.; van Baarlen, J.; Zhang, Y.S.; Jain, K.; Prakash, J. Microarchitectural mimicking of stroma-induced vasculature compression in pancreatic tumors using a 3D engineered model. Bioact. Mater. 2023, 22, 18–33. [Google Scholar] [CrossRef]
- Miyabayashi, K.; Baker, L.A.; Deschênes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal Transplantation Models of Human Pancreatic Ductal Adenocarcinoma Reveal Progressive Transition of Molecular Subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Furihata, K.; Naganuma, S.; Ogasawara, M.; Yoshioka, R.; Taniguchi, H.; Furihata, M.; Taniuchi, K. Establishment of a mouse model of pancreatic cancer using human pancreatic cancer cell line S2-013-derived organoid. Hum. Cell Off. J. Hum. Cell Res. Soc. 2022, 35, 735–744. [Google Scholar] [CrossRef]
- Raimondi, G.; Mato-Berciano, A.; Pascual-Sabater, S.; Rovira-Rigau, M.; Cuatrecasas, M.; Fondevila, C.; Sánchez-Cabús, S.; Begthel, H.; Boj, S.F.; Clevers, H.; et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 2020, 56, 102786. [Google Scholar] [CrossRef]
- Le Bras, A. Humanized mouse models of drug metabolism. Lab Anim. 2024, 53, 87. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes. Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e517. [Google Scholar] [CrossRef]
- Edgar, R.D.; Perrone, F.; Foster, A.R.; Payne, F.; Lewis, S.; Nayak, K.M.; Kraiczy, J.; Cenier, A.; Torrente, F.; Salvestrini, C.; et al. Culture-Associated DNA Methylation Changes Impact on Cellular Function of Human Intestinal Organoids. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 1295–1310. [Google Scholar] [CrossRef]
- Fang, Z.; Li, P.; Du, F.; Shang, L.; Li, L. The role of organoids in cancer research. Exp. Hematol. Oncol. 2023, 12, 69. [Google Scholar] [CrossRef]
- Peng, Z.; Lv, X.; Sun, H.; Zhao, L.; Huang, S. 3D tumor cultures for drug resistance and screening development in clinical applications. Mol. Cancer 2025, 24, 93. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.; Rozich, N.; Blair, A.; Thomas, D.L.; Teinor, J.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020, 272, 427–435. [Google Scholar] [CrossRef]
- Magouliotis, D.; Dimas, K.; Sakellaridis, N.; Ioannou, M.; Zacharouli, K.; Ntalagiorgos, A.; Fergadi, M.; Zacharoulis, D. Development of an Orthotopic Pancreatic Ductal Adenocarcinoma (PDAC) Patient Derived Xenografts (PDX) Preclinical Model and Characterization of Aquaporin 7 (AQP7) Expression. HPB 2022, 24, S304. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, F.; Chen, X.; Wan, J.; Wang, Y.; Li, T.; Wang, H. Self-Assembled Gemcitabine Prodrug Nanoparticles Show Enhanced Efficacy against Patient-Derived Pancreatic Ductal Adenocarcinoma. ACS Appl. Mater. Interfaces 2020, 12, 3327–3340. [Google Scholar] [CrossRef]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef]
- Zanella, E.R.; Grassi, E.; Trusolino, L. Towards precision oncology with patient-derived xenografts. Nat. Rev. Clin. Oncol. 2022, 19, 719–732. [Google Scholar] [CrossRef]
- Delitto, D.; Pham, K.; Vlada, A.C.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Liu, C.; Hughes, S.J.; Wallet, S.M.; Trevino, J.G. Patient-Derived Xenograft Models for Pancreatic Adenocarcinoma Demonstrate Retention of Tumor Morphology through Incorporation of Murine Stromal Elements. Am. J. Pathol. 2015, 185, 1297–1303. [Google Scholar] [CrossRef]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4–16. [Google Scholar] [CrossRef]
- Liu, X.; Xin, Z.; Wang, K. Patient-derived xenograft model in colorectal cancer basic and translational research. Anim. Models Exp. Med. 2023, 6, 26–40. [Google Scholar] [CrossRef] [PubMed]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Güven, E. Gene Expression Characteristics of Tumor and Adjacent Non-Tumor Tissues of Pancreatic Ductal Adenocarcinoma (PDAC) In-Silico. Iran. J. Biotechnol. 2022, 20, e3092. [Google Scholar] [CrossRef]
- Zaccagnino, A.; Pilarsky, C.; Tawfik, D.; Sebens, S.; Trauzold, A.; Novak, I.; Schwab, A.; Kalthoff, H. In silico analysis of the transportome in human pancreatic ductal adenocarcinoma. Eur. Biophys. J. 2016, 45, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bhardwaj, V. Therapeutic resistance in pancreatic ductal adenocarcinoma: Current challenges and future opportunities. World J. Gastroenterol. 2021, 27, 6527–6550. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015, 34, 643–690. [Google Scholar] [CrossRef]
- Yanovsky, R.L.; Bartenstein, D.W.; Rogers, G.S.; Isakoff, S.J.; Chen, S.T. Photodynamic therapy for solid tumors: A review of the literature. Photodermatol. Photoimmunol. Photomed. 2019, 35, 295–303. [Google Scholar] [CrossRef]
- Schuitmaker, J.J.; Baas, P.; van Leengoed, H.L.; van der Meulen, F.W.; Star, W.M.; van Zandwijk, N. Photodynamic therapy: A promising new modality for the treatment of cancer. J. Photochem. Photobiol. B 1996, 34, 3–12. [Google Scholar] [CrossRef]
- Kim, T.E.; Chang, J.E. Recent Studies in Photodynamic Therapy for Cancer Treatment: From Basic Research to Clinical Trials. Pharmaceutics 2023, 15, 2257. [Google Scholar] [CrossRef]
- Reiniers, M.J.; van Golen, R.F.; Bonnet, S.; Broekgaarden, M.; van Gulik, T.M.; Egmond, M.R.; Heger, M. Preparation and Practical Applications of 2′,7′-Dichlorodihydrofluorescein in Redox Assays. Anal. Chem. 2017, 89, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; de Kroon, A.I.; Gulik, T.M.; Heger, M. Development and in vitro proof-of-concept of interstitially targeted zinc-phthalocyanine liposomes for photodynamic therapy. Curr. Med. Chem. 2014, 21, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.J.; Chien, K.Y.; Yang, I.F.; Lee, I.N.; Wu, C.C.; Huang, T.Y.; Yu, J.S. Oxidation of protein-bound methionine in Photofrin-photodynamic therapy-treated human tumor cells explored by methionine-containing peptide enrichment and quantitative proteomics approach. Sci. Rep. 2017, 7, 1370. [Google Scholar] [CrossRef]
- Sakharov, D.V.; Elstak, E.D.; Chernyak, B.; Wirtz, K.W. Prolonged lipid oxidation after photodynamic treatment. Study with oxidation-sensitive probe C11-BODIPY581/591. FEBS Lett. 2005, 579, 1255–1260. [Google Scholar] [CrossRef]
- Kanamori, T.; Kaneko, S.; Hamamoto, K.; Yuasa, H. Mapping the diffusion pattern of (1)O(2) along DNA duplex by guanine photooxidation with an appended biphenyl photosensitizer. Sci. Rep. 2023, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- Weijer, R.; Broekgaarden, M.; Kos, M.; van Vught, R.; Rauws, E.A.; Breukink, E.J.; van Gulik, T.M.; Storm, G.; Heger, M. Enhancing photodynamic therapy of refractory solid cancers: Combining second-generation photosensitizers with multi-targeted liposomal delivery. J. Photochem. Photobiol. C 2015, 23, 103–131. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two-cellular signaling, cell metabolism and modes of cell death. Photodiagnosis Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef]
- Weijer, R.; Clavier, S.; Zaal, E.A.; Pijls, M.M.; van Kooten, R.T.; Vermaas, K.; Leen, R.; Jongejan, A.; Moerland, P.D.; van Kampen, A.H.; et al. Multi-OMIC profiling of survival and metabolic signaling networks in cells subjected to photodynamic therapy. Cell Mol. Life Sci. 2017, 74, 1133–1151. [Google Scholar] [CrossRef]
- Dias, L.M.; Sharifi, F.; de Keijzer, M.J.; Mesquita, B.; Desclos, E.; Kochan, J.A.; de Klerk, D.J.; Ernst, D.; de Haan, L.R.; Franchi, L.P.; et al. Attritional evaluation of lipophilic and hydrophilic metallated phthalocyanines for oncological photodynamic therapy. J. Photochem. Photobiol. B 2021, 216, 112146. [Google Scholar] [CrossRef]
- Mishchenko, T.; Balalaeva, I.; Gorokhova, A.; Vedunova, M.; Krysko, D.V. Which cell death modality wins the contest for photodynamic therapy of cancer? Cell Death Dis. 2022, 13, 455. [Google Scholar] [CrossRef] [PubMed]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Zhou, Z.; Song, J.; Nie, L.; Chen, X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 2016, 45, 6597–6626. [Google Scholar] [CrossRef] [PubMed]
- Weijer, R.; Broekgaarden, M.; van Golen, R.F.; Bulle, E.; Nieuwenhuis, E.; Jongejan, A.; Moerland, P.D.; van Kampen, A.H.; van Gulik, T.M.; Heger, M. Low-power photodynamic therapy induces survival signaling in perihilar cholangiocarcinoma cells. BMC Cancer 2015, 15, 1014. [Google Scholar] [CrossRef]
- De Silva, P.; Bano, S.; Pogue, B.W.; Wang, K.K.; Maytin, E.V.; Hasan, T. Photodynamic priming with triple-receptor targeted nanoconjugates that trigger T cell-mediated immune responses in a 3D in vitro heterocellular model of pancreatic cancer. Nanophotonics 2021, 10, 3199–3214. [Google Scholar] [CrossRef]
- Seshadri, M.; Spernyak, J.A.; Mazurchuk, R.; Camacho, S.H.; Oseroff, A.R.; Cheney, R.T.; Bellnier, D.A. Tumor vascular response to photodynamic therapy and the antivascular agent 5,6-dimethylxanthenone-4-acetic acid: Implications for combination therapy. Clin. Cancer Res. 2005, 11, 4241–4250. [Google Scholar] [CrossRef]
- Wang, W.; Moriyama, L.T.; Bagnato, V.S. Photodynamic therapy induced vascular damage: An overview of experimental PDT. Laser Phys. Lett. 2013, 10, 023001. [Google Scholar] [CrossRef]
- Bano, S.; Alburquerque, J.Q.; Roberts, H.J.; Pang, S.; Huang, H.C.; Hasan, T. Minocycline and photodynamic priming significantly improve chemotherapy efficacy in heterotypic spheroids of pancreatic ductal adenocarcinoma. J. Photochem. Photobiol. B 2024, 255, 112910. [Google Scholar] [CrossRef]
- Kleinovink, J.W.; van Driel, P.B.; Snoeks, T.J.; Prokopi, N.; Fransen, M.F.; Cruz, L.J.; Mezzanotte, L.; Chan, A.; Lowik, C.W.; Ossendorp, F. Combination of Photodynamic Therapy and Specific Immunotherapy Efficiently Eradicates Established Tumors. Clin. Cancer Res. 2016, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Schroder, T.; Chen, I.W.; Sperling, M.; Bell, R.H., Jr.; Brackett, K.; Joffe, S.N. Hematoporphyrin derivative uptake and photodynamic therapy in pancreatic carcinoma. J. Surg. Oncol. 1988, 38, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Chatlani, P.T.; Nuutinen, P.J.; Toda, N.; Barr, H.; MacRobert, A.J.; Bedwell, J.; Bown, S.G. Selective necrosis in hamster pancreatic tumours using photodynamic therapy with phthalocyanine photosensitization. Br. J. Surg. 1992, 79, 786–790. [Google Scholar] [CrossRef]
- Mikvy, P.; Messman, H.; MacRobert, A.J.; Pauer, M.; Sams, V.R.; Davies, C.L.; Stewart, J.C.; Bown, S.G. Photodynamic therapy of a transplanted pancreatic cancer model using meta-tetrahydroxyphenylchlorin (mTHPC). Br. J. Cancer 1997, 76, 713–718. [Google Scholar] [CrossRef]
- Hajri, A.; Coffy, S.; Vallat, F.; Evrard, S.; Marescaux, J.; Aprahamian, M. Human pancreatic carcinoma cells are sensitive to photodynamic therapy in vitro and in vivo. Br. J. Surg. 1999, 86, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhu, Q.; Li, T.; Saeed, M.; Xu, Z.; Zhong, F.; Song, R.; Huai, M.; Zheng, M.; Xie, C.; et al. Regulating Glucose Metabolism with Prodrug Nanoparticles for Promoting Photoimmunotherapy of Pancreatic Cancer. Adv. Sci. 2021, 8, 2002746. [Google Scholar] [CrossRef]
- De Silva, P.; Saad, M.A.; Camargo, A.P.; Swain, J.; Palanasami, A.; Obaid, G.; Shetty, S.; Hasan, T. Abstract A17: Enhanced immune infiltration and antitumor immune reactivity in response to optical priming in pancreatic cancer. Cancer Immunol. Res. 2020, 8, A17. [Google Scholar] [CrossRef]
- Huang, H.C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018, 78, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Weijer, R.; Broekgaarden, M.; Krekorian, M.; Alles, L.K.; van Wijk, A.C.; Mackaaij, C.; Verheij, J.; van der Wal, A.C.; van Gulik, T.M.; Storm, G.; et al. Inhibition of hypoxia inducible factor 1 and topoisomerase with acriflavine sensitizes perihilar cholangiocarcinomas to photodynamic therapy. Oncotarget 2016, 7, 3341–3356. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; Krekorian, M.; van den Ijssel, B.; Kos, M.; Alles, L.K.; van Wijk, A.C.; Bikadi, Z.; Hazai, E.; van Gulik, T.M.; et al. Inhibition of hypoxia-inducible factor 1 with acriflavine sensitizes hypoxic tumor cells to photodynamic therapy with zinc phthalocyanine-encapsulating cationic liposomes. Nano Res. 2016, 9, 1639–1662. [Google Scholar] [CrossRef]
- de Keijzer, M.J.; de Klerk, D.J.; de Haan, L.R.; van Kooten, R.T.; Franchi, L.P.; Dias, L.M.; Kleijn, T.G.; van Doorn, D.J.; Heger, M.; on behalf of the Photodynamic Therapy Study Group. Inhibition of the HIF-1 Survival Pathway as a Strategy to Augment Photodynamic Therapy Efficacy. Methods Mol. Biol. 2022, 2451, 285–403. [Google Scholar] [CrossRef]
- Conte, M.; Cauda, V. Multimodal Therapies against Pancreatic Ductal Adenocarcinoma: A Review on Synergistic Approaches toward Ultimate Nanomedicine Treatments. Adv. Ther. 2022, 5, 2200079. [Google Scholar] [CrossRef]
- Anbil, S.; Pigula, M.; Huang, H.C.; Mallidi, S.; Broekgaarden, M.; Baglo, Y.; De Silva, P.; Simeone, D.M.; Mino-Kenudson, M.; Maytin, E.V.; et al. Vitamin D Receptor Activation and Photodynamic Priming Enables Durable Low-dose Chemotherapy. Mol. Cancer Ther. 2020, 19, 1308–1319. [Google Scholar] [CrossRef]
- Obaid, G.; Bano, S.; Mallidi, S.; Broekgaarden, M.; Kuriakose, J.; Silber, Z.; Bulin, A.L.; Wang, Y.; Mai, Z.; Jin, W.; et al. Impacting Pancreatic Cancer Therapy in Heterotypic in Vitro Organoids and in Vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett. 2019, 19, 7573–7587. [Google Scholar] [CrossRef]
- Obaid, G.; Bano, S.; Thomsen, H.; Callaghan, S.; Shah, N.; Swain, J.W.R.; Jin, W.; Ding, X.; Cameron, C.G.; McFarland, S.A.; et al. Remediating Desmoplasia with EGFR-Targeted Photoactivable Multi-Inhibitor Liposomes Doubles Overall Survival in Pancreatic Cancer. Adv. Sci. 2022, 9, e2104594. [Google Scholar] [CrossRef]
- Grunwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Zhou, X.; An, J.; Peccerella, T.; Hu, K.; Springfeld, C.; Buchler, M.; Neoptolemos, J.P. Refining the Treatment of Pancreatic Cancer From Big Data to Improved Individual Survival. Function 2023, 4, zqad011. [Google Scholar] [CrossRef]
- Karimnia, V.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodestruction of Stromal Fibroblasts Enhances Tumor Response to PDT in 3D Pancreatic Cancer Coculture Models. Photochem. Photobiol. 2021, 97, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Cai, H.; Wei, Q.; Tang, X.; Zhang, Q.; Kopytynski, M.; Yang, J.; Yi, Y.; Zhang, H.; Gong, Q.; et al. Enhanced chemo-photodynamic therapy of an enzyme-responsive prodrug in bladder cancer patient-derived xenograft models. Biomaterials 2021, 277, 121061. [Google Scholar] [CrossRef]
- Murayama, T.; Gotoh, N. Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells 2019, 8, 621. [Google Scholar] [CrossRef]
- Chitrangi, S.; Vaity, P.; Jamdar, A.; Bhatt, S. Patient-derived organoids for precision oncology: A platform to facilitate clinical decision making. BMC Cancer 2023, 23, 689. [Google Scholar] [CrossRef] [PubMed]
- Bubin, R.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7030. [Google Scholar] [CrossRef]
- Gurung, P.; Lim, J.; Shrestha, R.; Kim, Y.W. Chlorin e6-associated photodynamic therapy enhances abscopal antitumor effects via inhibition of PD-1/PD-L1 immune checkpoint. Sci. Rep. 2023, 13, 4647. [Google Scholar] [CrossRef]
- Lou, J.; Aragaki, M.; Bernards, N.; Chee, T.; Gregor, A.; Hiraishi, Y.; Ishiwata, T.; Leung, C.; Ding, L.; Kitazawa, S.; et al. Repeated photodynamic therapy mediates the abscopal effect through multiple innate and adaptive immune responses with and without immune checkpoint therapy. Biomaterials 2023, 292, 121918. [Google Scholar] [CrossRef]
- Quilbe, A.; Morales, O.; Baydoun, M.; Kumar, A.; Mustapha, R.; Murakami, T.; Leroux, B.; de Schutter, C.; Thecua, E.; Ziane, L.; et al. An Efficient Photodynamic Therapy Treatment for Human Pancreatic Adenocarcinoma. J. Clin. Med. 2020, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Zhou, L.; Lu, J.; Jiang, B.; Liu, C.; Guo, J. Photodynamic therapy of pancreatic cancer: Where have we come from and where are we going? Photodiagnosis Photodyn. Ther. 2020, 31, 101876. [Google Scholar] [CrossRef]
- Dorst, D.N.; Smeets, E.M.M.; Klein, C.; Frielink, C.; Geijs, D.; Trajkovic-Arsic, M.; Cheung, P.F.Y.; Stommel, M.W.J.; Gotthardt, M.; Siveke, J.T.; et al. Fibroblast Activation Protein-Targeted Photodynamic Therapy of Cancer-Associated Fibroblasts in Murine Models for Pancreatic Ductal Adenocarcinoma. Mol. Pharm. 2023, 20, 4319–4330. [Google Scholar] [CrossRef]
- Tomas-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Foglizzo, V.; Cocco, E.; Marchio, S. Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology. Cancers 2022, 14, 3692. [Google Scholar] [CrossRef]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Hubrecht, R.C.; Carter, E. The 3Rs and Humane Experimental Technique: Implementing Change. Animals 2019, 9, 754. [Google Scholar] [CrossRef]
- Saad, M.A.; Zhung, W.; Stanley, M.E.; Formica, S.; Grimaldo-Garcia, S.; Obaid, G.; Hasan, T. Photoimmunotherapy Retains Its Anti-Tumor Efficacy with Increasing Stromal Content in Heterotypic Pancreatic Cancer Spheroids. Mol. Pharm. 2022, 19, 2549–2563. [Google Scholar] [CrossRef] [PubMed]
- Bulin, A.L.; Broekgaarden, M.; Simeone, D.; Hasan, T. Low dose photodynamic therapy harmonizes with radiation therapy to induce beneficial effects on pancreatic heterocellular spheroids. Oncotarget 2019, 10, 2625–2643. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Alkhateeb, A.; Bano, S.; Bulin, A.L.; Obaid, G.; Rizvi, I.; Hasan, T. Cabozantinib Inhibits Photodynamic Therapy-Induced Auto- and Paracrine MET Signaling in Heterotypic Pancreatic Microtumors. Cancers 2020, 12, 1401. [Google Scholar] [CrossRef]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Benton, G.; George, J.; Kleinman, H.K.; Arnaoutova, I.P. Advancing science and technology via 3D culture on basement membrane matrix. J. Cell Physiol. 2009, 221, 18–25. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, S.; Clement, M.V. Tumor intracellular redox status and drug resistance--serendipity or a causal relationship? Curr. Pharm. Des. 2004, 10, 1969–1977. [Google Scholar] [CrossRef]
- Onodera, Y.; Teramura, T.; Takehara, T.; Shigi, K.; Fukuda, K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio 2015, 5, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival. Cells 2021, 10, 2401. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- de Haan, L.R.; Reiniers, M.J.; Reeskamp, L.F.; Belkouz, A.; Ao, L.; Cheng, S.; Ding, B.; van Golen, R.F.; Heger, M. Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH(2)-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status. Antioxidants 2022, 11, 1424. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Bulin, A.L.; Frederick, J.; Mai, Z.; Hasan, T. Tracking Photodynamic- and Chemotherapy-Induced Redox-State Perturbations in 3D Culture Models of Pancreatic Cancer: A Tool for Identifying Therapy-Induced Metabolic Changes. J. Clin. Med. 2019, 8, 1399. [Google Scholar] [CrossRef]
- Carigga Gutierrez, N.M.; Le Clainche, T.; Coll, J.L.; Sancey, L.; Broekgaarden, M. Generating Large Numbers of Pancreatic Microtumors on Alginate-Gelatin Hydrogels for Quantitative Imaging of Tumor Growth and Photodynamic Therapy Optimization. Methods Mol. Biol. 2022, 2451, 91–105. [Google Scholar] [CrossRef]
- Cramer, G.M.; Jones, D.P.; El-Hamidi, H.; Celli, J.P. ECM Composition and Rheology Regulate Growth, Motility, and Response to Photodynamic Therapy in 3D Models of Pancreatic Ductal Adenocarcinoma. Mol. Cancer Res. 2017, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Cramer, G.M.; Celli, J.P. Modulation of Extracellular Matrix Rigidity Via Riboflavin-mediated Photocrosslinking Regulates Invasive Motility and Treatment Response in a 3D Pancreatic Tumor Model. Photochem. Photobiol. 2020, 96, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Auk-Emblem, P.; Dornish, M. 3D Cell Culture in Alginate Hydrogels. Microarrays 2015, 4, 133–161. [Google Scholar] [CrossRef]
- Abdelrahim, A.A.; Hong, S.; Song, J.M. Integrative In Situ Photodynamic Therapy-Induced Cell Death Measurement of 3D-Bioprinted MCF-7 Tumor Spheroids. Anal. Chem. 2022, 94, 13936–13943. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Lehnert, L.; Trost, H.; Schmiegel, W.; Roder, C.; Kalthoff, H. Hollow-spheres: A new model for analyses of differentiation of pancreatic duct epithelial cells. Ann. N. Y. Acad. Sci. 1999, 880, 83–93. [Google Scholar] [CrossRef]
- Sipos, B.; Moser, S.; Kalthoff, H.; Torok, V.; Lohr, M.; Kloppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Arlt, A.; Duttmann, A.; Ungefroren, H.; Schafer, H.; Kalthoff, H.; Kruse, M.L. Characterisation of FAP-1 expression and CD95 mediated apoptosis in the A818-6 pancreatic adenocarcinoma differentiation system. Differentiation 2012, 83, 148–157. [Google Scholar] [CrossRef]
- Algarni, A.; Greenman, J.; Madden, L.A. PO-48—Assessment of the procoagulant potential state of tumour-MP in cancer patients. Thromb. Res. 2016, 140 (Suppl. 1), S194. [Google Scholar] [CrossRef]
- Lee, W.T.; Lee, J.; Kim, H.; Nguyen, N.T.; Lee, E.S.; Oh, K.T.; Choi, H.G.; Youn, Y.S. Photoreactive-proton-generating hyaluronidase/albumin nanoparticles-loaded PEG-hydrogel enhances antitumor efficacy and disruption of the hyaluronic acid extracellular matrix in AsPC-1 tumors. Mater. Today Bio 2021, 12, 100164. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice with Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734.e726. [Google Scholar] [CrossRef] [PubMed]
- Hye Jeong, J.; Park, S.; Lee, S.; Kim, Y.; Kyong Shim, I.; Jeong, S.Y.; Kyung Choi, E.; Kim, J.; Jun, E. Orthotopic model of pancreatic cancer using CD34+ humanized mice and generation of tumor organoids from humanized tumors. Int. Immunopharmacol. 2023, 121, 110451. [Google Scholar] [CrossRef] [PubMed]
- Er, O.; Tuncel, A.; Ocakoglu, K.; Ince, M.; Kolatan, E.H.; Yilmaz, O.; Aktas, S.; Yurt, F. Radiolabeling, In Vitro Cell Uptake, and In Vivo Photodynamic Therapy Potential of Targeted Mesoporous Silica Nanoparticles Containing Zinc Phthalocyanine. Mol. Pharm. 2020, 17, 2648–2659. [Google Scholar] [CrossRef]
- Samkoe, K.S.; Chen, A.; Rizvi, I.; O’Hara, J.A.; Hoopes, P.J.; Pereira, S.P.; Hasan, T.; Pogue, B.W. Imaging tumor variation in response to photodynamic therapy in pancreatic cancer xenograft models. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 251–259. [Google Scholar] [CrossRef]
- Yu, L.S.; Jhunjhunwala, M.; Hong, S.Y.; Yu, L.Y.; Lin, W.R.; Chen, C.S. Tissue Architecture Influences the Biological Effectiveness of Boron Neutron Capture Therapy in In Vitro/In Silico Three-Dimensional Self-Assembly Cell Models of Pancreatic Cancers. Cancers 2021, 13, 4058. [Google Scholar] [CrossRef]
- Karnevi, E.; Rosendahl, A.H.; Hilmersson, K.S.; Saleem, M.A.; Andersson, R. Impact by pancreatic stellate cells on epithelial-mesenchymal transition and pancreatic cancer cell invasion: Adding a third dimension in vitro. Exp. Cell Res. 2016, 346, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, A.V.; Zhou, L.; Ralff, M.D.; Lee, Y.S.; Navaraj, A.; Carneiro, B.A.; Safran, H.; Prabhu, V.V.; Ross, E.A.; Lee, S.; et al. Combination of ONC201 and TLY012 induces selective, synergistic apoptosis in vitro and significantly delays PDAC xenograft growth in vivo. Cancer Biol. Ther. 2021, 22, 607–618. [Google Scholar] [CrossRef]
- Lee, J.; Han, S.; Thapa Magar, T.B.; Gurung, P.; Lee, J.; Seong, D.; Park, S.; Kim, Y.W.; Jeon, M.; Kim, J. Efficient Assessment of Tumor Vascular Shutdown by Photodynamic Therapy on Orthotopic Pancreatic Cancer Using High-Speed Wide-Field Waterproof Galvanometer Scanner Photoacoustic Microscopy. Int. J. Mol. Sci. 2024, 25, 3457. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Kuroda, Y.; Oda, T.; Shimomura, O.; Hashimoto, S.; Akashi, Y.; Miyazaki, Y.; Furuya, K.; Furuta, T.; Nakahashi, H.; Louphrasitthiphol, P.; et al. Lectin-based phototherapy targeting cell surface glycans for pancreatic cancer. Int. J. Cancer 2023, 152, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Benzing, C.; Lam, H.; Tsang, C.M.; Rimmer, A.; Arroyo-Berdugo, Y.; Calle, Y.; Wells, C.M. TIMP-2 secreted by monocyte-like cells is a potent suppressor of invadopodia formation in pancreatic cancer cells. BMC Cancer 2019, 19, 1214. [Google Scholar] [CrossRef] [PubMed]
- Shapoval, O.; Vetvicka, D.; Patsula, V.; Engstova, H.; Kockova, O.; Konefal, M.; Kabesova, M.; Horak, D. Temoporfin-Conjugated Upconversion Nanoparticles for NIR-Induced Photodynamic Therapy: Studies with Pancreatic Adenocarcinoma Cells In Vitro and In Vivo. Pharmaceutics 2023, 15, 2694. [Google Scholar] [CrossRef]
- Gaviraghi, M.; Tunici, P.; Valensin, S.; Rossi, M.; Giordano, C.; Magnoni, L.; Dandrea, M.; Montagna, L.; Ritelli, R.; Scarpa, A.; et al. Pancreatic cancer spheres are more than just aggregates of stem marker-positive cells. Biosci. Rep. 2011, 31, 45–55. [Google Scholar] [CrossRef]
- Nishino, H.; Takano, S.; Yoshitomi, H.; Suzuki, K.; Kagawa, S.; Shimazaki, R.; Shimizu, H.; Furukawa, K.; Miyazaki, M.; Ohtsuka, M. Grainyhead-like 2 (GRHL2) regulates epithelial plasticity in pancreatic cancer progression. Cancer Med. 2017, 6, 2686–2696. [Google Scholar] [CrossRef]
- Anthiya, S.; Ozturk, S.C.; Yanik, H.; Tavukcuoglu, E.; Sahin, A.; Datta, D.; Charisse, K.; Alvarez, D.M.; Loza, M.I.; Calvo, A.; et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J. Control. Release 2023, 357, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yuan, H.; Meng, Z.; Yang, C.; Li, Z.; Li, M.; Zhang, Z.; Gan, Y.; Tu, H. Cadherin 13 Inhibits Pancreatic Cancer Progression and Epithelial-mesenchymal Transition by Wnt/beta-Catenin Signaling. J. Cancer 2020, 11, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Lin, C.C. Viscoelastic stiffening of gelatin hydrogels for dynamic culture of pancreatic cancer spheroids. Acta Biomater. 2024, 177, 203–215. [Google Scholar] [CrossRef]
- Kettler, B.; Trauzold, A.; Roder, C.; Egberts, J.H.; Kalthoff, H. Topology impacts TRAIL therapy: Differences in primary cancer growth and liver metastasis between orthotopic and subcutaneous xenotransplants of pancreatic ductal adenocarcinoma cells. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 279–284. [Google Scholar] [CrossRef]
- Feng, H.; Ou, B.C.; Zhao, J.K.; Yin, S.; Lu, A.G.; Oechsle, E.; Thasler, W.E. Homogeneous pancreatic cancer spheroids mimic growth pattern of circulating tumor cell clusters and macrometastases: Displaying heterogeneity and crater-like structure on inner layer. J. Cancer Res. Clin. Oncol. 2017, 143, 1771–1786. [Google Scholar] [CrossRef]
- Fiore, P.F.; Di Pace, A.L.; Conti, L.A.; Tumino, N.; Besi, F.; Scaglione, S.; Munari, E.; Moretta, L.; Vacca, P. Different effects of NK cells and NK-derived soluble factors on cell lines derived from primary or metastatic pancreatic cancers. Cancer Immunol. Immunother. 2023, 72, 1417–1428. [Google Scholar] [CrossRef]
- Curley, R.C.; Burke, C.S.; Gkika, K.S.; Noorani, S.; Walsh, N.; Keyes, T.E. Phototoxicity of Tridentate Ru(II) Polypyridyl Complex with Expanded Bite Angles toward Mammalian Cells and Multicellular Tumor Spheroids. Inorg. Chem. 2023, 62, 13089–13102. [Google Scholar] [CrossRef]
- Gagliano, N.; Celesti, G.; Tacchini, L.; Pluchino, S.; Sforza, C.; Rasile, M.; Valerio, V.; Laghi, L.; Conte, V.; Procacci, P. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: Characterization in a 3D-cell culture model. World J. Gastroenterol. 2016, 22, 4466–4483. [Google Scholar] [CrossRef] [PubMed]
- Gang, J.; Park, S.B.; Hyung, W.; Choi, E.H.; Wen, J.; Kim, H.S.; Shul, Y.G.; Haam, S.; Song, S.Y. Magnetic poly epsilon-caprolactone nanoparticles containing Fe3O4 and gemcitabine enhance anti-tumor effect in pancreatic cancer xenograft mouse model. J. Drug Target. 2007, 15, 445–453. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Al-Katib, A.; Pettit, G.R.; Vaitkevicius, V.K.; Joshi, U.; Adsay, V.; Majumdar, A.P.; Sarkar, F.H. An orthotopic model of human pancreatic cancer in severe combined immunodeficient mice: Potential application for preclinical studies. Clin. Cancer Res. 1998, 4, 887–894. [Google Scholar]
- Usha, L.; Klapko, O.; Edassery, S. Xenogeneic fibroblasts inhibit the growth of the breast and ovarian cancer cell lines in co-culture. Neoplasma 2021, 68, 1265–1271. [Google Scholar] [CrossRef]
- Chen, S.T.; Kuo, T.C.; Liao, Y.Y.; Lin, M.C.; Tien, Y.W.; Huang, M.C. Silencing of MUC20 suppresses the malignant character of pancreatic ductal adenocarcinoma cells through inhibition of the HGF/MET pathway. Oncogene 2018, 37, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Cheung, E.C.; Zarei, M.; Preet, R.; Chand, S.N.; Mambelli-Lisboa, N.C.; Romeo, C.; Stout, M.C.; Londin, E.; Goetz, A.; et al. CRISPR Knockout of the HuR Gene Causes a Xenograft Lethal Phenotype. Mol. Cancer Res. 2017, 15, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Fredebohm, J.; Boettcher, M.; Eisen, C.; Gaida, M.M.; Heller, A.; Keleg, S.; Tost, J.; Greulich-Bode, K.M.; Hotz-Wagenblatt, A.; Lathrop, M.; et al. Establishment and characterization of a highly tumourigenic and cancer stem cell enriched pancreatic cancer cell line as a well defined model system. PLoS ONE 2012, 7, e48503. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Dugan, M.C.; Mohamed, A.N.; Almatchy, V.P.; Flake, T.M.; Dergham, S.T.; Shields, A.F.; Al-Katib, A.A.; Vaitkevicius, V.K.; Sarkar, F.H. Establishment of a human pancreatic tumor xenograft model: Potential application for preclinical evaluation of novel therapeutic agents. Pancreas 1998, 16, 19–25. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ishiwata, T.; Kawamoto, Y.; Kawahara, K.; Peng, W.X.; Yamamoto, T.; Naito, Z. Morphological and cytoskeletal changes of pancreatic cancer cells in three-dimensional spheroidal culture. Med. Mol. Morphol. 2010, 43, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Murota, Y.; Nagane, M.; Wu, M.; Santra, M.; Venkateswaran, S.; Tanaka, S.; Bradley, M.; Taga, T.; Tabu, K. A niche-mimicking polymer hydrogel-based approach to identify molecular targets for tackling human pancreatic cancer stem cells. Inflamm. Regen. 2023, 43, 46. [Google Scholar] [CrossRef]
- Hollevoet, K.; Mason-Osann, E.; Liu, X.F.; Imhof-Jung, S.; Niederfellner, G.; Pastan, I. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol. Cancer Ther. 2014, 13, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.; Zhang, J.; Khanal, M.; Hong, J.; Venugopalan, A.; Jiang, Q.; Sengupta, M.; Miettinen, M.; Li, N.; Pastan, I.; et al. Development of Highly Effective Anti-Mesothelin hYP218 Chimeric Antigen Receptor T Cells with Increased Tumor Infiltration and Persistence for Treating Solid Tumors. Mol. Cancer Ther. 2022, 21, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Ezaki, M.; Hayashi, I.; Yasuda, D.; Nakayama, K.; Kono, A. Establishment and characterization of human pancreatic cancer cell lines in tissue culture and in nude mice. Jpn. J. Cancer Res. 1990, 81, 987–993. [Google Scholar] [CrossRef]
- Shichi, Y.; Gomi, F.; Hasegawa, Y.; Nonaka, K.; Shinji, S.; Takahashi, K.; Ishiwata, T. Artificial intelligence-based analysis of time-lapse images of sphere formation and process of plate adhesion and spread of pancreatic cancer cells. Front. Cell Dev. Biol. 2023, 11, 1290753. [Google Scholar] [CrossRef]
- Xiong, W.; Friese-Hamim, M.; Johne, A.; Stroh, C.; Klevesath, M.; Falchook, G.S.; Hong, D.S.; Girard, P.; El Bawab, S. Translational pharmacokinetic-pharmacodynamic modeling of preclinical and clinical data of the oral MET inhibitor tepotinib to determine the recommended phase II dose. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 428–440. [Google Scholar] [CrossRef]
- Ghosh, S.; Lovell, J.F. Two Laser Treatments Can Improve Tumor Ablation Efficiency of Chemophototherapy. Pharmaceutics 2021, 13, 2183. [Google Scholar] [CrossRef]
- Heike, M.; Rohrig, O.; Gabbert, H.E.; Moll, R.; Meyer zum Buschenfelde, K.H.; Dippold, W.G.; Knuth, A. New cell lines of gastric and pancreatic cancer: Distinct morphology, growth characteristics, expression of epithelial and immunoregulatory antigens. Virchows Arch. 1995, 426, 375–384. [Google Scholar] [CrossRef]
- Hlavaty, J.; Petznek, H.; Holzmuller, H.; Url, A.; Jandl, G.; Berger, A.; Salmons, B.; Gunzburg, W.H.; Renner, M. Evaluation of a gene-directed enzyme-product therapy (GDEPT) in human pancreatic tumor cells and their use as in vivo models for pancreatic cancer. PLoS ONE 2012, 7, e40611. [Google Scholar] [CrossRef]
- Kalinina, T.; Gungor, C.; Thieltges, S.; Moller-Krull, M.; Penas, E.M.; Wicklein, D.; Streichert, T.; Schumacher, U.; Kalinin, V.; Simon, R.; et al. Establishment and characterization of a new human pancreatic adenocarcinoma cell line with high metastatic potential to the lung. BMC Cancer 2010, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, K.; Kubo, T.; Mihara, K.; Kuwata, T.; Ochiai, A.; Seyama, T.; Yokozaki, H. Development and Biological Analysis of a Novel Orthotopic Peritoneal Dissemination Mouse Model Generated Using a Pancreatic Ductal Adenocarcinoma Cell Line. Pancreas 2019, 48, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Blackham, A.U.; Northrup, S.A.; Willingham, M.; Sirintrapun, J.; Russell, G.B.; Lyles, D.S.; Stewart, J.H. Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma. J. Surg. Res. 2014, 187, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blazquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Hallden, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer Ther. 2018, 17, 575–587. [Google Scholar] [CrossRef]
- Xu, Y.; Fu, J.; Henderson, M.; Lee, F.; Jurcak, N.; Henn, A.; Wahl, J.; Shao, Y.; Wang, J.; Lyman, M.; et al. CLDN18.2 BiTE Engages Effector and Regulatory T Cells for Antitumor Immune Response in Preclinical Models of Pancreatic Cancer. Gastroenterology 2023, 165, 1219–1232. [Google Scholar] [CrossRef]
- Salem, A.F.; Bonuccelli, G.; Bevilacqua, G.; Arafat, H.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Caveolin-1 promotes pancreatic cancer cell differentiation and restores membranous E-cadherin via suppression of the epithelial-mesenchymal transition. Cell Cycle 2011, 10, 3692–3700. [Google Scholar] [CrossRef]
- Kaye, E.G.; Kailass, K.; Sadovski, O.; Beharry, A.A. A Green-Absorbing, Red-Fluorescent Phenalenone-Based Photosensitizer as a Theranostic Agent for Photodynamic Therapy. ACS Med. Chem. Lett. 2021, 12, 1295–1301. [Google Scholar] [CrossRef]
- Shen, Y.J.; Cao, J.; Sun, F.; Cai, X.L.; Li, M.M.; Zheng, N.N.; Qu, C.Y.; Zhang, Y.; Shen, F.; Zhou, M.; et al. Effect of photodynamic therapy with (17R,18R)-2-(1-hexyloxyethyl)-2-devinyl chlorine E6 trisodium salt on pancreatic cancer cells in vitro and in vivo. World J. Gastroenterol. 2018, 24, 5246–5258. [Google Scholar] [CrossRef]
- Alves, F.; Contag, S.; Missbach, M.; Kaspareit, J.; Nebendahl, K.; Borchers, U.; Heidrich, B.; Streich, R.; Hiddemann, W. An orthotopic model of ductal adenocarcinoma of the pancreas in severe combined immunodeficient mice representing all steps of the metastatic cascade. Pancreas 2001, 23, 227–235. [Google Scholar] [CrossRef]
- Vankova, K.; Markova, I.; Jasprova, J.; Dvorak, A.; Subhanova, I.; Zelenka, J.; Novosadova, I.; Rasl, J.; Vomastek, T.; Sobotka, R.; et al. Chlorophyll-Mediated Changes in the Redox Status of Pancreatic Cancer Cells Are Associated with Its Anticancer Effects. Oxid. Med. Cell. Longev. 2018, 2018, 4069167. [Google Scholar] [CrossRef]
- Zhao, X.; Li, D.C.; Zhu, X.G.; Gan, W.J.; Li, Z.; Xiong, F.; Zhang, Z.X.; Zhang, G.B.; Zhang, X.G.; Zhao, H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int. J. Mol. Med. 2013, 31, 283–291. [Google Scholar] [CrossRef]
- Chen, J.; Liu, T.H.; Guo, X.Y.; Ye, S.F. Two new human exocrine pancreatic adenocarcinoma cell lines in vitro and in vivo. Chin. Med. J. 1990, 103, 369–375. [Google Scholar] [PubMed]
- Wei, H.J.; Yin, T.; Zhu, Z.; Shi, P.F.; Tian, Y.; Wang, C.Y. Expression of CD44, CD24 and ESA in pancreatic adenocarcinoma cell lines varies with local microenvironment. Hepatobiliary Pancreat. Dis. Int. 2011, 10, 428–434. [Google Scholar] [CrossRef]
- Kumar, M.; Liu, Z.R.; Thapa, L.; Wang, D.Y.; Tian, R.; Qin, R.Y. Mechanisms of inhibition of growth of human pancreatic carcinoma implanted in nude mice by somatostatin receptor subtype 2. Pancreas 2004, 29, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liang, Z.Y.; Ren, X.Y.; Liu, T.H. Small interfering RNAs targeting mutant K-ras inhibit human pancreatic carcinoma cells growth in vitro and in vivo. Cancer Biol. Ther. 2006, 5, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ishikura, H.; Kato, H.; Ogawa, Y.; Kondo, S.; Kato, H.; Yoshiki, T. Vaccination effect of interleukin-6-producing pancreatic cancer cells in nude mice: A model of tumor prevention and treatment in immune-compromised patients. Jpn. J. Cancer Res. 2001, 92, 83–87. [Google Scholar] [CrossRef]
- Shichinohe, T.; Senmaru, N.; Furuuchi, K.; Ogiso, Y.; Ishikura, H.; Yoshiki, T.; Takahashi, T.; Kato, H.; Kuzumaki, N. Suppression of pancreatic cancer by the dominant negative ras mutant, N116Y. J. Surg. Res. 1996, 66, 125–130. [Google Scholar] [CrossRef]
- Du, X.; He, K.; Huang, Y.; Xu, Z.; Kong, M.; Zhang, J.; Cao, J.; Teng, L. Establishment of a novel human cell line retaining the characteristics of the original pancreatic adenocarcinoma, and evaluation of MEK as a therapeutic target. Int. J. Oncol. 2020, 56, 761–771. [Google Scholar] [CrossRef]
- Rahman, A.; Matsuyama, M.; Ebihara, A.; Shibayama, Y.; Hasan, A.U.; Nakagami, H.; Suzuki, F.; Sun, J.; Kobayashi, T.; Hayashi, H.; et al. Antiproliferative Effects of Monoclonal Antibodies against (Pro)Renin Receptor in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2020, 19, 1844–1855. [Google Scholar] [CrossRef]
- Shichi, Y.; Gomi, F.; Ueda, Y.; Nonaka, K.; Hasegawa, F.; Hasegawa, Y.; Hinata, N.; Yoshimura, H.; Yamamoto, M.; Takahashi, K.; et al. Multiple cystic sphere formation from PK-8 cells in three-dimensional culture. Biochem. Biophys. Rep. 2022, 32, 101339. [Google Scholar] [CrossRef]
- Hoshida, T.; Sunamura, M.; Duda, D.G.; Egawa, S.; Miyazaki, S.; Shineha, R.; Hamada, H.; Ohtani, H.; Satomi, S.; Matsuno, S. Gene therapy for pancreatic cancer using an adenovirus vector encoding soluble flt-1 vascular endothelial growth factor receptor. Pancreas 2002, 25, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Suemizu, H.; Monnai, M.; Ohnishi, Y.; Ito, M.; Tamaoki, N.; Nakamura, M. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/gammacnull (NOG) mice. Int. J. Oncol. 2007, 31, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Mustafa, A.; Fischer, J.W.; Singh, A.; Zhong, W.; Shekhani, M.O.; Meske, L.; Havighurst, T.; Kim, K.; Verma, A.K. alpha-Mangostin: A dietary antioxidant derived from the pericarp of Garcinia mangostana L. inhibits pancreatic tumor growth in xenograft mouse model. Antioxid. Redox Signal. 2014, 21, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Cykowiak, M.; Kleszcz, R.; Kucinska, M.; Paluszczak, J.; Szaefer, H.; Plewinski, A.; Piotrowska-Kempisty, H.; Murias, M.; Krajka-Kuzniak, V. Attenuation of Pancreatic Cancer In Vitro and In Vivo via Modulation of Nrf2 and NF-kappaB Signaling Pathways by Natural Compounds. Cells 2021, 10, 3556. [Google Scholar] [CrossRef]
- Brancato, V.; Comunanza, V.; Imparato, G.; Cora, D.; Urciuolo, F.; Noghero, A.; Bussolino, F.; Netti, P.A. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017, 49, 152–166. [Google Scholar] [CrossRef]
- Cherubini, G.; Kallin, C.; Mozetic, A.; Hammaren-Busch, K.; Muller, H.; Lemoine, N.R.; Hallden, G. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011, 18, 1157–1165. [Google Scholar] [CrossRef]
- Dandawate, P.; Ghosh, C.; Palaniyandi, K.; Paul, S.; Rawal, S.; Pradhan, R.; Sayed, A.A.A.; Choudhury, S.; Standing, D.; Subramaniam, D.; et al. The Histone Demethylase KDM3A, Increased in Human Pancreatic Tumors, Regulates Expression of DCLK1 and Promotes Tumorigenesis in Mice. Gastroenterology 2019, 157, 1646–1659.e1611. [Google Scholar] [CrossRef]
- Lachowski, D.; Matellan, C.; Cortes, E.; Saiani, A.; Miller, A.F.; Del Rio Hernandez, A.E. Self-Assembling Polypeptide Hydrogels as a Platform to Recapitulate the Tumor Microenvironment. Cancers 2021, 13, 3286. [Google Scholar] [CrossRef]
- Taniguchi, S.; Iwamura, T.; Katsuki, T. Correlation between spontaneous metastatic potential and type I collagenolytic activity in a human pancreatic cancer cell line (SUIT-2) and sublines. Clin. Exp. Metastasis 1992, 10, 259–266. [Google Scholar] [CrossRef]
- Hennig, R.; Ventura, J.; Segersvard, R.; Ward, E.; Ding, X.Z.; Rao, S.M.; Jovanovic, B.D.; Iwamura, T.; Talamonti, M.S.; Bell, R.H., Jr.; et al. LY293111 improves efficacy of gemcitabine therapy on pancreatic cancer in a fluorescent orthotopic model in athymic mice. Neoplasia 2005, 7, 417–425. [Google Scholar] [CrossRef]
- Kramer, B.; Haan, L.; Vermeer, M.; Olivier, T.; Hankemeier, T.; Vulto, P.; Joore, J.; Lanz, H.L. Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. Int. J. Mol. Sci. 2019, 20, 4647. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Bohl, C.; Natsugoe, S.; Nishizono, Y.; Harihar, S.; Sharma, R.; Iwakuma, T.; Welch, D.R. Suppression of pancreatic cancer growth and metastasis by HMP19 identified through genome-wide shRNA screen. Int. J. Cancer 2016, 139, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Kawaguchi, M.; Haruyama, Y.; Kanemaru, A.; Fukushima, T.; Yamamoto, K.; Lin, C.Y.; Kataoka, H. Loss of hepatocyte growth factor activator inhibitor type 1 participates in metastatic spreading of human pancreatic cancer cells in a mouse orthotopic transplantation model. Cancer Sci. 2014, 105, 44–51. [Google Scholar] [CrossRef]
- Diaz, V.M.; Planaguma, J.; Thomson, T.M.; Reventos, J.; Paciucci, R. Tissue plasminogen activator is required for the growth, invasion, and angiogenesis of pancreatic tumor cells. Gastroenterology 2002, 122, 806–819. [Google Scholar] [CrossRef]
- Liu, C.; Deng, S.; Jin, K.; Gong, Y.; Cheng, H.; Fan, Z.; Qian, Y.; Huang, Q.; Ni, Q.; Luo, G.; et al. Lewis antigen-negative pancreatic cancer: An aggressive subgroup. Int. J. Oncol. 2020, 56, 900–908. [Google Scholar] [CrossRef]
- Yanagihara, K.; Takigahira, M.; Tanaka, H.; Arao, T.; Aoyagi, Y.; Oda, T.; Ochiai, A.; Nishio, K. Establishment and molecular profiling of a novel human pancreatic cancer panel for 5-FU. Cancer Sci. 2008, 99, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef]
- Kawano, K.; Iwamura, T.; Yamanari, H.; Seo, Y.; Suganuma, T.; Chijiiwa, K. Establishment and characterization of a novel human pancreatic cancer cell line (SUIT-4) metastasizing to lymph nodes and lungs in nude mice. Oncology 2004, 66, 458–467. [Google Scholar] [CrossRef]
- Takahashi, N.; Aoyama, F.; Sawaguchi, A. Three-dimensional culture of a pancreatic cancer cell line, SUIT-58, with air exposure can reflect the intrinsic features of the original tumor through electron microscopy. Microscopy 2021, 70, 192–200. [Google Scholar] [CrossRef]
- Sang, M.; Nakamura, M.; Ogata, T.; Sun, D.; Shimozato, O.; Nikaido, T.; Ozaki, T. Impact of RUNX2 gene silencing on the gemcitabine sensitivity of p53-mutated pancreatic cancer MiaPaCa-2 spheres. Oncol. Rep. 2018, 39, 2749–2758. [Google Scholar] [CrossRef]
- Li, M.M.; Cao, J.; Yang, J.C.; Shen, Y.J.; Cai, X.L.; Chen, Y.W.; Qu, C.Y.; Zhang, Y.; Shen, F.; Xu, L.M. Effects of arginine-glycine-aspartic acid peptide-conjugated quantum dots-induced photodynamic therapy on pancreatic carcinoma in vivo. Int. J. Nanomed. 2017, 12, 2769–2779. [Google Scholar] [CrossRef]
- Weber, H.L.; Gidekel, M.; Werbajh, S.; Salvatierra, E.; Rotondaro, C.; Sganga, L.; Haab, G.A.; Curiel, D.T.; Cafferata, E.G.; Podhajcer, O.L. A Novel CDC25B Promoter-Based Oncolytic Adenovirus Inhibited Growth of Orthotopic Human Pancreatic Tumors in Different Preclinical Models. Clin. Cancer Res. 2015, 21, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Okabe, T.; Yamaguchi, N.; Ohsawa, N. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer 1983, 51, 662–668. [Google Scholar] [CrossRef]
- Saxena, S.; Purohit, A.; Varney, M.L.; Hayashi, Y.; Singh, R.K. Semaphorin-5A maintains epithelial phenotype of malignant pancreatic cancer cells. BMC Cancer 2018, 18, 1283. [Google Scholar] [CrossRef]
- Stefano, E.; Cossa, L.G.; De Castro, F.; De Luca, E.; Vergaro, V.; My, G.; Rovito, G.; Migoni, D.; Muscella, A.; Marsigliante, S.; et al. Evaluation of the Antitumor Effects of Platinum-Based [Pt(η1-C2H4-OR)(DMSO)(phen)]+ (R = Me, Et) Cationic Organometallic Complexes on Chemoresistant Pancreatic Cancer Cell Lines. Bioinorg. Chem. Appl. 2023, 2023, 5564624. [Google Scholar] [CrossRef]
- Neureiter, D.; Zopf, S.; Dimmler, A.; Stintzing, S.; Hahn, E.G.; Kirchner, T.; Herold, C.; Ocker, M. Different capabilities of morphological pattern formation and its association with the expression of differentiation markers in a xenograft model of human pancreatic cancer cell lines. Pancreatology 2005, 5, 387–397. [Google Scholar] [CrossRef]
- Heger, M. Editor’s inaugural issue foreword: Perspectives on translational and clinical research. J. Clin. Transl. Res. 2015, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Snow, C.J.; Simmers, M.B.; Hoang, S.A.; Figler, R.A.; Allende, J.A.; Roller, D.G.; Parsons, J.T.; Wulfkuhle, J.D.; Petricoin, E.F.; et al. Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells. Lab Chip 2019, 19, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef]
- Walters, D.M.; Stokes, J.B.; Adair, S.J.; Stelow, E.B.; Borgman, C.A.; Lowrey, B.T.; Xin, W.; Blais, E.M.; Lee, J.K.; Papin, J.A.; et al. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PLoS ONE 2013, 8, e77065. [Google Scholar] [CrossRef]
- Gorg, C.; Seifart, U.; Gorg, K.; Zugmaier, G. Color Doppler sonographic mapping of pulmonary lesions: Evidence of dual arterial supply by spectral analysis. J. Ultrasound Med. 2003, 22, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Baronzio, G.; Schwartz, L.; Kiselevsky, M.; Guais, A.; Sanders, E.; Milanesi, G.; Baronzio, M.; Freitas, I. Tumor interstitial fluid as modulator of cancer inflammation, thrombosis, immunity and angiogenesis. Anticancer Res. 2012, 32, 405–414. [Google Scholar]
- Majumder, S.; Islam, M.T.; Righetti, R. Non-invasive imaging of interstitial fluid transport parameters in solid tumors in vivo. Sci. Rep. 2023, 13, 7132. [Google Scholar] [CrossRef]
- Di Maggio, F.; Arumugam, P.; Delvecchio, F.R.; Batista, S.; Lechertier, T.; Hodivala-Dilke, K.; Kocher, H.M. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 995–1004. [Google Scholar] [CrossRef]
- Korbelik, M. Induction of tumor immunity by photodynamic therapy. J. Clin. Laser Med. Surg. 1996, 14, 329–334. [Google Scholar] [CrossRef]
- van Duijnhoven, F.H.; Aalbers, R.I.; Rovers, J.P.; Terpstra, O.T.; Kuppen, P.J. The immunological consequences of photodynamic treatment of cancer, a literature review. Immunobiology 2003, 207, 105–113. [Google Scholar] [CrossRef]
- Canti, G.; De Simone, A.; Korbelik, M. Photodynamic therapy and the immune system in experimental oncology. Photochem. Photobiol. Sci. 2002, 1, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kousis, P.C.; Henderson, B.W.; Maier, P.G.; Gollnick, S.O. Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res. 2007, 67, 10501–10510. [Google Scholar] [CrossRef]
- Gollnick, S.O.; Evans, S.S.; Baumann, H.; Owczarczak, B.; Maier, P.; Vaughan, L.; Wang, W.C.; Unger, E.; Henderson, B.W. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer 2003, 88, 1772–1779. [Google Scholar] [CrossRef]
- Hwang, H.S.; Shin, H.; Han, J.; Na, K. Combination of photodynamic therapy (PDT) and anti-tumor immunity in cancer therapy. J. Pharm. Investig. 2018, 48, 143–151. [Google Scholar] [CrossRef]
- Korbelik, M.; Krosl, G.; Krosl, J.; Dougherty, G.J. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996, 56, 5647–5652. [Google Scholar] [PubMed]
- Korbelik, M.; Dougherty, G.J. Photodynamic therapy-mediated immune response against subcutaneous mouse tumors. Cancer Res. 1999, 59, 1941–1946. [Google Scholar]
- Hendrzak-Henion, J.A.; Knisely, T.L.; Cincotta, L.; Cincotta, E.; Cincotta, A.H. Role of the immune system in mediating the antitumor effect of benzophenothiazine photodynamic therapy. Photochem. Photobiol. 1999, 69, 575–581. [Google Scholar] [CrossRef]
- Castano, A.P.; Liu, Q.; Hamblin, M.R. A green fluorescent protein-expressing murine tumour but not its wild-type counterpart is cured by photodynamic therapy. Br. J. Cancer 2006, 94, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Uenaka, A.; Nakayama, E. Murine leukemia RL male 1 and sarcoma Meth A antigens recognized by cytotoxic T lymphocytes (CTL). Cancer Sci. 2003, 94, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Kos, M.; Jurg, F.A.; van Beek, A.A.; van Gulik, T.M.; Heger, M. Inhibition of NF-kappaB in Tumor Cells Exacerbates Immune Cell Activation Following Photodynamic Therapy. Int. J. Mol. Sci. 2015, 16, 19960–19977. [Google Scholar] [CrossRef]
- Chen, J.; Liao, S.; Xiao, Z.; Pan, Q.; Wang, X.; Shen, K.; Wang, S.; Yang, L.; Guo, F.; Liu, H.F.; et al. The development and improvement of immunodeficient mice and humanized immune system mouse models. Front. Immunol. 2022, 13, 1007579. [Google Scholar] [CrossRef]
- Partecke, I.L.; Kaeding, A.; Sendler, M.; Albers, N.; Kuhn, J.P.; Speerforck, S.; Roese, S.; Seubert, F.; Diedrich, S.; Kuehn, S.; et al. In vivo imaging of pancreatic tumours and liver metastases using 7 Tesla MRI in a murine orthotopic pancreatic cancer model and a liver metastases model. BMC Cancer 2011, 11, 40. [Google Scholar] [CrossRef]
- Courtin, A.; Richards, F.M.; Bapiro, T.E.; Bramhall, J.L.; Neesse, A.; Cook, N.; Krippendorff, B.F.; Tuveson, D.A.; Jodrell, D.I. Anti-tumour efficacy of capecitabine in a genetically engineered mouse model of pancreatic cancer. PLoS ONE 2013, 8, e67330. [Google Scholar] [CrossRef]
- Blaauboer, A.; Van Koetsveld, P.M.; Mustafa, D.A.M.; Dumas, J.; Dogan, F.; Van Zwienen, S.; Van Eijck, C.H.J.; Hofland, L.J. Immunomodulatory antitumor effect of interferon-beta combined with gemcitabine in pancreatic cancer. Int. J. Oncol. 2022, 61, 97. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, E.; Student, M.; Roth, K.; Siti Utami, N.; Huber, M.; Buchholz, M.; Gress, T.M.; Bauer, C. IL18 Receptor Signaling Inhibits Intratumoral CD8+ T-Cell Migration in a Murine Pancreatic Cancer Model. Cells 2023, 12, 456. [Google Scholar] [CrossRef]
- Czaplinska, D.; Ialchina, R.; Andersen, H.B.; Yao, J.; Stigliani, A.; Dannesboe, J.; Flinck, M.; Chen, X.; Mitrega, J.; Gnosa, S.P.; et al. Crosstalk between tumor acidosis, p53 and extracellular matrix regulates pancreatic cancer aggressiveness. Int. J. Cancer 2023, 152, 1210–1225. [Google Scholar] [CrossRef]
- Fukushima, H.; Furusawa, A.; Kato, T.; Wakiyama, H.; Takao, S.; Okuyama, S.; Choyke, P.L.; Kobayashi, H. Intratumoral IL15 Improves Efficacy of Near-Infrared Photoimmunotherapy. Mol. Cancer Ther. 2023, 22, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, J.; Zhang, Y.; Liu, M.; Lang, M.L.; Li, M.; Chen, W.R. Local Phototherapy Synergizes with Immunoadjuvant for Treatment of Pancreatic Cancer through Induced Immunogenic Tumor Vaccine. Clin. Cancer Res. 2018, 24, 5335–5346. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: Applications in diagnosis and therapy. PLoS ONE 2013, 8, e80580. [Google Scholar] [CrossRef]
- Benali, N.; Cordelier, P.; Calise, D.; Pages, P.; Rochaix, P.; Nagy, A.; Esteve, J.P.; Pour, P.M.; Schally, A.V.; Vaysse, N.; et al. Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (sst2) gene expression and administration of cytotoxic somatostatin analog AN-238. Proc. Natl. Acad. Sci. USA 2000, 97, 9180–9185. [Google Scholar] [CrossRef]
- Hirota, M.; Egami, H.; Mogaki, M.; Kazakoff, K.; Chaney, W.G.; Pour, P.M. Relationship between blood group-A antigen expression and malignant potential in hamster pancreatic cancers. Teratog. Carcinog. Mutagen. 1993, 13, 217–224. [Google Scholar] [CrossRef]
- Chang, B.K.; Gutman, R. Chemotherapy of pancreatic adenocarcinoma: Initial report on two transplantable models in the Syrian hamster. Cancer Res. 1982, 42, 2666–2670. [Google Scholar]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef]
- Sitnik, T.M.; Hampton, J.A.; Henderson, B.W. Reduction of tumour oxygenation during and after photodynamic therapy in vivo: Effects of fluence rate. Br. J. Cancer 1998, 77, 1386–1394. [Google Scholar] [CrossRef]
- Debefve, E.; Pegaz, B.; van den Bergh, H.; Wagnieres, G.; Lange, N.; Ballini, J.P. Video monitoring of neovessel occlusion induced by photodynamic therapy with verteporfin (Visudyne), in the CAM model. Angiogenesis 2008, 11, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three-Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kedzierska, E.; Knap-Czop, K.; Kotlinska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.M.; de Keijzer, M.J.; Ernst, D.; Sharifi, F.; de Klerk, D.J.; Kleijn, T.G.; Desclos, E.; Kochan, J.A.; de Haan, L.R.; Franchi, L.P.; et al. Metallated phthalocyanines and their hydrophilic derivatives for multi-targeted oncological photodynamic therapy. J. Photochem. Photobiol. B 2022, 234, 112500. [Google Scholar] [CrossRef]
- Xie, Q.; Li, Z.; Liu, Y.; Zhang, D.; Su, M.; Niitsu, H.; Lu, Y.; Coffey, R.J.; Bai, M. Translocator protein-targeted photodynamic therapy for direct and abscopal immunogenic cell death in colorectal cancer. Acta Biomater. 2021, 134, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Aragaki, M.; Bernards, N.; Kinoshita, T.; Mo, J.; Motooka, Y.; Ishiwata, T.; Gregor, A.; Chee, T.; Chen, Z.; et al. Repeated porphyrin lipoprotein-based photodynamic therapy controls distant disease in mouse mesothelioma via the abscopal effect. Nanophotonics 2021, 10, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Osuna de la Pena, D.; Trabulo, S.M.D.; Collin, E.; Liu, Y.; Sharma, S.; Tatari, M.; Behrens, D.; Erkan, M.; Lawlor, R.T.; Scarpa, A.; et al. Bioengineered 3D models of human pancreatic cancer recapitulate in vivo tumour biology. Nat. Commun. 2021, 12, 5623. [Google Scholar] [CrossRef]
- Godier, C.; Baka, Z.; Lamy, L.; Gribova, V.; Marchal, P.; Lavalle, P.; Gaffet, E.; Bezdetnaya, L.; Alem, H. A 3D Bio-Printed-Based Model for Pancreatic Ductal Adenocarcinoma. Diseases 2024, 12, 206. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; Sun, M.; Ke, X.; Li, C.; Jin, B.; Pang, M.; Wang, Y.; Jiang, S.; Du, L.; et al. Developing Patient-Derived 3D-Bioprinting models of pancreatic cancer. J. Adv. Res. 2024, in press. [Google Scholar] [CrossRef]
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Sgarminato, V.; Marasso, S.L.; Cocuzza, M.; Scordo, G.; Ballesio, A.; Ciardelli, G.; Tonda-Turo, C. PDAC-on-chip for in vitro modeling of stromal and pancreatic cancer cell crosstalk. Biomater. Sci. 2022, 11, 208–224. [Google Scholar] [CrossRef]
- Goluba, K.; Parfejevs, V.; Rostoka, E.; Jekabsons, K.; Blake, I.; Neimane, A.; Ule, A.A.; Rimsa, R.; Vangravs, R.; Pcolkins, A.; et al. Personalized PDAC chip with functional endothelial barrier for tumour biomarker detection: A platform for precision medicine applications. Mater. Today Bio 2024, 29, 101262. [Google Scholar] [CrossRef]
- Geyer, M.; Schreyer, D.; Gaul, L.M.; Pfeffer, S.; Pilarsky, C.; Queiroz, K. A microfluidic-based PDAC organoid system reveals the impact of hypoxia in response to treatment. Cell Death Discov. 2023, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Nakahashi, H.; Louphrasitthiphol, P.; Kuroda, Y.; Miyazaki, Y.; Shimomura, O.; Hashimoto, S.; Akashi, Y.; Mathis, B.J.; Kim, J.; et al. Hypoxia at 3D organoid establishment selects essential subclones within heterogenous pancreatic cancer. Front. Cell Dev. Biol. 2024, 12, 1327772. [Google Scholar] [CrossRef]
- Abou Khouzam, R.; Lehn, J.M.; Mayr, H.; Clavien, P.A.; Wallace, M.B.; Ducreux, M.; Limani, P.; Chouaib, S. Hypoxia, a Targetable Culprit to Counter Pancreatic Cancer Resistance to Therapy. Cancers 2023, 15, 1235. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; van Wijk, A.C.; Cox, R.C.; Egmond, M.R.; Hoebe, R.; van Gulik, T.M.; Heger, M. Photodynamic Therapy with Liposomal Zinc Phthalocyanine and Tirapazamine Increases Tumor Cell Death via DNA Damage. J. Biomed. Nanotechnol. 2017, 13, 204–220. [Google Scholar] [CrossRef]
- Cros, J.; Raffenne, J.; Couvelard, A.; Pote, N. Tumor Heterogeneity in Pancreatic Adenocarcinoma. Pathobiology 2018, 85, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.M.; Vudatha, V.; Peixoto, M.L.; Madan, E. Tumor heterogeneity: An oncogenic driver of PDAC progression and therapy resistance under stress conditions. Adv. Cancer Res. 2023, 159, 203–249. [Google Scholar] [CrossRef]
- Evan, T.; Wang, V.M.; Behrens, A. The roles of intratumour heterogeneity in the biology and treatment of pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 4686–4695. [Google Scholar] [CrossRef]
- Roos, E.; Soer, E.C.; Klompmaker, S.; Meijer, L.L.; Besselink, M.G.; Giovannetti, E.; Heger, M.; Kazemier, G.; Klumpen, H.J.; Takkenberg, R.B.; et al. Crossing borders: A systematic review with quantitative analysis of genetic mutations of carcinomas of the biliary tract. Crit. Rev. Oncol. Hematol. 2019, 140, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Cholangiocarcinoma—Controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 189–200. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- de Klerk, D.J.; de Keijzer, M.J.; Dias, L.M.; Heemskerk, J.; de Haan, L.R.; Kleijn, T.G.; Franchi, L.P.; Heger, M.; Photodynamic Therapy Study, G. Strategies for Improving Photodynamic Therapy Through Pharmacological Modulation of the Immediate Early Stress Response. Methods Mol. Biol. 2022, 2451, 405–480. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.M.; Siccardi, M. Optimizing nanomedicine pharmacokinetics using physiologically based pharmacokinetics modelling. Br. J. Pharmacol. 2014, 171, 3963–3979. [Google Scholar] [CrossRef]
- Kadam, R.S.; Bourne, D.W.; Kompella, U.B. Nano-advantage in enhanced drug delivery with biodegradable nanoparticles: Contribution of reduced clearance. Drug Metab. Dispos. 2012, 40, 1380–1388. [Google Scholar] [CrossRef]
- Zhang, A.; Meng, K.; Liu, Y.; Pan, Y.; Qu, W.; Chen, D.; Xie, S. Absorption, distribution, metabolism, and excretion of nanocarriers in vivo and their influences. Adv. Colloid. Interface Sci. 2020, 284, 102261. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhu, Y.; Ye, L.; Zhu, Y.; Wang, Y. Comparison of tumor angiogenesis in subcutaneous and orthotopic LNCaP mouse models using contrast-enhanced ultrasound imaging. Transl. Cancer Res. 2021, 10, 3268–3277. [Google Scholar] [CrossRef]
- Guerin, M.V.; Finisguerra, V.; Van den Eynde, B.J.; Bercovici, N.; Trautmann, A. Preclinical murine tumor models: A structural and functional perspective. Elife 2020, 9, e50740. [Google Scholar] [CrossRef]
- Fung, A.S.; Lee, C.; Yu, M.; Tannock, I.F. The effect of chemotherapeutic agents on tumor vasculature in subcutaneous and orthotopic human tumor xenografts. BMC Cancer 2015, 15, 112. [Google Scholar] [CrossRef]
- Li, M.; Bosman, E.D.C.; Smith, O.M.; Lintern, N.; de Klerk, D.J.; Sun, H.; Cheng, S.; Pan, W.; Storm, G.; Khaled, Y.S.; et al. Comparative analysis of whole cell-derived vesicular delivery systems for photodynamic therapy of extrahepatic cholangiocarcinoma. J. Photochem. Photobiol. B 2024, 254, 112903. [Google Scholar] [CrossRef]
- Price, O.T.; Lau, C.; Zucker, R.M. Quantitative fluorescence of 5-FU-treated fetal rat limbs using confocal laser scanning microscopy and Lysotracker Red. Cytom. A 2003, 53, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Farber, H.; Assaraf, Y.G. LysoTracker and MitoTracker Red are transport substrates of P-glycoprotein: Implications for anticancer drug design evading multidrug resistance. J. Cell. Mol. Med. 2018, 22, 2131–2141. [Google Scholar] [CrossRef]
- Heger, M.; Salles, I.I.; van Vuure, W.; Hamelers, I.H.; de Kroon, A.I.; Deckmyn, H.; Beek, J.F. On the interaction of fluorophore-encapsulating PEGylated lecithin liposomes with hamster and human platelets. Microvasc. Res. 2009, 78, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E.; Karras, J.G.; Murphy, T.F.; Barton, A.; Huang, H.F. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol. Cancer Ther. 2004, 3, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Reiniers, M.J.; de Haan, L.R.; Reeskamp, L.F.; Broekgaarden, M.; van Golen, R.F.; Heger, M. Analysis and Optimization of Conditions for the Use of 2′,7′-Dichlorofluorescein Diacetate in Cultured Hepatocytes. Antioxidants 2021, 10, 674. [Google Scholar] [CrossRef]
- Harada, M.; Woodhams, J.; MacRobert, A.J.; Feneley, M.R.; Kato, H.; Bown, S.G. The vascular response to photodynamic therapy with ATX-S10Na(II) in the normal rat colon. J. Photochem. Photobiol. B 2005, 79, 223–230. [Google Scholar] [CrossRef]
- Suzuki, T.; Tanaka, M.; Sasaki, M.; Ichikawa, H.; Nishie, H.; Kataoka, H. Vascular Shutdown by Photodynamic Therapy Using Talaporfin Sodium. Cancers 2020, 12, 2369. [Google Scholar] [CrossRef]
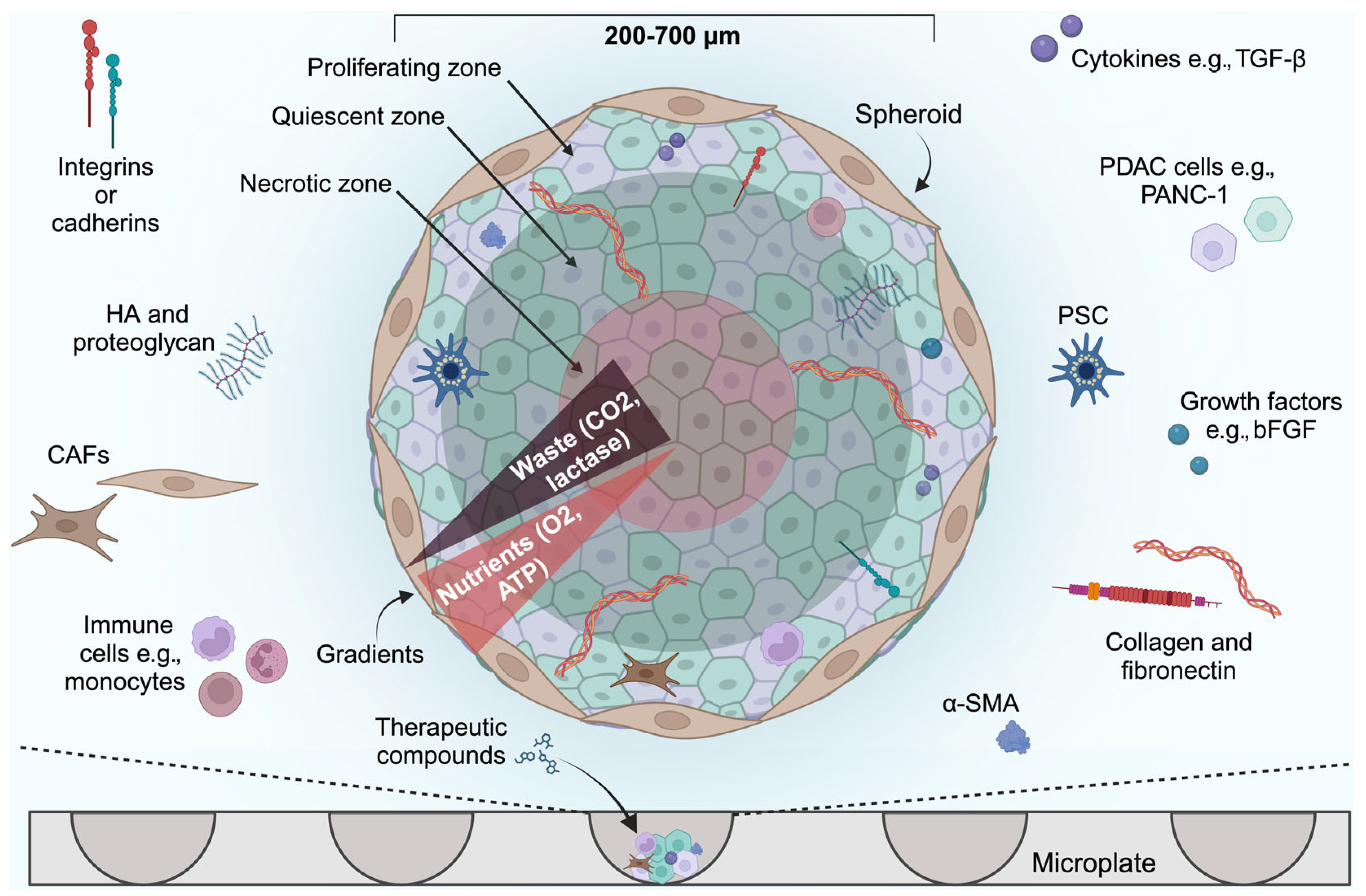
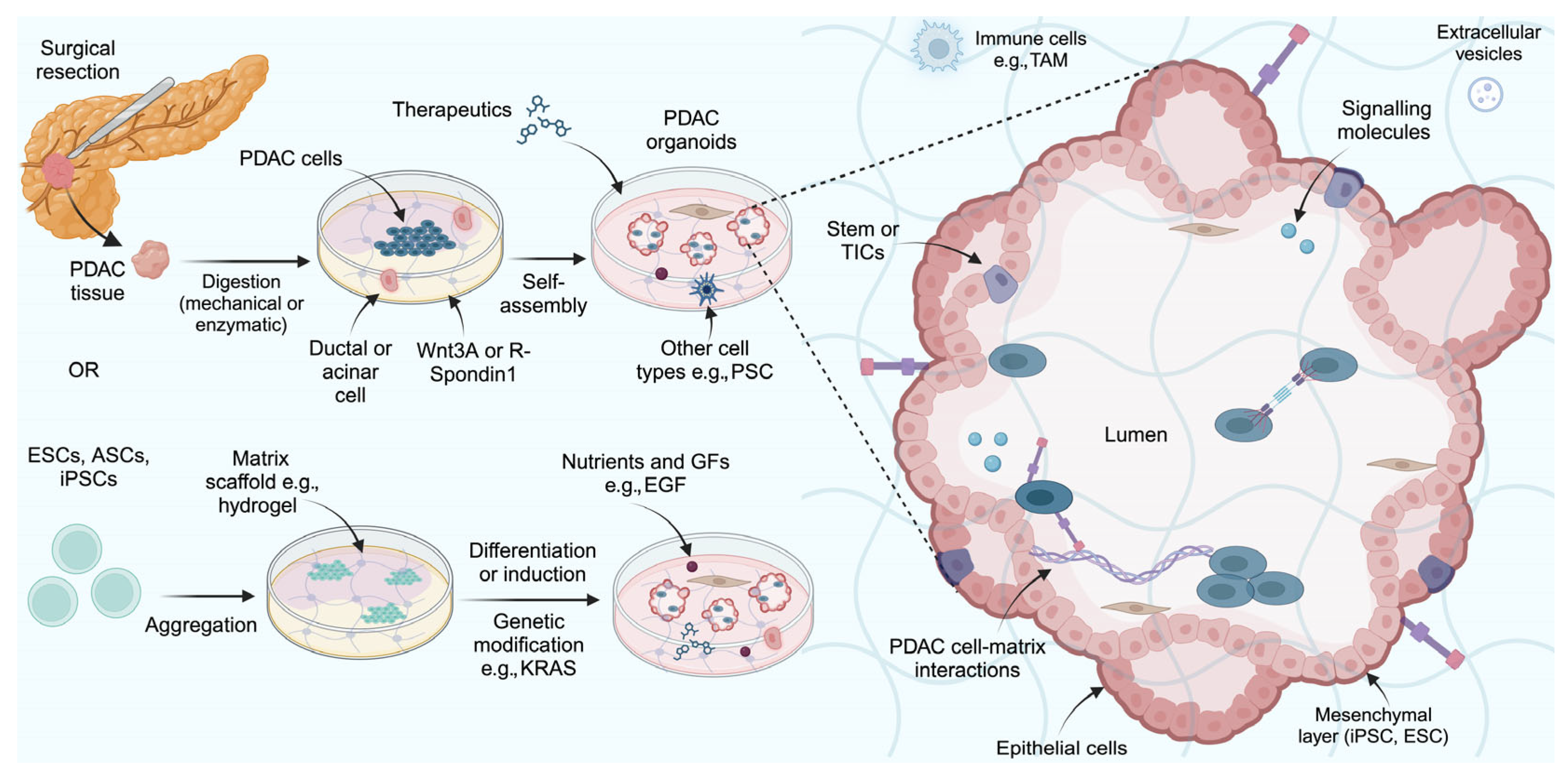
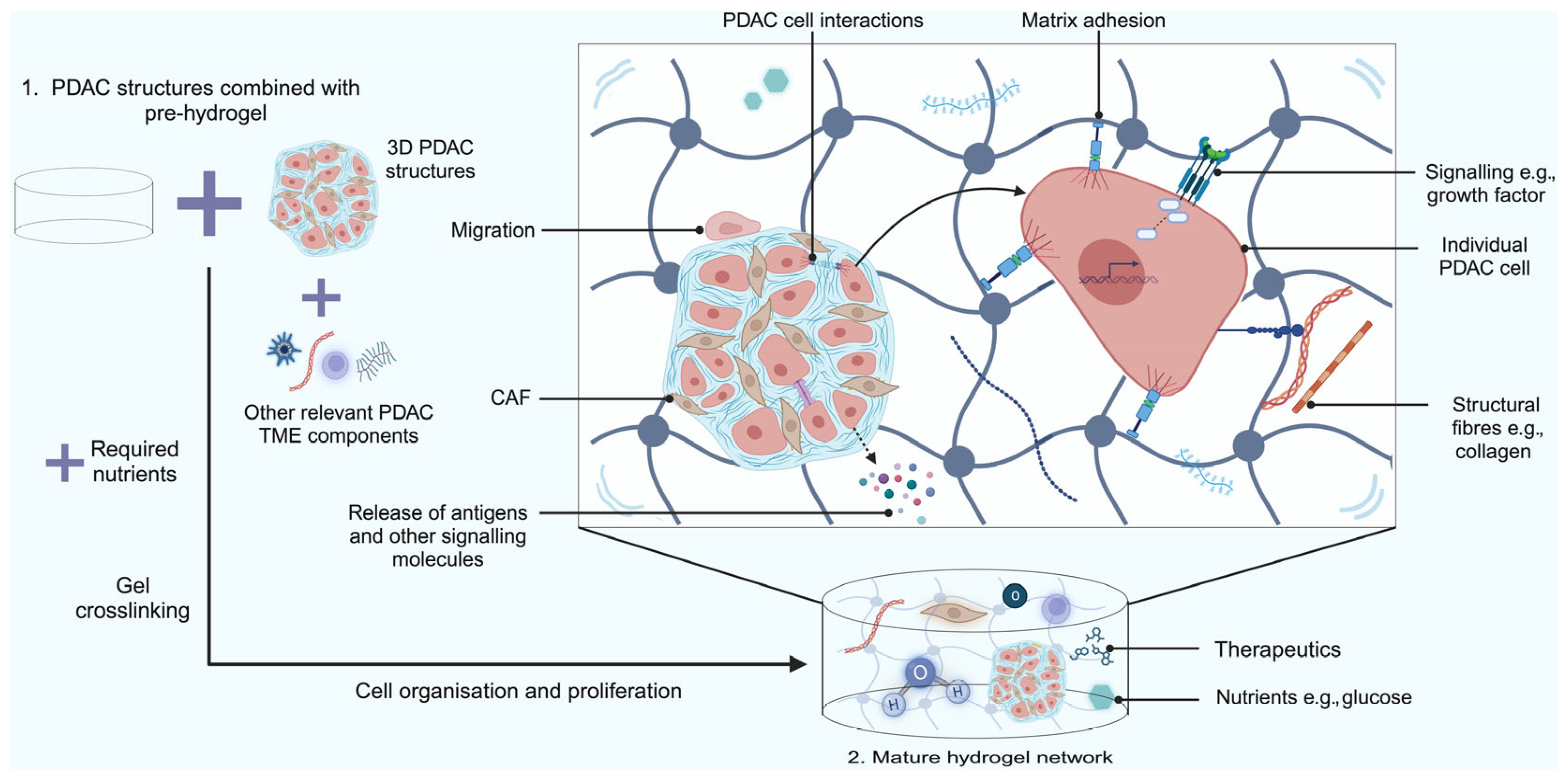
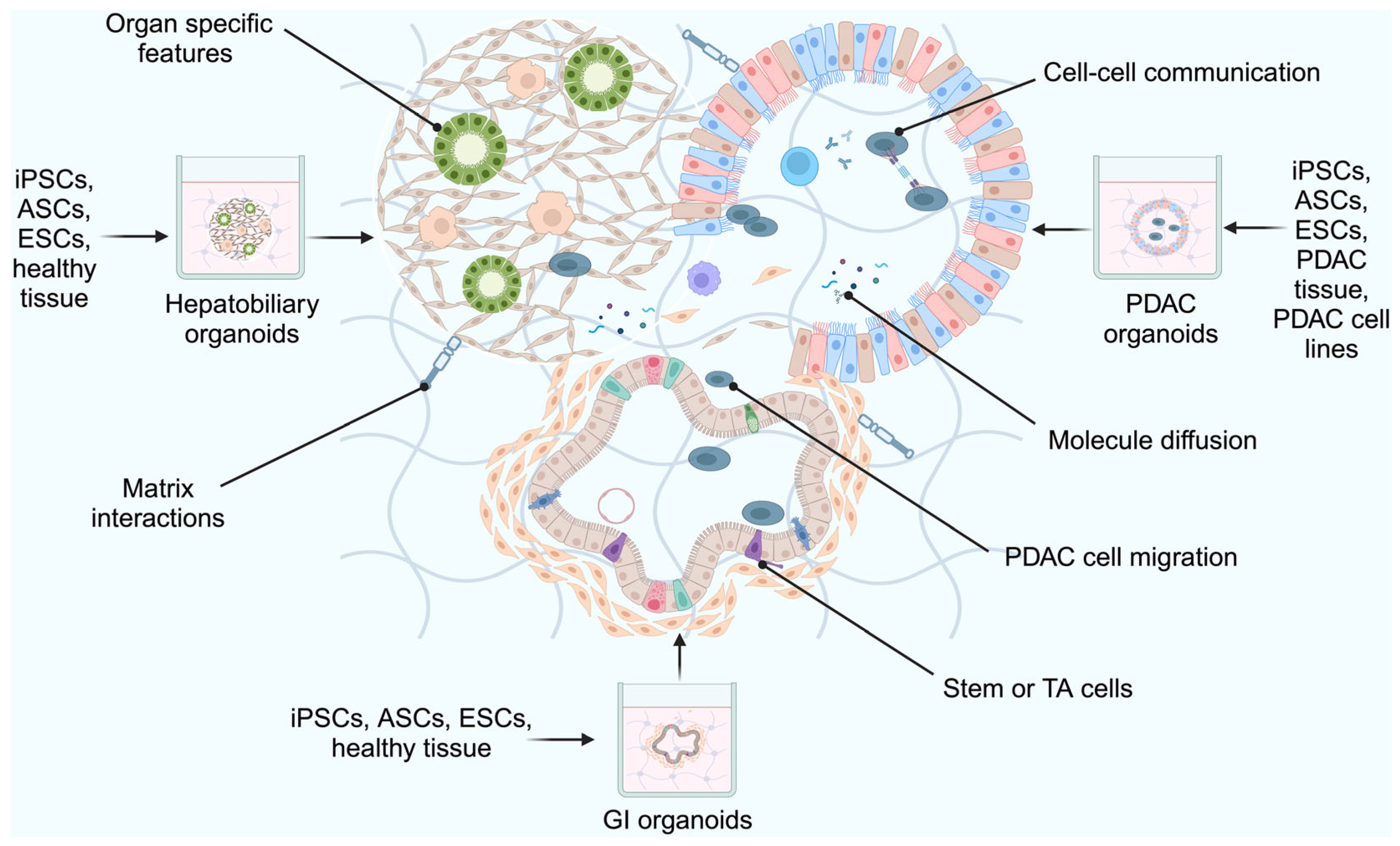
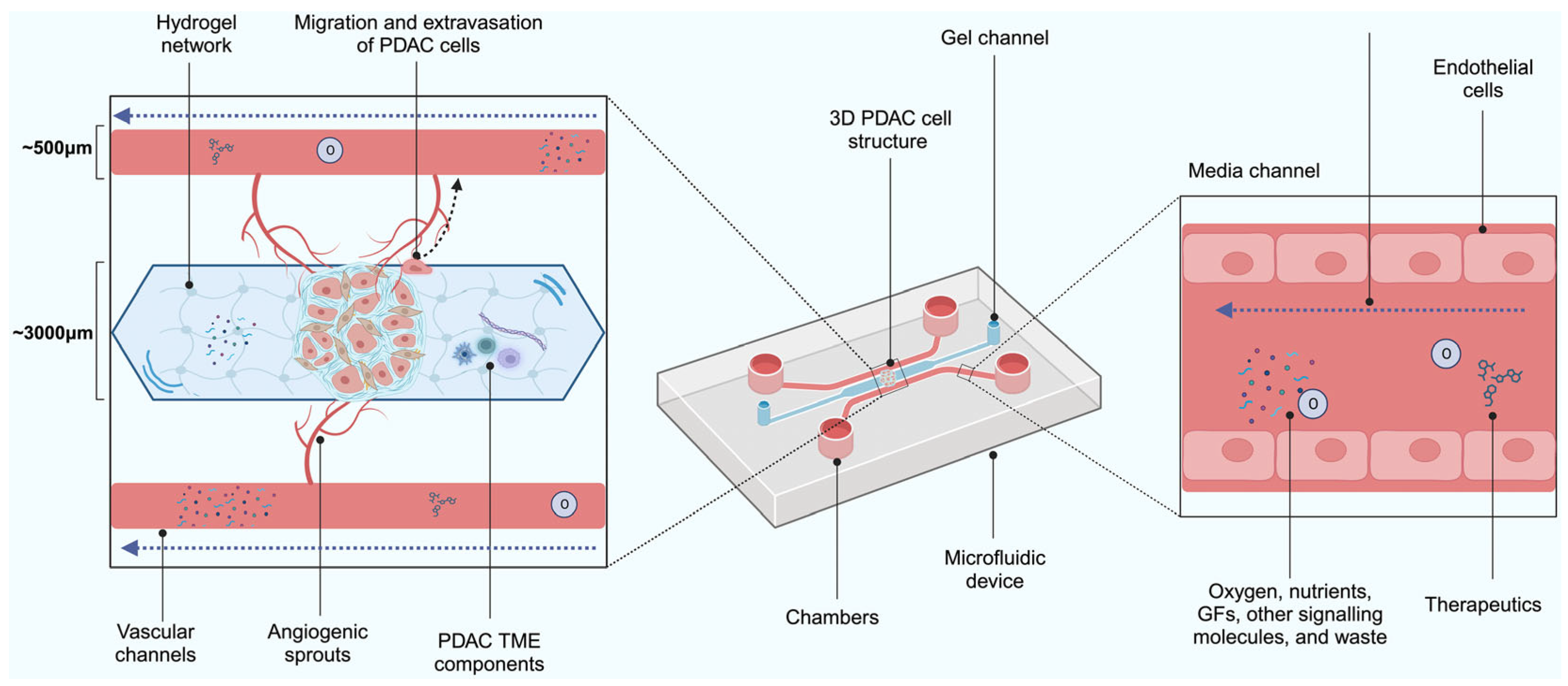
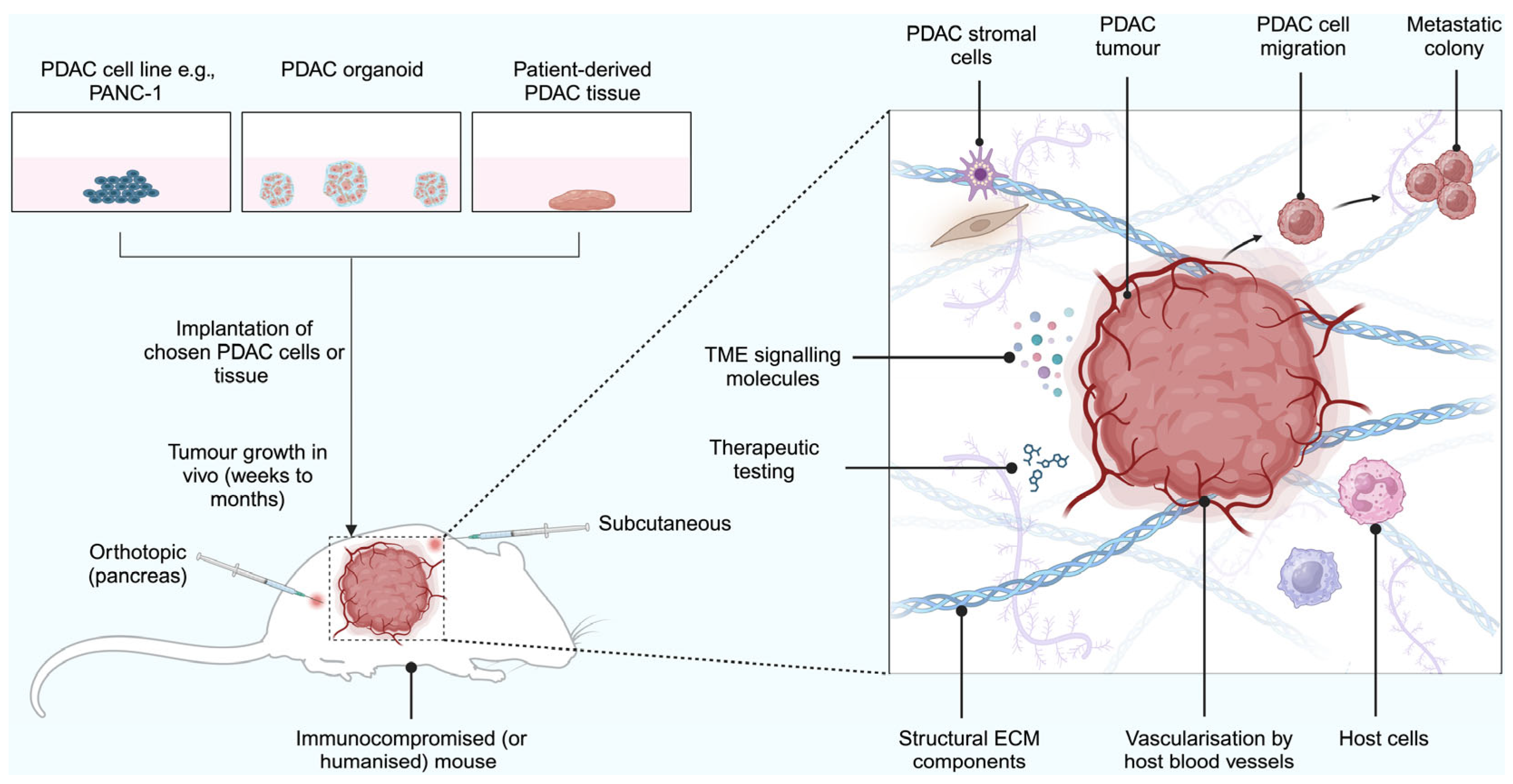

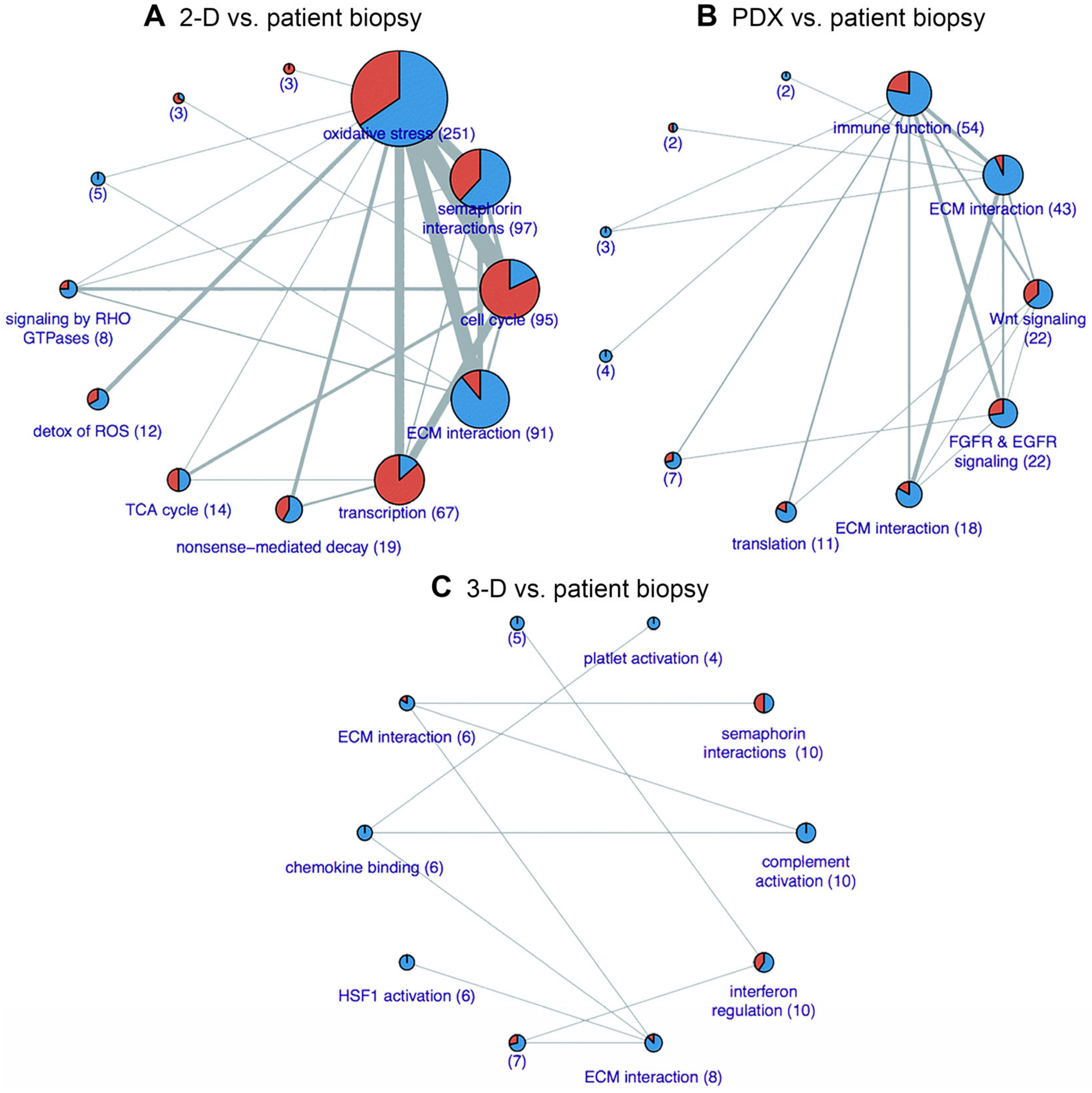
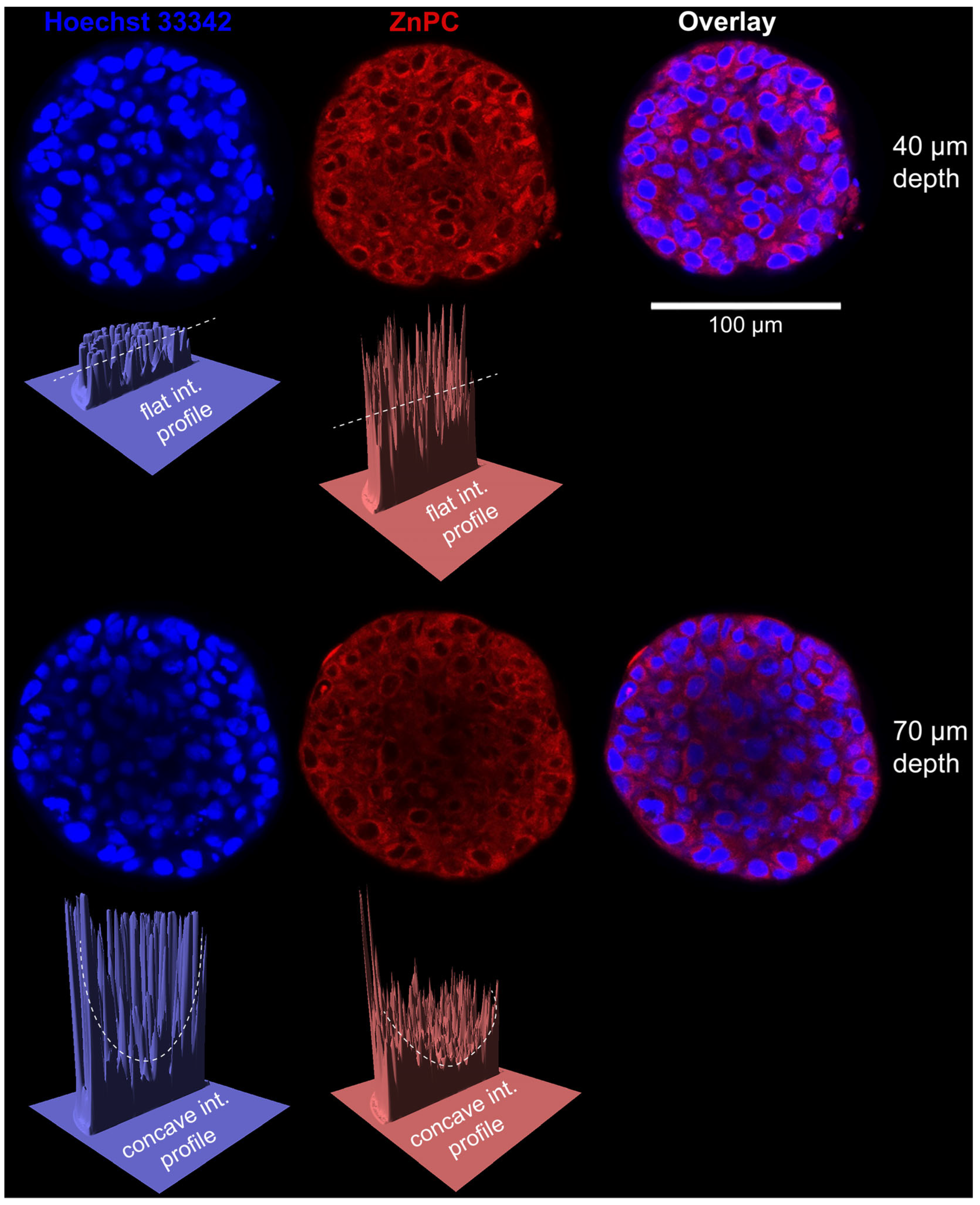
| Model | Spheroid | Co-Cultured Spheroids | Microfluidic Spheroid Models | Hydrogel Scaffold-Based Models | Organoids | Assembloids | Organoid-Based Xenografts | Patient-Derived Xenografts | Cell Line-Derived Xenografts |
|---|---|---|---|---|---|---|---|---|---|
| Application | Investigating PDAC drug toxicity | Investigating PDAC drug toxicity | Industrial, large-scale PDAC drug testing | Testing PDAC drugs where loss of cell viability is not an issue | Investigating the physiology of PDAC | Investigating physiological changes in multiple organs | Investigating disease progression in PDAC | Investigating the efficacy of PDAC therapeutics (TME is well represented) | More convenient than PDXs, but do not fully represent the TME |
| Cost | Low | Moderate | Low | Moderate-high | Moderate | Moderate-high | Moderate-high | High | Moderate-high |
| Time | ~3 to 5 days | ~3 to 5 days | Within 7 days | ~10 days | ~4 days | ≥7 days | 1–2 months | Up to 6 months | 1–2 months |
| Specimen | Cell lines | Cell lines | Cell lines | Cell lines | Tumour tissue, tumour cells, or stem cells | Tumour tissue, tumour cells, or stem cells | Tumour tissue, tumour cells, or stem cells and immunocompromised mice | Resected tumour tissue and immunocompromised mice | Cell lines and immunocompromised mice |
| Cell Line | Disease | Source | Models | Methods | Tested in PDT | Ref. |
|---|---|---|---|---|---|---|
| Mouse | ||||||
| 6606PDA | PDAC | Primary | Cell line-derived xenografts | male C57BL/6 mice (6–8 wk), orthotopic | No | [349] |
| K8484 | PDAC | Primary | Cell line-derived xenografts | KPC mice, subcutaneous | No | [350] |
| KPC3 | PN | Primary | Cell line-derived xenografts | male C57BL/6 mice (8–10 wk), subcutaneous | No | [351] |
| Panc02 | PDAC | Primary | Co-culture spheroids | Matrigel, co-culture with CD8+ cytotoxic T cells | No | [352] |
| Hydrogel-based spheroids | Matrigel and complete DMEM mixture; Matrigel and collagen type I mixture | No | [352,353] | |||
| Cell line-derived xenografts | female C57BL/6 mice (6–8 wk), subcutaneous; C57BL/6 mice (6–8 wk), orthotopic; PDT regimen: IR700-conjugated anti-CD44 monoclonal antibody, 690 nm, 150 mW/cm2, 50 J/cm2; PTT regimen: 980 nm, 850 mW/cm2, 50 J/cm2, 600 s | Yes | [354,355] | |||
| Cell line-derived xenografts | C57BL/6 mice (6–8 wk), orthotopic; PTT regimen: 980 nm, 850 mW/cm2, 50 J/cm2, 600 s | PTT | [355] | |||
| UN-KC-6141 | PDAC | Primary | Cell line-derived xenografts | C57BL/6 mice, orthotopic | No | [356] |
| UN-KPC-960 | PDAC | Primary | Cell line-derived xenografts | B6.129 mice, orthotopic | No | [356] |
| UN-KPC-961 | PDAC | Primary | Cell line-derived xenografts | B6.129 mice, subcutaneous and orthotopic | No | [356] |
| Hamster | ||||||
| HaP-T1 | PDAC | Primary | Cell line-derived xenografts | male Syrian golden hamsters (5 wk), orthotopic | No | [322] |
| PC-1.0 | PDAC | Primary | Cell line-derived xenografts | male Syrian golden hamsters (5 wk), orthotopic | No | [357] |
| PC-1.2 | PDAC | Primary | Cell line-derived xenografts | Syrian golden hamsters (8 wk), orthotopic | No | [358] |
| WD PaCa | PDAC | Primary | No | [359] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, O.M.; Lintern, N.; Tian, J.; Mesquita, B.M.; Oliveira, S.; Vymetalkova, V.; Prakash, J.; Smith, A.M.; Jayne, D.G.; Heger, M.; et al. Biomimetic Tumour Model Systems for Pancreatic Ductal Adenocarcinoma in Relation to Photodynamic Therapy. Int. J. Mol. Sci. 2025, 26, 6388. https://doi.org/10.3390/ijms26136388
Smith OM, Lintern N, Tian J, Mesquita BM, Oliveira S, Vymetalkova V, Prakash J, Smith AM, Jayne DG, Heger M, et al. Biomimetic Tumour Model Systems for Pancreatic Ductal Adenocarcinoma in Relation to Photodynamic Therapy. International Journal of Molecular Sciences. 2025; 26(13):6388. https://doi.org/10.3390/ijms26136388
Chicago/Turabian StyleSmith, Olivia M., Nicole Lintern, Jiahao Tian, Bárbara M. Mesquita, Sabrina Oliveira, Veronika Vymetalkova, Jai Prakash, Andrew M. Smith, David G. Jayne, Michal Heger, and et al. 2025. "Biomimetic Tumour Model Systems for Pancreatic Ductal Adenocarcinoma in Relation to Photodynamic Therapy" International Journal of Molecular Sciences 26, no. 13: 6388. https://doi.org/10.3390/ijms26136388
APA StyleSmith, O. M., Lintern, N., Tian, J., Mesquita, B. M., Oliveira, S., Vymetalkova, V., Prakash, J., Smith, A. M., Jayne, D. G., Heger, M., & Khaled, Y. S., on behalf of the Photodynamic Therapy Study Group. (2025). Biomimetic Tumour Model Systems for Pancreatic Ductal Adenocarcinoma in Relation to Photodynamic Therapy. International Journal of Molecular Sciences, 26(13), 6388. https://doi.org/10.3390/ijms26136388