3.1. Materials and General Methods
All chemicals and dry solvents were purchased from ABCR, ACROS Organics, Alfa Aesar, Fisher Scientific, Fluorochem, Fluka, Merck, Sigma Aldrich, TCI, or VWR chemicals, and they were used as received without further purification unless stated otherwise. The addition of solvents and liquid reagents was performed via syringe or cannula. The Injekt syringes used were plastic (Braun) or Teflon (Hamilton) with Sterican needles. For low-temperature reactions, we used water/ice, CO2/acetone baths, or a Cryocool Haake EK90 Immersion Cooler.
All reactions involving non-aqueous solvents were carried out in flame-dried glassware sealed with rubber septa and under argon unless otherwise stated. Reactions were stirred using magnetic stirring bars, monitored by TLC performed on Merck Silica Gel 60 F254 aluminum sheets and visualized with UV (254 nm) fluorescence quenching and p-anisaldehyde stain (p-anisaldehyde (4.2 mL), AcOH (3.75 mL), conc. H2SO4 (12.5 mL) and MeOH (338 mL)). Concentrations under reduced pressure were performed by rotary evaporation at 40 °C at the appropriate pressure using Büchi R-II, R-200, and R-300. Chromatographic purification was performed as flash chromatography using silica gel Silicycle SiliaFlash® P60 230–400 mesh under pressure. Unless otherwise stated, the yields given refer to chromatographically purified and spectroscopically pure compounds.
NMR spectra were recorded in CDCl3 at 298 K on Bruker ARX400 and AVANCE DPX400 spectrometers. All chemical shifts are reported in ppm with the residual solvent peak as the standard (CHCl3, δH = 7.26 ppm and δC = 77.2 ppm; CD3OH, δH = 4.87 ppm and δC = 49.0 ppm). The DEPT-135 pulse sequence was used to aid in the assignment of signals in the 13C NMR spectra. Peaks are reported as follows: δ (multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or unresolved, br = broad signal), coupling constant(s) in Hz, integration and assignment).
MS analyses were recorded on FTMS APEXIII and microTOF-Focus Mass Spectrometers by the mass spectrometry service of the Centro de Apoio Científico e Tecnolóxico á Investigación (CACTI) at the University of Vigo by Dr. Nieves Atanes and Dr. Manuel Marcos.
IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer, equipped with an ATR using a Smart Orbit diamond crystal and/or a Nicolet Continuum Microscope by the Vigo Food Safety Service of CACTI at University of Vigo by Dr. Estefanía López.
Melting point (m.p.) was determined using the Stuart SMP10 melting point apparatus.
HPLC analysis was carried out on a Hewlett Packard 1050 system equipped with a DAD detector (254 nm), using an HPLC Phenomenex column (Luna 5μm Silica(2) 100 Å, 250 × 4.6 mm, isocratic mode; iPrOH/hexane). The analysis was performed by the Vigo Food Safety Service of CACTI at University of Vigo by Dr. Alberto Acuña.
XRD analysis was performed at 100 K using a Bruker D8 Venture diffractometer with a Photon II CMOS detector and Mo-Kα radiation by the Single Crystal X-Ray Diffraction Unit of CACTI at the University of Vigo by Dr. Berta Covelo.
3.2. Experimental Procedures
Synthesis of (8β)-(16β)-(17
E)-Des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]pregn-17(20)-en-16-ol (
4) (
Scheme 10).
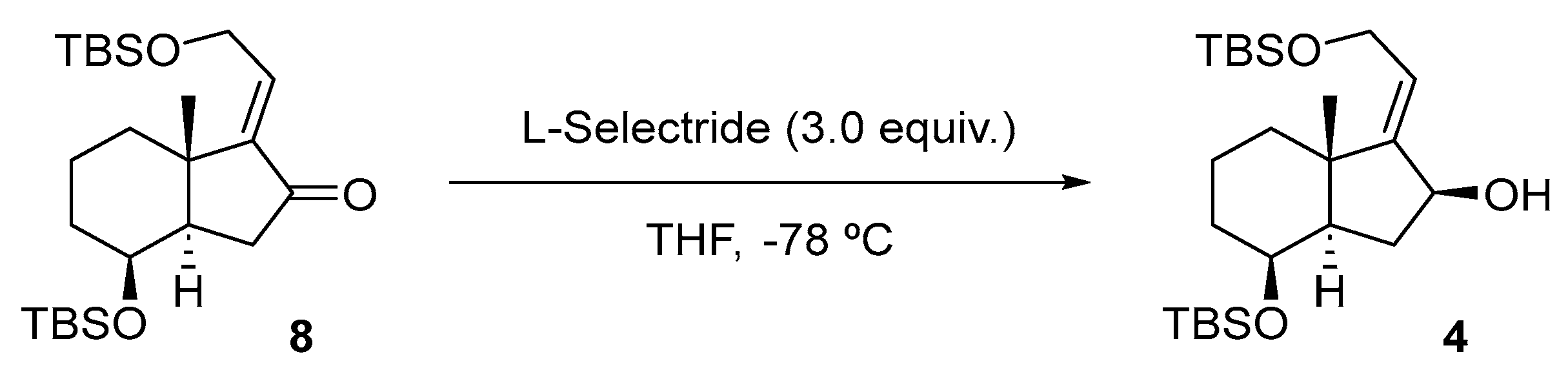
To a solution of 8 (250 mg, 0.57 mmol, 1.0 equiv.) in MeOH (5.0 mL) at rt was added CeCl3·7H2O (339 mg, 0.91 mmol, 1.6 equiv.). The mixture was cooled to −78 °C, NaBH4 (35 mg, 0.91 mmol, 1.6 equiv.) was added, and the reaction was stirred at this temperature for 10 min. Then, the mixture was allowed to reach rt and was left standing for 19 h. The mixture reaction was quenched by the addition of H2O (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 8 mL), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by FCC (SiO2; using 2.5% EtOAc/hexane), affording compound 4 as a colorless oil (150 mg, 60%).
Compound 4:
1H-NMR (400 MHz, CDCl
3): δ 5.47 (td,
J = 6.1, 1.7 Hz, 1H, H-20), 4.39 (ddd,
J = 7.3, 6.7, 1.4 Hz, 2H, CH
2-21), 4.28 (ddd,
J = 13.3, 6.2, 1.6 Hz, 1H, H-16), 4.03 (c,
J = 2.4 Hz, 1H, H-8), 2.05–1.96 (m, 1H, H-14), 1.95–1.81 (m, 2H), 1.77–1.62 (m, 3H), 1.51–1.32 (m, 4H), 1.25 (s, 3H, CH
3-18), 0.90 (s, 9H, CH
3-
tBu), 0.89 (s, 9H, CH
3-
tBu), 0.07 (s, 6H, CH
3-Si), 0.02 (s, 6H, CH
3-Si);
13C-NMR (101 MHz, CDCl
3): δ 155.2 (C-17), 124.4 (CH-20), 74.7 (CH-8), 69.1 (CH-16), 59.6 (CH
2-21), 48.3 (CH-14), 44.0 (C-13), 37.7 (CH
2), 34.3 (CH
2), 34.2 (CH
2), 26.2 (CH
3-
tBu), 26.0 (CH
3-
tBu), 21.2 (CH
3-18), 18.5 (C-
tBu), 18.1 (C-
tBu), 17.8 (CH
2), −4.7 (CH
3-Si), −4.9 (CH
3-Si), −4.9 (CH
3-Si), −5.0 (CH
3-Si);
IR (ATR, cm
−1): ν 3360, 2950, 2929, 2856, 1471, 1254, 1080, 835, 773;
MS (ESI):
m/
z (%) 463.3 ([M + Na]+, 4), 423.3 ([M-OH]+, 100);
HRMS (ESI):
m/
z calculated for C
24H
48NaO
3Si
2 [M + Na]
+ 463.3034, found463.3033;
TLC (SiO
2; 10% EtOAc/hexane): R
f = 0.53. (
Figure 4).
Synthesis of (8β)-(16β)-(17
E)-Des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]pregn-17(20)-en-16-ol (
4) (
Scheme 11).
To a solution of 8 (399 mg, 0.91 mmol, 1.0 equiv.) in anhydrous THF (3.0 mL) at −78 °C was added L-Selectride (1.0 M in THF, 2.7 mL, 2.70 mmol, 3.0 equiv.) dropwise, and the solution was stirred at this temperature for 16 h. The mixture reaction was quenched with sat. aq. NH4Cl solution (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by FCC (SiO2; using 5% EtOAc/hexane), affording the product as a colorless oil (225 mg, 56%), whose physical constants and spectroscopic data virtually coincide with those described for this compound, using reduction with NaBH4.
Synthesis of Phenyl (8β)-(20
S)-Des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]-24-nor-22-thiachol-16-en-23-oate (
5) (
Scheme 12).
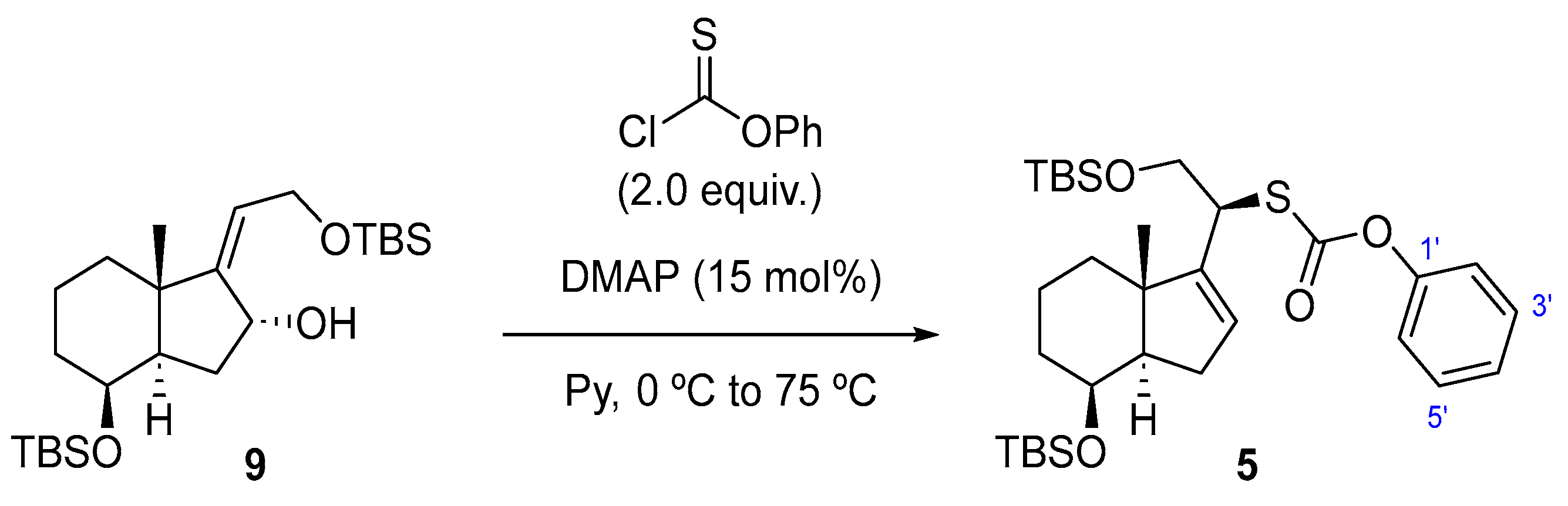
To a solution of 4 (150 mg, 0.34 mmol, 1.0 equiv.) in dry Py (2.2 mL) at rt was added DMAP (6.2 mg, 51 µmol, 15 mol%). The mixture was cooled to 0 °C, O-phenyl chlorothionoformate (95 µL, 0.68 mmol, 2.0 equiv.) was added, and the reaction was stirred at this temperature for 30 min. Then, the mixture was allowed to warm up to rt and was left standing for 16 h. The mixture reaction was quenched with H2O (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were washed with 15% w/v aq. CuSO4 solution (2 × 8 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified (for purification prior to FCC, it is necessary to reduce the co-eluting impurity, O,O-diphenyl carbonothioate, using NaBH4 in MeOH at 0 °C.) by FCC (SiO2; using 0.5% EtOAc/hexane), affording compound 5 as a yellow oil (140 mg, 72%).
Compound 5:
1H-NMR (400 MHz, Methanol-
d4): δ 7.45–7.39 (m, 2H, H-3′, H-5′), 7.30–7.26 (m, 1H, H-4′), 7.16–7.12 (m, 2H, H-2’, H-6’), 5.77–5.73 (m, 1H, H-16), 4.18 (q,
J = 2.6 Hz, 1H, H-8), 4.02–3.87 (m, 3H, H-20, CH
2-21), 2.31 (ddt,
J = 13.3, 11.7, 1.5 Hz, 1H, H-14), 2.04–1.88 (m, 2H), 1.76 (dtd,
J = 11.9, 5.0, 3.6, 1.9 Hz, 3H), 1.66–1.52 (m, 3H), 1.11 (s, 3H, CH
3-18), 0.93 (s, 9H, CH
3-
tBu), 0.92 (s, 9H, CH
3-
tBu), 0.11 (s, 3H, CH
3-Si), 0.10 (s, 3H, CH
3-Si), 0.08 (s, 3H, CH
3-Si), 0.07 (s, 3H, CH
3-Si);
13C-NMR (101 MHz, CDCl
3): δ 170.0 (C=O), 151.4 (C-17), 150.1 (C-1’), 129.6 (CH-3’, CH-5’), 127.1 (CH-4’), 126.2 (CH-16), 121.5 (CH-2’, CH-6’), 68.9 (CH-8), 65.2 (CH
2-21), 54.6 (CH-14/20), 46.9 (C-13), 44.7 (CH-14/20), 35.0 (CH
2), 34.7 (CH
2), 31.3 (CH
2), 26.0 (CH
3-
tBu), 25.9 (CH
3-
tBu), 18.9 (CH
3-18), 18.4 (C-
tBu), 18.1 (C-
tBu), 18.0 (CH
2), −4.7 (CH
3-Si), −5.0 (CH
3-Si), −5.2 (CH
3-Si), −5.3 (CH
3-Si);
IR (ATR, cm
−1): ν 2995, 2952, 2927, 2855, 1770, 1462, 1382, 1374, 1246, 1097, 1057, 927, 853, 837, 776;
MS (ESI):
m/
z (%) 599.3 ([M + Na]
+, 26), 577.3 ([M + H]
+, 100), 445.2 ([M-OTBS]
+, 25);
HRMS (ESI):
m/
z calculated for C
31H
52NaO
4SSi
2 [M + Na]
+ 599.3017, found 599.3017;
TLC (SiO
2; 10% EtOAc/hexane): R
f = 0.86. (
Figure 5).
Synthesis of Phenyl (8β)-(20
S)-Des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]-24-nor-22-thiachol-16-en-23-oate (
5) (
Scheme 13).
To a solution of 9 (820 mg, 1.86 mmol, 1.0 equiv.) in dry Py (10.0 mL) at rt was added DMAP (34 mg, 0.28 mmol, 15 mol%). The mixture was cooled to 0 °C, O-phenyl chlorothionoformate (1.0 mL, 7.44 mmol, 4.0 equiv.) was added, and the reaction was stirred at this temperature for 30 min. Then, the orange mixture was allowed to warm up to 75 °C and was left standing for 6 h. The mixture reaction was quenched with H2O (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 5 mL), and the combined organic extracts were washed with 15% w/v aq. CuSO4 solution (2 × 8 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by FCC (SiO2; using 0.5% EtOAc/hexane), affording the product as a yellow oil (750 mg, 70%), whose physical constants and spectroscopic data virtually coincide with those described for this compound when the reaction was carried out at rt.
Synthesis of Methyl (8β)-(20
S)-des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]-26,27-dinor-22-thiacholest-16-en-25-oate (
6) and (8β)-(20
S)-Des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]-26,27-dinor-22-thiacholest-16-en-25-oic acid (
11) (
Scheme 14).
To a solution of 5 (300 mg, 0.52 mmol, 1.0 equiv.) in 10% w/v KOH/MeOH solution (4.2 mL) at rt was added ethyl 3-bromopropionate (660 µL, 5.19 mmol, 10.0 equiv.), and the resulting mixture was stirred at this temperature for 22 h. No progress was observed in the reaction with time, the mixture reaction was quenched by the addition of H2O (5 mL), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 8 mL), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by FCC (SiO2; using 0.5–20% EtOAc/hexane), affording starting material 5 in the first fractions as a yellow oil (56 mg, 19%), ester 6 in the intermediate fractions as a colorless oil (165 mg, 60%), and carboxylic acid 11 in the last fractions as a colorless oil (23 mg, 8%).
Compound 6:
1H-NMR (400 MHz, CDCl
3): δ 5.48–5.41 (m, 1H, H-16), 4.10–4.04 (m, 1H, H-8), 3.86–3.80 (m, 1H, CH
2-21), 3.79–3.74 (m, 1H, CH
2-21), 3.66 (s, 3H, CH
3-OMe), 3.19 (t,
J = 6.9 Hz, 1H, H-20), 2.94–2.72 (m, 2H, CH
2-23), 2.63–2.52 (m, 2H, CH
2-24), 2.28–2.17 (m, 1H, H-14), 1.97–1.76 (m, 2H), 1.67 (dd,
J = 11.6, 4.3 Hz, 3H), 1.61–1.40 (m, 3H), 1.00 (s, 3H, CH
3-18), 0.86 (s, 18H, CH
3-
tBu), 0.04 (s, 3H, CH
3-Si), 0.03 (s, 3H, CH
3-Si), 0.00 (s, 3H, CH
3-Si), −0.00 (s, 3H, CH
3-Si);
13C-NMR (101 MHz, CDCl
3): δ 172.6 (C=O), 151.4 (C-17), 124.5 (CH-16), 68.9 (CH-8), 67.5 (CH
2-21), 54.5 (CH-14/20), 51.8 (CH
3-OMe), 46.7 (C-13), 43.4 (CH-14/20), 35.1 (CH
2), 35.1 (CH
2), 34.7 (CH
2), 31.1 (CH
2), 26.7 (CH
2), 26.0 (CH
3-
tBu), 25.8 (CH
3-
tBu), 18.9 (CH
3-18), 18.4 (C-
tBu), 18.1 (C-
tBu), 18.0 (CH
2), −4.7 (CH
3-Si), −5.1 (CH
3-Si), −5.2 (CH
3-Si), −5.3 (CH
3-Si);
IR (ATR, cm
−1): ν 2950, 2927, 2885, 2855, 1742, 1471, 1463, 1436, 1360, 1251, 1140, 1110, 1080, 1025, 1005, 972, 926, 833, 772;
MS (ESI):
m/
z (%) 565.3 ([M + Na]
+, 17), 543.3 ([M + H]
+, 63), 411.2 ([M-OTBS]
+, 100);
HRMS (ESI):
m/
z calculated for C
28H
55O
4SSi
2 [M + H]
+ 543.3354, found 543.3364;
TLC (SiO
2; 10% EtOAc/hexane): R
f = 0.58.
Figure 6.
Compound 11:
1H-NMR (400 MHz, CDCl
3): δ 5.52–5.41 (m, 1H, H-16), 4.13–4.07 (m, 1H, H-8), 3.88–3.80 (m, 1H, CH
2-21), 3.84–3.76 (m, 1H, CH
2-21), 3.21 (t,
J = 6.9 Hz, 1H, H-20), 2.98–2.72 (m, 2H, CH
2-23), 2.68–2.54 (m, 2H, CH
2-24), 2.28–2.19 (m, 1H, H-14), 1.98–1.79 (m, 2H), 1.73–1.64 (m, 3H), 1.60 (dd,
J = 12.7, 3.4 Hz, 3H), 1.56–1.41 (m, 2H), 1.01 (s, 3H, CH
3-18), 0.88 (s, 18H, CH
3-
tBu), 0.06 (s, 3H, CH
3-Si), 0.05 (s, 3H, CH
3-Si), 0.02 (s, 3H, CH
3-Si), −0.01 (s, 3H, CH
3-Si);
13C-NMR (101 MHz, CDCl
3): δ 178.4 (C=O), 151.3 (C-17), 124.7 (CH-16), 69.0 (CH-8), 67.6 (CH
2-21), 54.5 (CH-14/20), 46.8 (C-13), 43.6 (CH-14/20), 35.2 (CH
2), 34.7 (CH
2), 31.2 (CH
2), 26.4 (CH
2), 26.0 (CH
3-
tBu), 25.9 (CH
3-
tBu), 19.0 (CH
3-18), 18.5 (C-
tBu), 18.1 (C-
tBu), 18.0 (CH
2), −4.7 (CH
3-Si), −5.1 (CH
3-Si), −5.2 (CH
3-Si), −5.3 (CH
3-Si);
IR (ATR, cm
−1): ν 2956, 2925, 2854, 1713, 1463, 1255, 1111, 1081, 1027, 836;
MS (ESI):
m/
z (%) 591.2 ([M + NaCH
2CN]
+, 100), 551.3 ([M + Na]
+, 9), 397.2 ([M-OTBS]
+, 21);
HRMS (ESI):
m/
z calculated for C
24H
45O
2SSi [M + H]
+ 529.3198, found 529.3197;
TLC (SiO
2; 10% EtOAc/hexane): R
f = 0.18.
Figure 7.
Synthesis of Methyl (8β)-(20
S)-des-
A,
B-8,21-bis[(
tert-butyldimethyl)silyloxy]-26,27-dinor-22-thiacholest-16-en-25-oate (
6) (
Scheme 15).
To a solution of 11 (70 mg, 0.13 mmol, 1.0 equiv.) in dry CH2Cl2 (1.0 mL) at rt was added DMAP (2.4 mg, 19.5 µmol, 15 mol%) and DIC (24 µL, 0.16 mmol, 1.2 equiv.). The solution was stirred for 30 min and then added dry MeOH (38 µL, 0.66 mmol, 5.0 equiv.), and the mixture was left stirring for 28 h. The mixture reaction was diluted with Et2O (5 mL) and washed with sat. aq. NaHCO3 solution (2 × 5 mL) and brine (2 × 5 mL). The organic phase was dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified by FCC (SiO2; using 1% EtOAc/hexane), affording the product as a colorless oil (64 mg, 90%), whose physical constants and spectroscopic data virtually coincide with those already described for this compound.






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
