


Molecular Determinants of TMC Protein Biogenesis and Trafficking

 ,
,
Abstract
1. Introduction
2. Results
2.1. MmTMC1 and MmTMC2 Do Not Localize to the Plasma Membrane of HEK 293T Cells
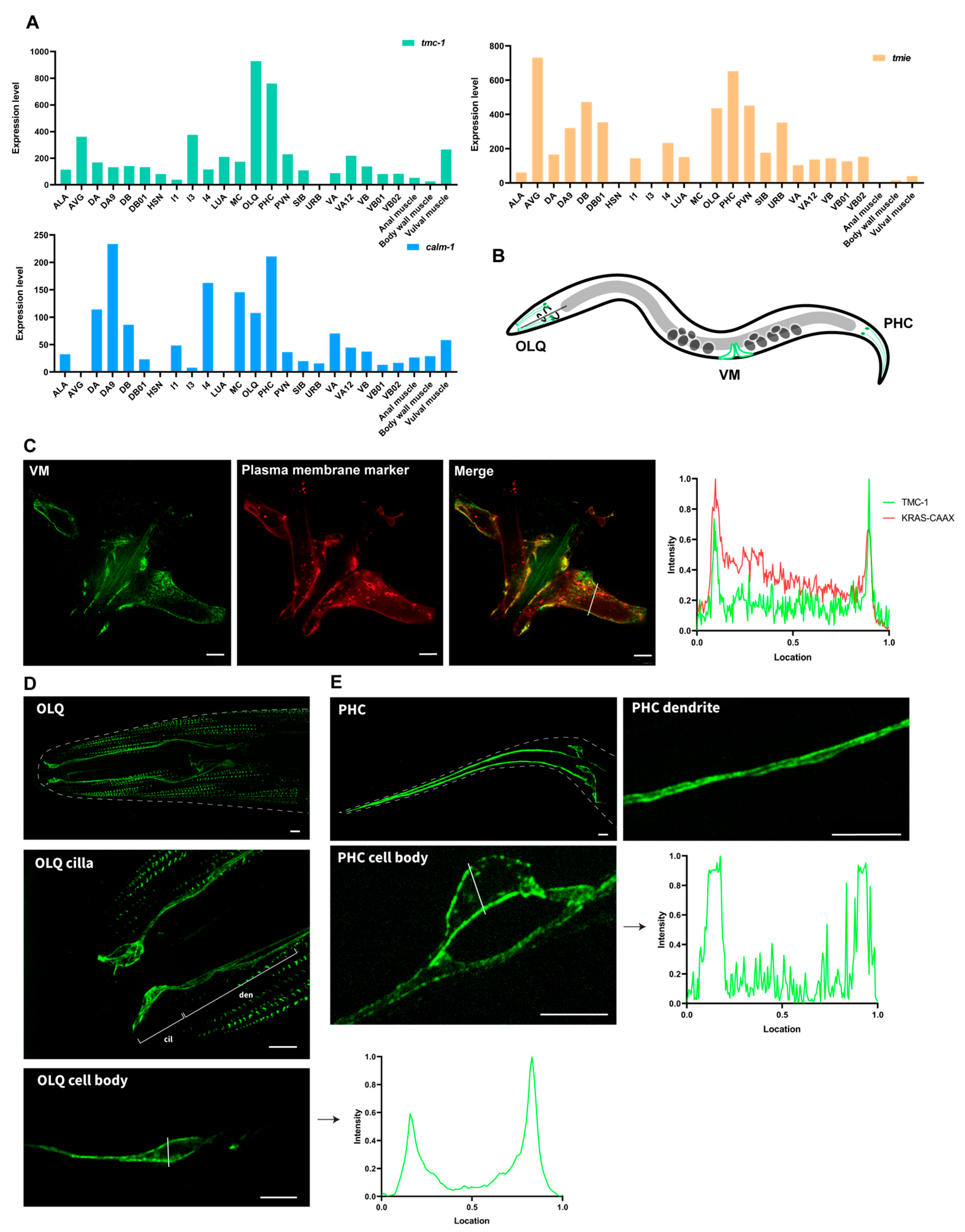
2.2. Intense Plasma Membrane Localization of TMC-1 in C. elegans Neurons and Muscle Cells
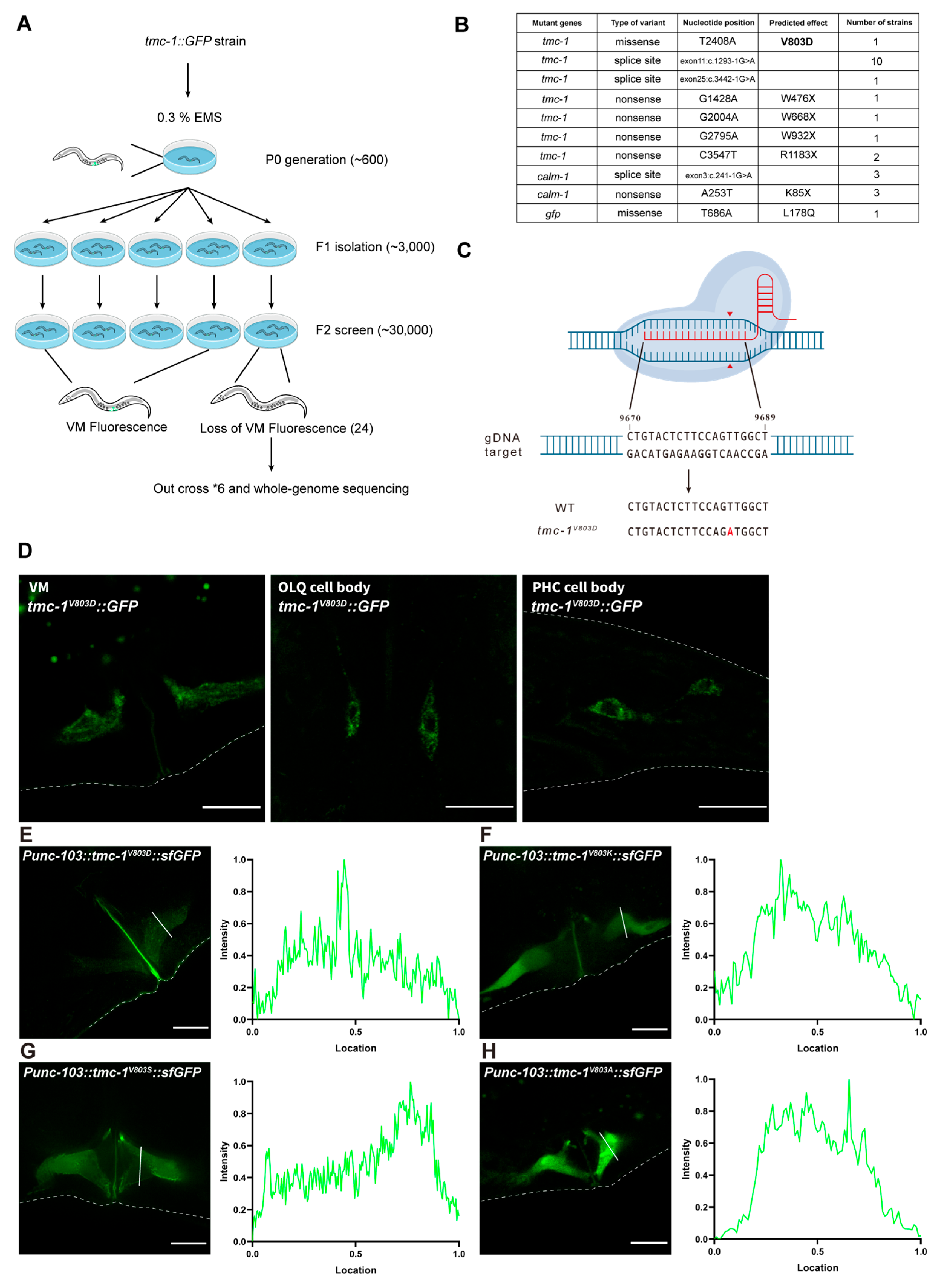
2.3. Forward Genetic Screen in C. elegans Identifies Critical Residues for the Plasma Membrane Localization of TMC-1
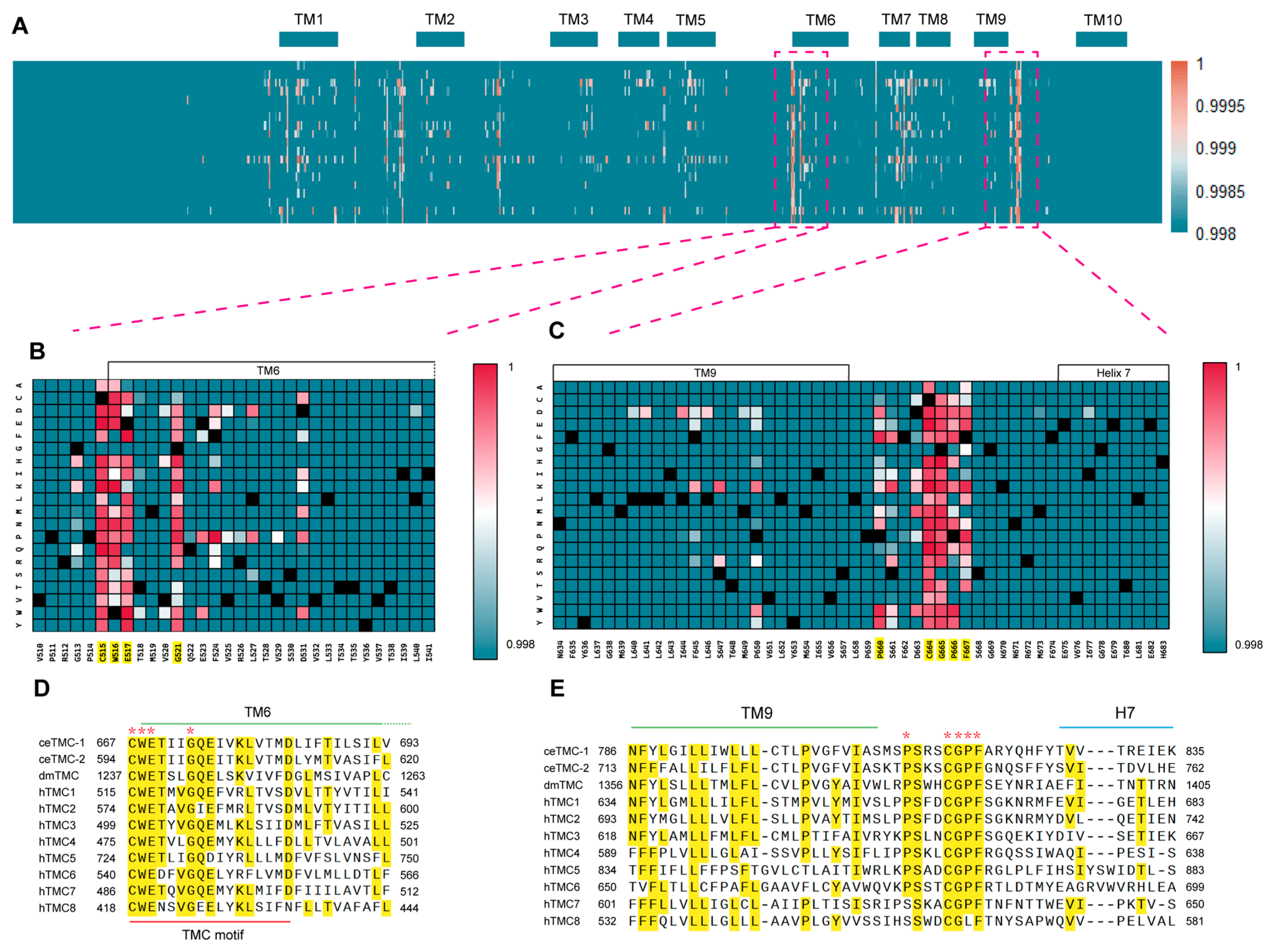
2.4. Human TMC1 TM9–TM10 Extracellular-Loop Mutation Hotspot and Its Conserved Equivalents in C. elegans TMC-1
2.5. AlphaMissense Reveals Two Structurally Connected Functional Hotspots in Human TMC1
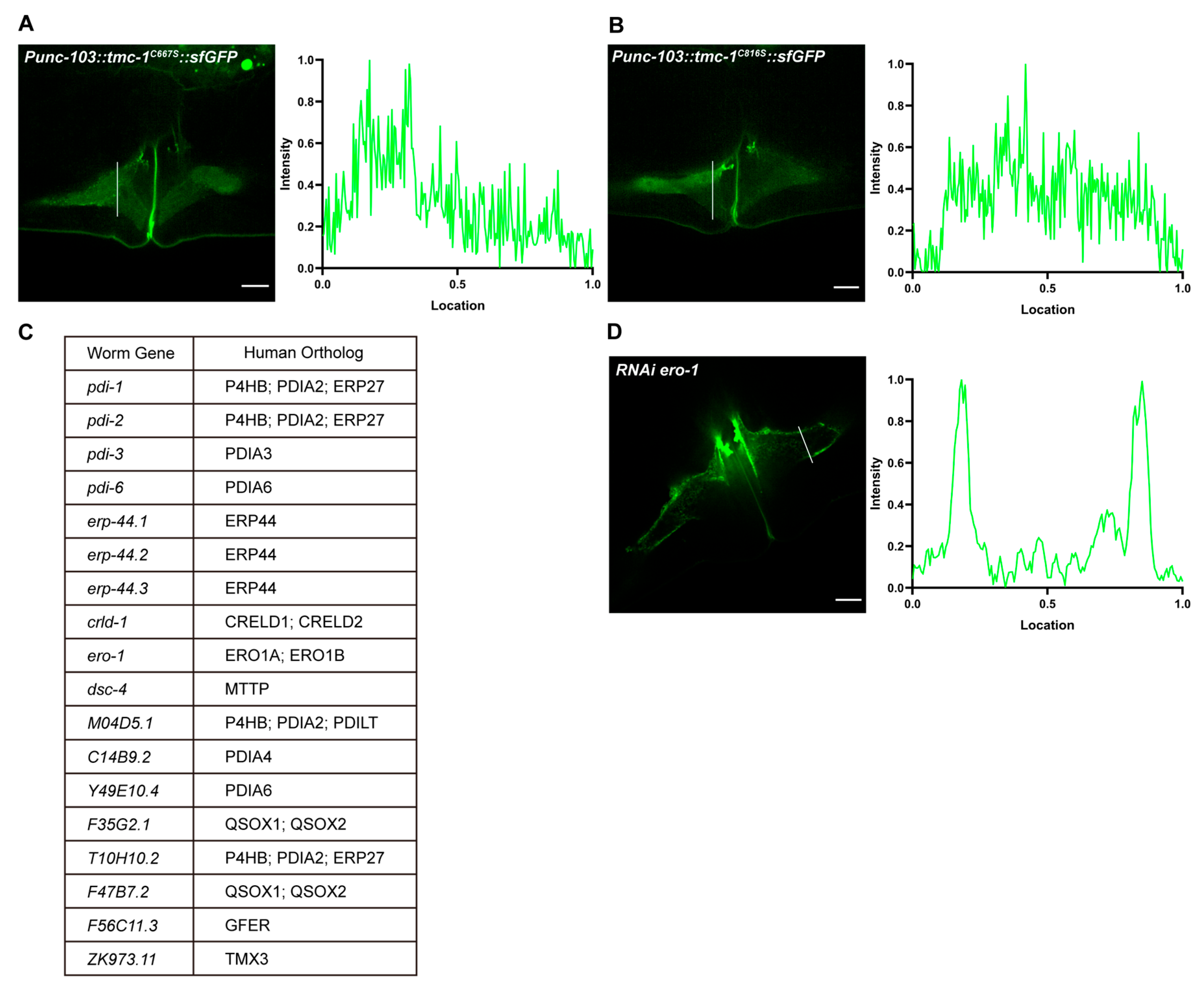
2.6. Disruption of the Disulfide Bond Impairs TMC-1 Trafficking
2.7. TMC-1 Disulfide Bond Formation May Involve Unknown Chaperones
3. Discussion
3.1. The TMC Motif and Disulfide Bond: Key Determinants of TMC Protein Folding and Trafficking
3.2. Disulfide-Bond-Connected Functional Hotspots and TMC-Associated Diseases
3.3. ER Redox Regulation of TMC Protein Folding
4. Materials and Methods
4.1. Experimental Worm Strains
4.2. Plasmids Construction
4.3. Immunofluorescence and Super-Resolution Imaging
4.4. EMS Mutagenesis and Genetic Mapping
4.5. Generation of CRISPR/Cas9-Mediated V803D Mutant Strains
4.6. RNA Interference (RNAi) Feeding
4.7. Protein Structure Accession Numbers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hudspeth, A.J.; Corey, D.P. Sensitivity, polarity, and conductance change in the response of vertebrate hair cells to controlled mechanical stimuli. Proc. Natl. Acad. Sci. USA 1977, 74, 2407–2411. [Google Scholar] [PubMed]
- Corey, D.P.; Hudspeth, A.J. Response latency of vertebrate hair cells. Biophys. J. 1979, 26, 499–506. [Google Scholar] [PubMed]
- Corey, D.P.; Hudspeth, A.J. Ionic basis of the receptor potential in a vertebrate hair cell. Nature 1979, 281, 675–677. [Google Scholar] [PubMed]
- Kawashima, Y.; Geleoc, G.S.G.; Kurima, K.; Labay, V.; Lelli, A.; Asai, Y.; Makishima, T.; Wu, D.K.; Della Santina, C.C.; Holt, J.R.; et al. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J. Clin. Investig. 2011, 121, 4796–4809. [Google Scholar]
- Pan, B.; Geleoc, G.S.; Asai, Y.; Horwitz, G.C.; Kurima, K.; Ishikawa, K.; Kawashima, Y.; Griffith, A.J.; Holt, J.R. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 2013, 79, 504–515. [Google Scholar]
- Pan, B.; Akyuz, N.; Liu, X.P.; Asai, Y.; Nist-Lund, C.; Kurima, K.; Derfler, B.H.; Gyorgy, B.; Limapichat, W.; Walujkar, S.; et al. TMC1 Forms the Pore of Mechanosensory Transduction Channels in Vertebrate Inner Ear Hair Cells. Neuron 2018, 99, 736–753 e6. [Google Scholar]
- Jia, Y.; Zhao, Y.; Kusakizako, T.; Wang, Y.; Pan, C.; Zhang, Y.; Nureki, O.; Hattori, M.; Yan, Z. TMC1 and TMC2 Proteins Are Pore-Forming Subunits of Mechanosensitive Ion Channels. Neuron 2020, 105, 310–321 e3. [Google Scholar]
- Jeong, H.; Clark, S.; Goehring, A.; Dehghani-Ghahnaviyeh, S.; Rasouli, A.; Tajkhorshid, E.; Gouaux, E. Structures of the TMC-1 complex illuminate mechanosensory transduction. Nature 2022, 610, 796–803. [Google Scholar]
- Kurima, K.; Peters, L.M.; Yang, Y.; Riazuddin, S.; Ahmed, Z.M.; Naz, S.; Arnaud, D.; Drury, S.; Mo, J.; Makishima, T.; et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat. Genet. 2002, 30, 277–284. [Google Scholar]
- Vreugde, S.; Erven, A.; Kros, C.J.; Marcotti, W.; Fuchs, H.; Kurima, K.; Wilcox, E.R.; Friedman, T.B.; Griffith, A.J.; Balling, R.; et al. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat. Genet. 2002, 30, 257–258. [Google Scholar]
- Boucher, S.; Tai, F.W.J.; Delmaghani, S.; Lelli, A.; Singh-Estivalet, A.; Dupont, T.; Niasme-Grare, M.; Michel, V.; Wolff, N.; Bahloul, A.; et al. Ultrarare heterozygous pathogenic variants of genes causing dominant forms of early-onset deafness underlie severe presbycusis. Proc. Natl. Acad. Sci. USA 2020, 117, 31278–31289. [Google Scholar] [PubMed]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [PubMed]
- Nist-Lund, C.A.; Pan, B.; Patterson, A.; Asai, Y.; Chen, T.; Zhou, W.; Zhu, H.; Romero, S.; Resnik, J.; Polley, D.B.; et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 2019, 10, 236. [Google Scholar] [PubMed]
- Yeh, W.H.; Shubina-Oleinik, O.; Levy, J.M.; Pan, B.; Newby, G.A.; Wornow, M.; Burt, R.; Chen, J.C.; Holt, J.R.; Liu, D.R. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 2020, 12, eaay9101. [Google Scholar]
- Wu, J.; Solanes, P.; Nist-Lund, C.; Spataro, S.; Shubina-Oleinik, O.; Marcovich, I.; Goldberg, H.; Schneider, B.L.; Holt, J.R. Single and Dual Vector Gene Therapy with AAV9-PHP.B Rescues Hearing in Tmc1 Mutant Mice. Mol. Ther. 2021, 29, 973–988. [Google Scholar]
- Zheng, Z.; Li, G.; Cui, C.; Wang, F.; Wang, X.; Xu, Z.; Guo, H.; Chen, Y.; Tang, H.; Wang, D.; et al. Preventing autosomal-dominant hearing loss in Bth mice with CRISPR/CasRx-based RNA editing. Signal Transduct. Target. Ther. 2022, 7, 79. [Google Scholar]
- Yu, X.; Zhao, Q.; Li, X.; Chen, Y.; Tian, Y.; Liu, S.; Xiong, W.; Huang, P. Deafness mutation D572N of TMC1 destabilizes TMC1 expression by disrupting LHFPL5 binding. Proc. Natl. Acad. Sci. USA 2020, 117, 29894–29903. [Google Scholar]
- Beurg, M.; Schimmenti, L.A.; Koleilat, A.; Amr, S.S.; Oza, A.; Barlow, A.J.; Ballesteros, A.; Fettiplace, R. New Tmc1 Deafness Mutations Impact Mechanotransduction in Auditory Hair Cells. J. Neurosci. 2021, 41, 4378–4391. [Google Scholar]
- Akyuz, N.; Karavitaki, K.D.; Pan, B.; Tamvakologos, P.I.; Brock, K.P.; Li, Y.; Marks, D.S.; Corey, D.P. Mechanical gating of the auditory transduction channel TMC1 involves the fourth and sixth transmembrane helices. Sci. Adv. 2022, 8, eabo1126. [Google Scholar]
- Ballesteros, A.; Swartz, K.J. Regulation of membrane homeostasis by TMC1 mechanoelectrical transduction channels is essential for hearing. Sci. Adv. 2022, 8, eabm5550. [Google Scholar]
- Fettiplace, R.; Furness, D.N.; Beurg, M. The conductance and organization of the TMC1-containing mechanotransducer channel complex in auditory hair cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2210849119. [Google Scholar]
- Beurg, M.; Schwalbach, E.T.; Fettiplace, R. LHFPL5 is a key element in force transmission from the tip link to the hair cell mechanotransducer channel. Proc. Natl. Acad. Sci. USA 2024, 121, e2318270121. [Google Scholar]
- Fu, S.; Pan, X.; Lu, M.; Dong, J.; Yan, Z. Human TMC1 and TMC2 are mechanically gated ion channels. Neuron 2025, 113, 411–425 e4. [Google Scholar] [PubMed]
- Yang, Z.; Zhou, S.H.; Zhang, Q.Y.; Song, Z.C.; Liu, W.W.; Sun, Y.; Wang, M.W.; Fu, X.L.; Zhu, K.K.; Guan, Y.; et al. A force-sensitive adhesion GPCR is required for equilibrioception. Cell Res. 2025, 35, 243–264. [Google Scholar]
- Chen, Y.; Li, Y.; Liu, Y.; Sun, J.; Feng, W.; Chen, Y.; Tian, Y.; Lei, T.; Huang, P. Ectopic mouse TMC1 and TMC2 alone form mechanosensitive channels that are potently modulated by TMIE. Proc. Natl. Acad. Sci. USA 2025, 122, e2403141122. [Google Scholar]
- Tang, Y.Q.; Lee, S.A.; Rahman, M.; Vanapalli, S.A.; Lu, H.; Schafer, W.R. Ankyrin Is An Intracellular Tether for TMC Mechanotransduction Channels. Neuron 2020, 107, 112–125 e10. [Google Scholar]
- Kurima, K.; Ebrahim, S.; Pan, B.; Sedlacek, M.; Sengupta, P.; Millis, B.A.; Cui, R.; Nakanishi, H.; Fujikawa, T.; Kawashima, Y.; et al. TMC1 and TMC2 Localize at the Site of Mechanotransduction in Mammalian Inner Ear Hair Cell Stereocilia. Cell Rep. 2015, 12, 1606–1617. [Google Scholar]
- Mahendrasingam, S.; Furness, D.N. Ultrastructural localization of the likely mechanoelectrical transduction channel protein, transmembrane-like channel 1 (TMC1) during development of cochlear hair cells. Sci. Rep. 2019, 9, 1274. [Google Scholar]
- Wang, P.; Miller, K.K.; He, E.; Dhawan, S.S.; Cunningham, C.L.; Grillet, N. LOXHD1 is indispensable for maintaining TMC1 auditory mechanosensitive channels at the site of force transmission. Nat. Commun. 2024, 15, 7865. [Google Scholar]
- Clark, S.; Jeong, H.; Posert, R.; Goehring, A.; Gouaux, E. The structure of the Caenorhabditis elegans TMC-2 complex suggests roles of lipid-mediated subunit contacts in mechanosensory transduction. Proc. Natl. Acad. Sci. USA 2024, 121, e2314096121. [Google Scholar]
- Taylor, S.R.; Santpere, G.; Weinreb, A.; Barrett, A.; Reilly, M.B.; Xu, C.; Varol, E.; Oikonomou, P.; Glenwinkel, L.; McWhirter, R.; et al. Molecular topography of an entire nervous system. Cell 2021, 184, 4329–4347.e23. [Google Scholar] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Zemgulyte, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [PubMed]
- Santos, R.L.; Wajid, M.; Khan, M.N.; McArthur, N.; Pham, T.L.; Bhatti, A.; Lee, K.; Irshad, S.; Mir, A.; Yan, K.; et al. Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Hum. Mutat. 2005, 26, 396. [Google Scholar]
- Kitajiri, S.I.; McNamara, R.; Makishima, T.; Husnain, T.; Zafar, A.U.; Kittles, R.A.; Ahmed, Z.M.; Friedman, T.B.; Riazuddin, S.; Griffith, A.J. Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clin. Genet. 2007, 72, 546–550. [Google Scholar]
- Sadeghian, L.; Tabatabaiefar, M.A.; Fattahi, N.; Pourreza, M.R.; Tahmasebi, P.; Alavi, Z.; Hashemzadeh Chaleshtori, M. Next-generation sequencing reveals a novel pathological mutation in the TMC1 gene causing autosomal recessive non-syndromic hearing loss in an Iranian kindred. Int. J. Pediatr. Otorhinolaryngol. 2019, 124, 99–105. [Google Scholar]
- Zardadi, S.; Razmara, E.; Asgaritarghi, G.; Jafarinia, E.; Bitarafan, F.; Rayat, S.; Almadani, N.; Morovvati, S.; Garshasbi, M. Novel homozygous variants in the TMC1 and CDH23 genes cause autosomal recessive nonsyndromic hearing loss. Mol. Genet. Genom. Med. 2020, 8, e1550. [Google Scholar]
- Tollefson, M.R.; Gogal, R.A.; Weaver, A.M.; Schaefer, A.M.; Marini, R.J.; Azaiez, H.; Kolbe, D.L.; Wang, D.; Weaver, A.E.; Casavant, T.L.; et al. Assessing variants of uncertain significance implicated in hearing loss using a comprehensive deafness proteome. Hum. Genet. 2023, 142, 819–834. [Google Scholar]
- Ramoz, N.; Rueda, L.A.; Bouadjar, B.; Montoya, L.S.; Orth, G.; Favre, M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 2002, 32, 579–581. [Google Scholar]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar]
- Rutkevich, L.A.; Williams, D.B. Vitamin K epoxide reductase contributes to protein disulfide formation and redox homeostasis within the endoplasmic reticulum. Mol. Biol. Cell 2012, 23, 2017–2027. [Google Scholar] [PubMed]
- Ushioda, R.; Nagata, K. Redox-Mediated Regulatory Mechanisms of Endoplasmic Reticulum Homeostasis. Cold Spring Harb. Perspect. Biol. 2019, 11, a033910. [Google Scholar]
- Wang, L.; Wang, C.C. Oxidative protein folding fidelity and redoxtasis in the endoplasmic reticulum. Trends Biochem. Sci. 2023, 48, 40–52. [Google Scholar] [PubMed]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar]
- Karnik, S.S.; Gogonea, C.; Patil, S.; Saad, Y.; Takezako, T. Activation of G-protein-coupled receptors: A common molecular mechanism. Trends Endocrinol. Metab. 2003, 14, 431–437. [Google Scholar]
- Tao, Y.X.; Conn, P.M. Chaperoning G protein-coupled receptors: From cell biology to therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar]
- Hwa, J.; Reeves, P.J.; Klein-Seetharaman, J.; Davidson, F.; Khorana, H.G. Structure and function in rhodopsin: Further elucidation of the role of the intradiscal cysteines, Cys-110, -185, and -187, in rhodopsin folding and function. Proc. Natl. Acad. Sci. USA 1999, 96, 1932–1935. [Google Scholar]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 2018, 293, 17464–17476. [Google Scholar]
- Fujii, S.; Ushioda, R.; Nagata, K. Redox states in the endoplasmic reticulum directly regulate the activity of calcium channel, inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2023, 120, e2216857120. [Google Scholar]
- Lee, Y.K.; Brewer, J.W.; Hellman, R.; Hendershot, L.M. BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol. Biol. Cell 1999, 10, 2209–2219. [Google Scholar]
- Meunier, L.; Usherwood, Y.K.; Chung, K.T.; Hendershot, L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 2002, 13, 4456–4469. [Google Scholar] [PubMed]
- Feige, M.J.; Groscurth, S.; Marcinowski, M.; Shimizu, Y.; Kessler, H.; Hendershot, L.M.; Buchner, J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 2009, 34, 569–579. [Google Scholar] [PubMed]
- Oliver, J.D.; Roderick, H.L.; Llewellyn, D.H.; High, S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell 1999, 10, 2573–2582. [Google Scholar] [PubMed]
- Mezghrani, A.; Fassio, A.; Benham, A.; Simmen, T.; Braakman, I.; Sitia, R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001, 20, 6288–6296. [Google Scholar]
- Sugiura, Y.; Araki, K.; Iemura, S.; Natsume, T.; Hoseki, J.; Nagata, K. Novel thioredoxin-related transmembrane protein TMX4 has reductase activity. J. Biol. Chem. 2010, 285, 7135–7142. [Google Scholar]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar]
- Zuryn, S.; Le Gras, S.; Jamet, K.; Jarriault, S. A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics 2010, 186, 427–430. [Google Scholar]
- Dokshin, G.A.; Ghanta, K.S.; Piscopo, K.M.; Mello, C.C. Robust Genome Editing with Short Single-Stranded and Long, Partial-ly Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics 2018, 210, 781–787. [Google Scholar]
- Kamath, R.S.; Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 2003, 30, 313–321. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Worm Strains | Source | Identifier |
|---|---|---|
| tmc-1(ok1859) | CGC | TYQ15 |
| yqt1[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR] | This study | TYQ28 |
| yqt5[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1V803D | This study | TYQ159 |
| yqt6[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1(splicing exon11:c.1293-1G>A) | This study | TYQ160 |
| yqt10[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1R1183* | This study | TYQ164 |
| yqt12[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1W668* | This study | TYQ166 |
| yqt13[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; calm-1(splicing exon3:c.241-1G>A) | This study | TYQ167 |
| yqt16[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; calm-1K85* | This study | TYQ170 |
| yqt21[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; GFPA110V | This study | TYQ175 |
| yqt24[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1W476* | This study | TYQ178 |
| yqt25[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1(splicing exon25:c.3442-1G>A) | This study | TYQ179 |
| yqt29[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]; tmc-1W932* | This study | TYQ183 |
| yqtEx138[Punc-103e::cetmc-1V803S::sfGFP(50ng/μ); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ277 |
| yqtEx139[Punc-103e::cetmc-1V803K::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ278 |
| yqtEx148[Punc-103e::cetmc-1P812L::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ289 |
| yqtEx157[Punc-103e::cetmc-1C816W::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ365 |
| yqtEx158[Punc-103e::cetmc-1E834Q::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ366 |
| yqtEx166[Punc-103e::cetmc-1V803D::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ374 |
| yqtEx175[Punc-103e::cetmc-1V829T::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ384 |
| yqtEx177[Punc-103e::cetmc-1P812L::sfGFP(50ng/μl);Ptmc-2-2kb::ctmie::mCherry(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ386 |
| yqtEx178[Punc-103e::cetmc-1C816W::sfGFP(50ng/μl);Ptmc-2-2kb::ctmie::mCherry(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ387 |
| yqtEx179[Punc-103e::cetmc-1C816S::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ408 |
| yqtEx180[Punc-103e::cetmc-1C667R::sfGFP(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ409 |
| yqtEx182[Ptmc-2-2kb::ctmie::mCherry(50ng/μl); Pmyo-2::GFP(2.5ng/μl); pcDNA3.1(to total 150ng/μl)); tmc-1(ok1859) | This study | TYQ411 |
| yqtEx183[Punc-103e::cetmc-1C816S::sfGFP(50ng/μl);Ptmc-2-2kb::ctmie::mCherry(50ng/μl);Pmyo-2::GFP(2.5ng/μl); pcDNA3.1(to total 150ng/μl)]; tmc-1(ok1859) | This study | TYQ412 |
| yqtEx184[Punc-103e::cetmc-1V828D::sfGFP(50ng/μl);Ptmc-2-2kb::ctmie::mCherry(50ng/μl); Pmyo-2::mCherry(5ng/μl); pcDNA3.1(to total 150ng/μl)] | This study | TYQ413 |
| yqtEx187[Punc-103e::mCherry::KRAS-CAAX(40ng/μl); Pmyo-2::GFP(2.5ng/μl); pcDNA3.1(to total 150ng/μl); yqt1[tmc-1::GFP;Prps-0::HygR::unc-54 3’UTR]] | This study | TYQ416 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, D.; Tan, J.; Fan, X.; Shu, Y.; Qu, Q.; Tang, Y.-Q. Molecular Determinants of TMC Protein Biogenesis and Trafficking. Int. J. Mol. Sci. 2025, 26, 6356. https://doi.org/10.3390/ijms26136356
Shao D, Tan J, Fan X, Shu Y, Qu Q, Tang Y-Q. Molecular Determinants of TMC Protein Biogenesis and Trafficking. International Journal of Molecular Sciences. 2025; 26(13):6356. https://doi.org/10.3390/ijms26136356
Chicago/Turabian StyleShao, Dedong, Jinru Tan, Xiaozhi Fan, Yilai Shu, Qianhui Qu, and Yi-Quan Tang. 2025. "Molecular Determinants of TMC Protein Biogenesis and Trafficking" International Journal of Molecular Sciences 26, no. 13: 6356. https://doi.org/10.3390/ijms26136356
APA StyleShao, D., Tan, J., Fan, X., Shu, Y., Qu, Q., & Tang, Y.-Q. (2025). Molecular Determinants of TMC Protein Biogenesis and Trafficking. International Journal of Molecular Sciences, 26(13), 6356. https://doi.org/10.3390/ijms26136356