

Methods for Mitochondrial DNA Damage and Depletion in Immortalized Trabecular Meshwork Cells


Abstract
1. Introduction
2. Results
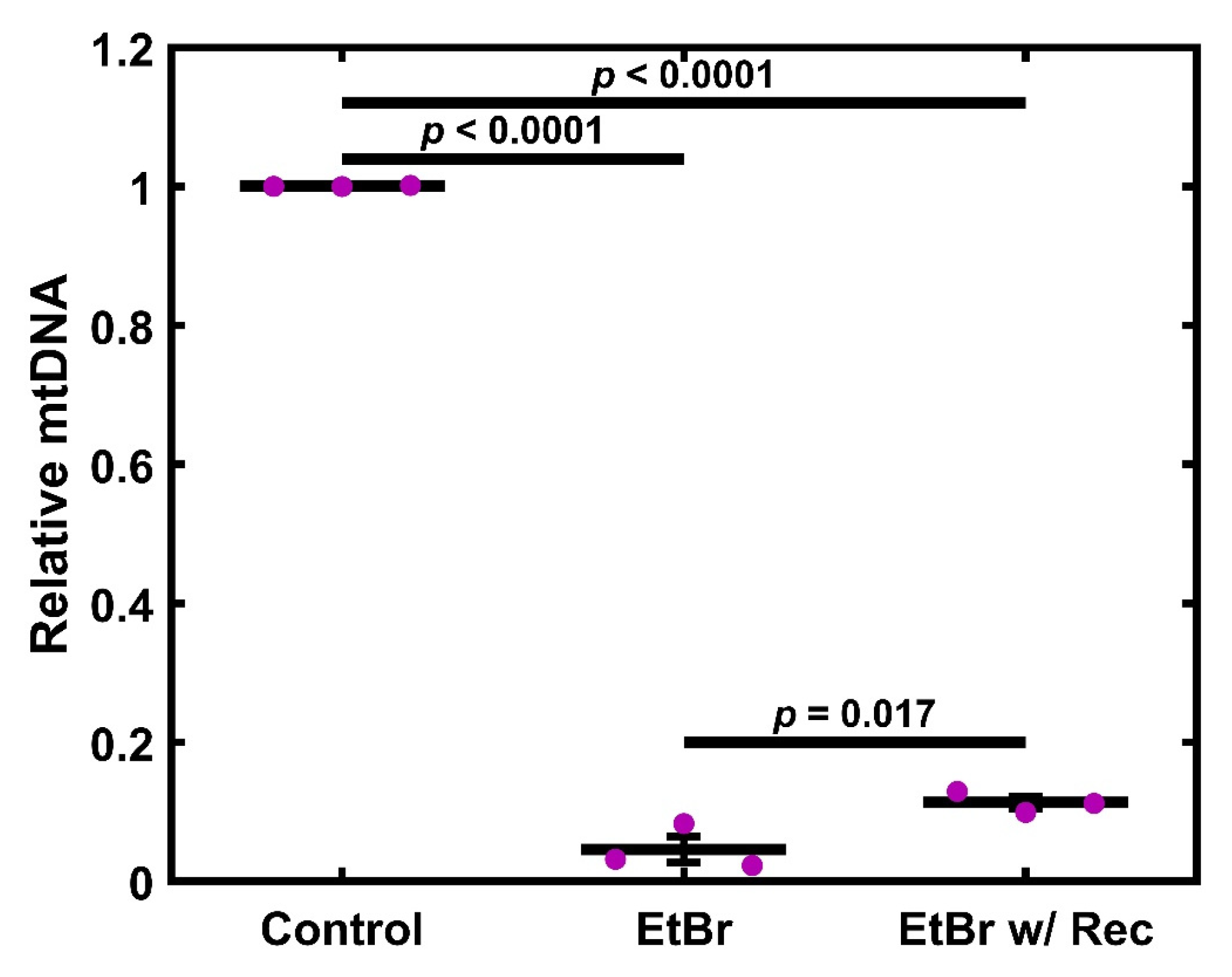
2.1. Ethidium Bromide Treatment Depletes mtDNA
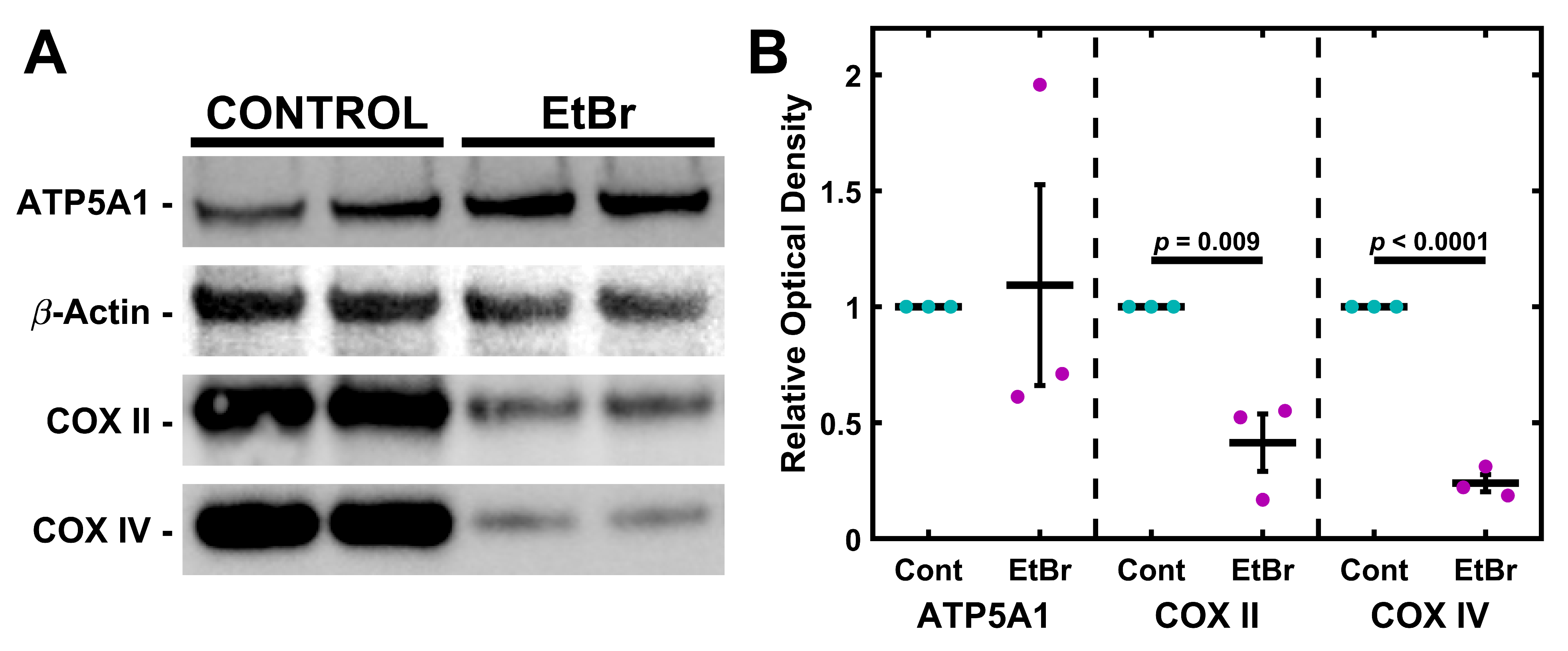
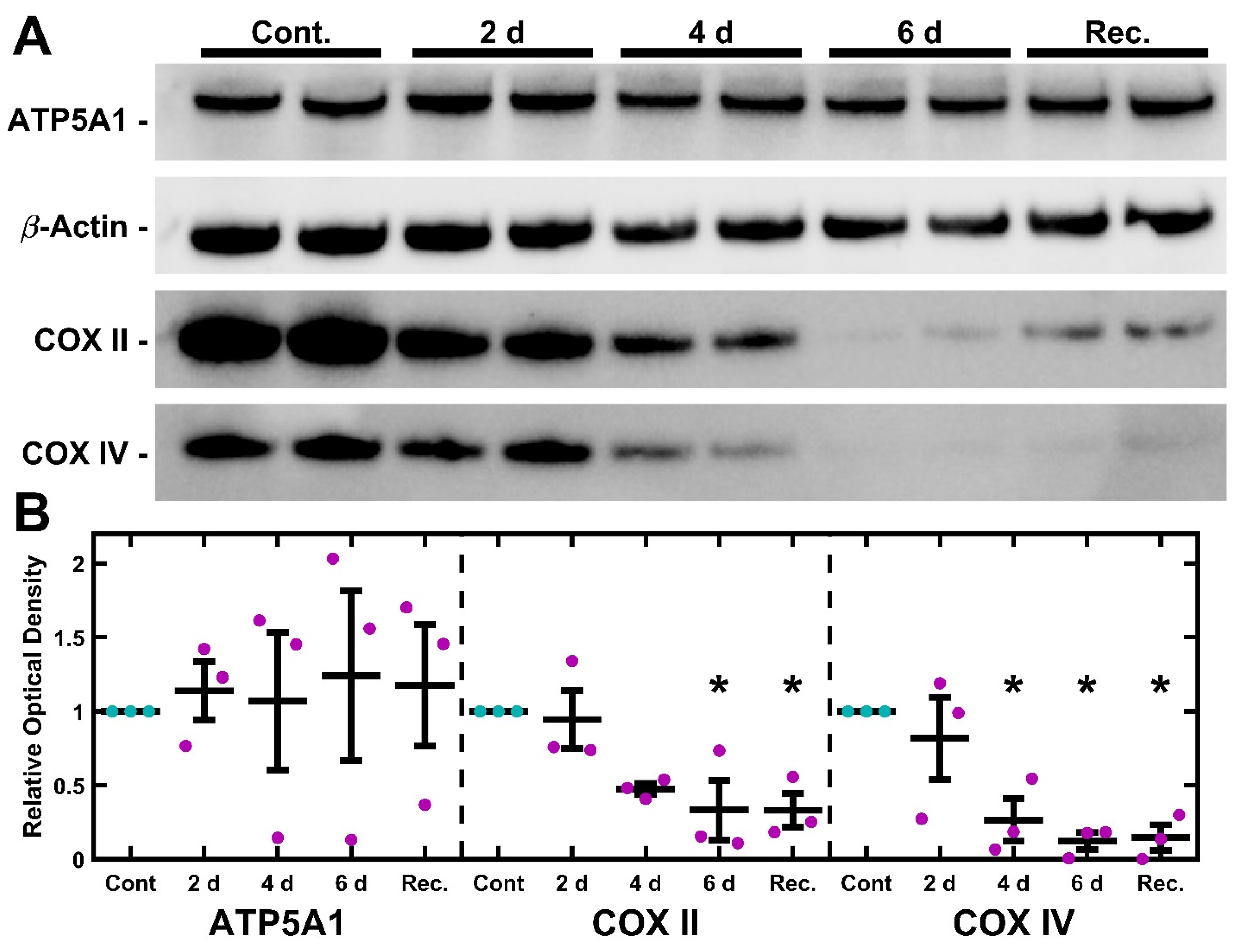
2.2. mtDNA Depletion Is Coupled to Loss of Cytochrome C Oxidase but Not ATP Synthase
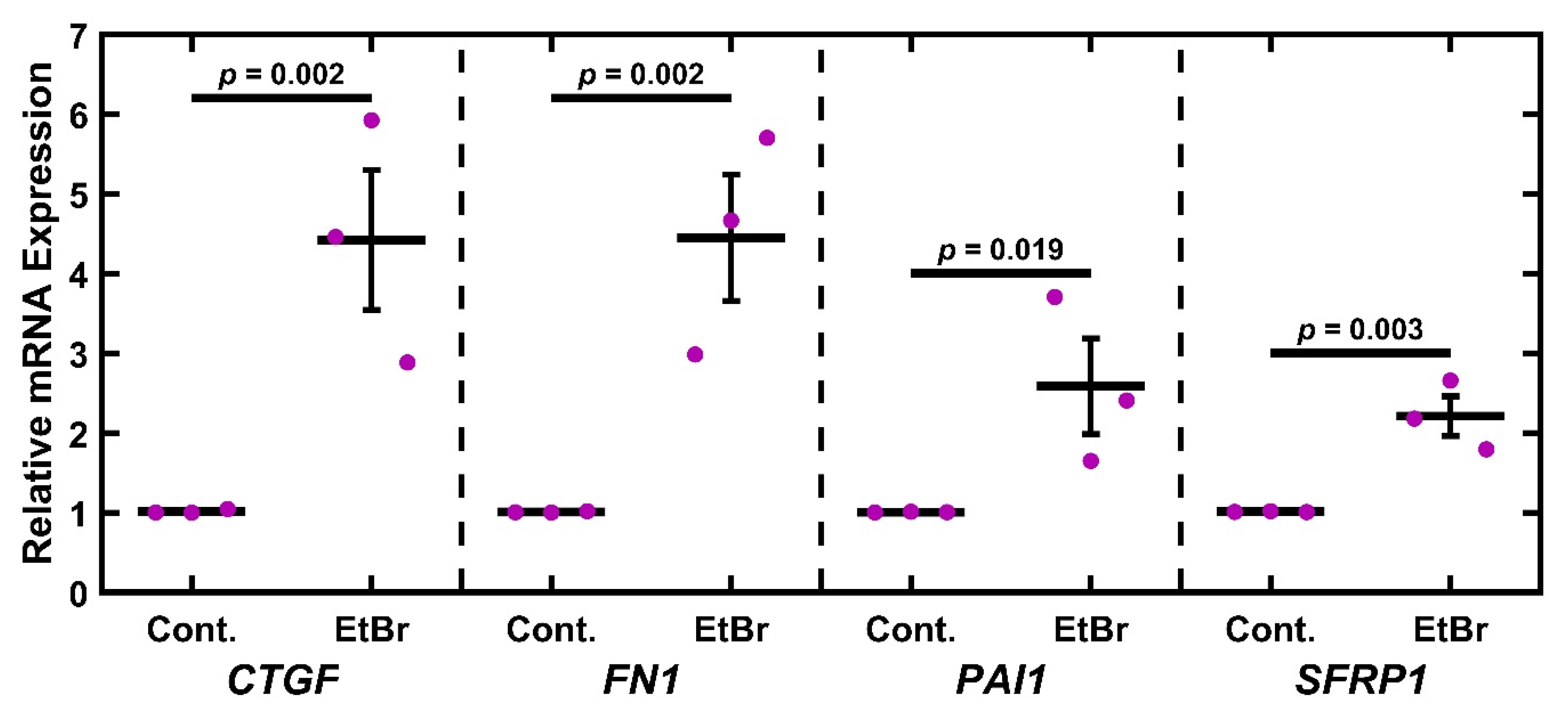
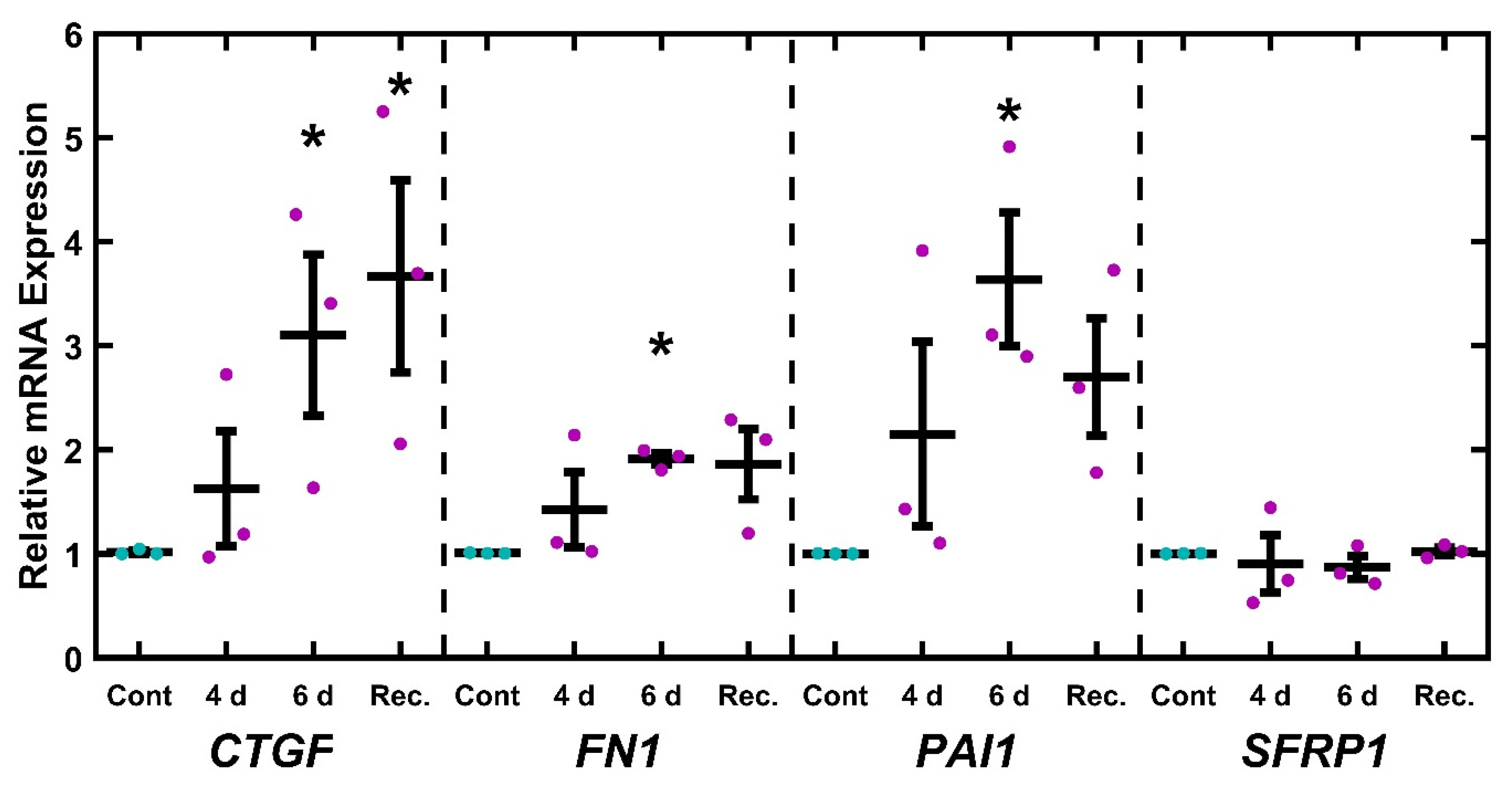
2.3. EtBr Treatment Increases Glaucoma-Associated Gene Expression
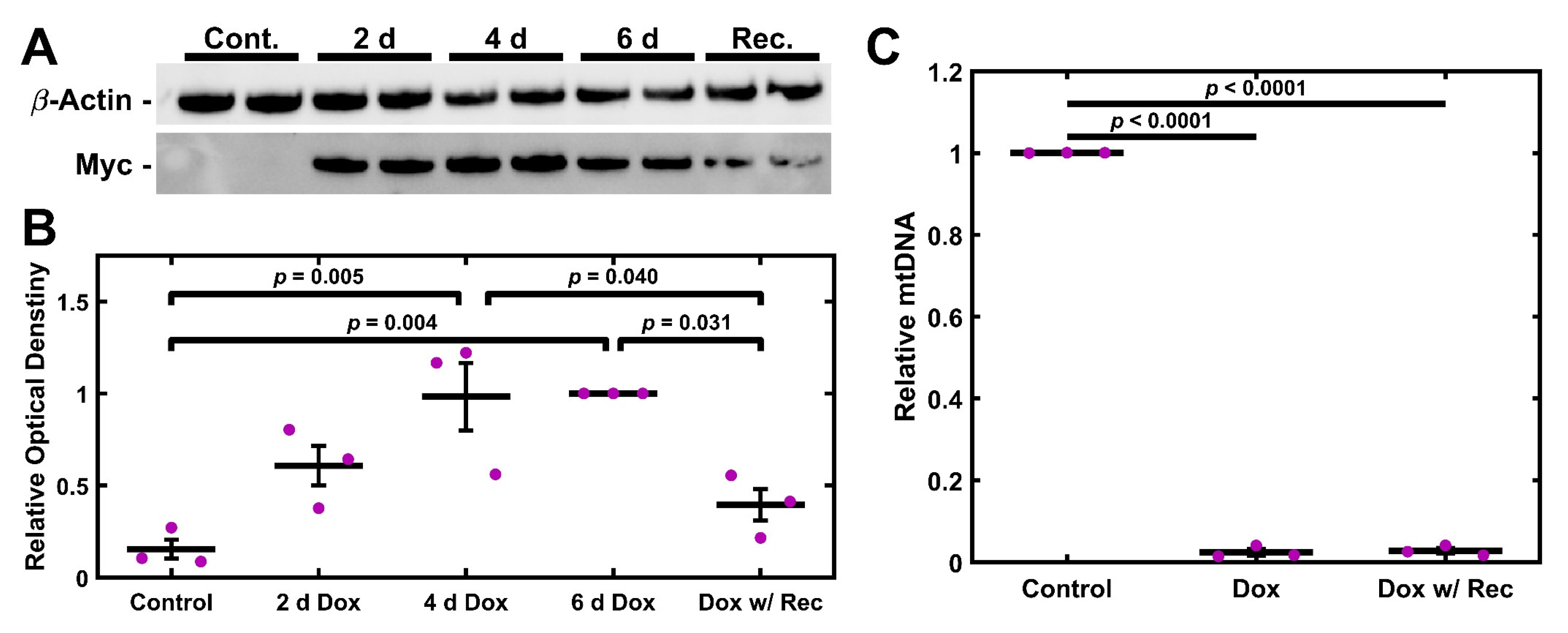
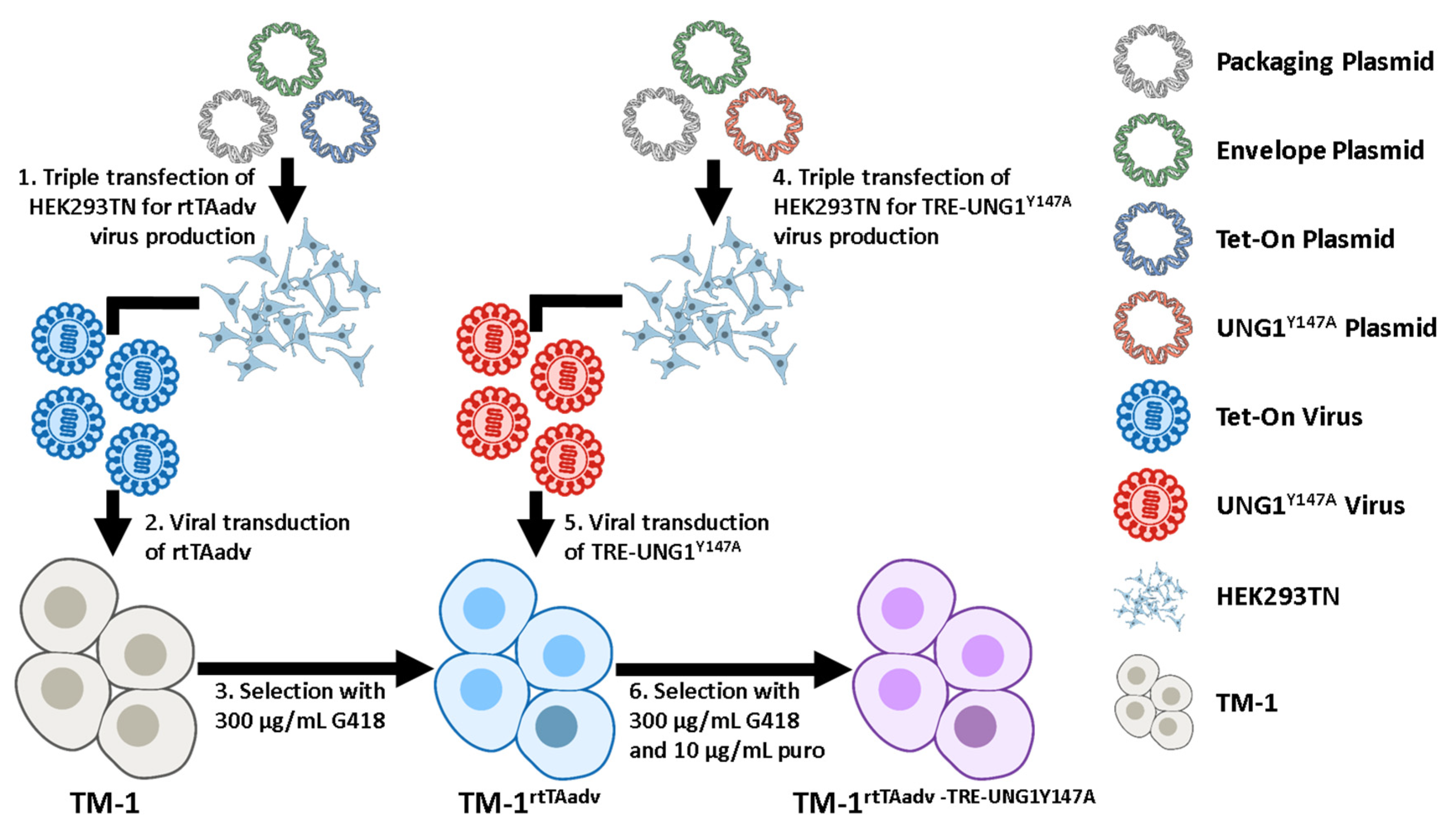
2.4. UNG1Y147A Expression in TM-1 Cells Depletes mtDNA
2.5. UNG1Y147A Meditated mtDNA Depletion Correlates to Loss of Cytochrome C Oxidase but Not ATP Synthase
2.6. UNG1Y147A Alters Glaucoma-Associated Gene Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Transfections and Viral Transduction
4.3. Mitochondrial Depletion Experimental Conditions
4.4. Isolation of DNA and RNA
4.5. Quantitative Polymerase Chain Reaction
4.6. Protein Isolation and Western Blotting
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A Systematic Review and Meta-Analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.V.; Quigley, H.A. Risk Factors and Open-Angle Glaucoma: Classification and Application. J. Glaucoma 2007, 16, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Glaucoma. Lancet 2011, 377, 1367–1377. [Google Scholar] [CrossRef]
- Stamer, W.D.; Acott, T.S. Current Understanding of Conventional Outflow Dysfunction in Glaucoma. Curr. Opin. Ophthalmol. 2012, 23, 135–143. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Izumchenko, E.; Kanherkar, R.R.; Teka, M.; Cantor, C.; Manaye, K.; Sidransky, D.; West, M.D.; Makarev, E.; Csoka, A.B. Pro-Fibrotic Pathway Activation in Trabecular Meshwork and Lamina Cribrosa Is the Main Driving Force of Glaucoma. Cell Cycle 2016, 15, 1643–1652. [Google Scholar] [CrossRef]
- Aktas, Z.; Karaca, E.E.; Gonul, I.I.; Hasanreisoglu, M.; Onol, M. Apoptosis in the Iris and Trabecular Meshwork of Medically Treated and Untreated Primary Open Angle Glaucoma Patients. Int. J. Ophthalmol. 2013, 6, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Yegorov, Y.E. Senescent Phenotype of Trabecular Meshwork Cells Displays Biomarkers in Primary Open-Angle Glaucoma. Curr. Mol. Med. 2011, 11, 528–552. [Google Scholar] [CrossRef]
- Iorga, R.E.; Moraru, A.D.; Costin, D.; Munteanu-Dănulescu, R.S.; Brănișteanu, D.C. Current Trends in Targeting the Oxidative Stress in Glaucoma (Review). Eur. J. Ophthalmol. 2024, 34, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative Stress in the Trabecular Meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef]
- Mehran, N.A.; Sinha, S.; Razeghinejad, R. New Glaucoma Medications: Latanoprostene Bunod, Netarsudil, and Fixed Combination Netarsudil-Latanoprost. Eye 2020, 34, 72–88. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial Damage in the Trabecular Meshwork Occurs Only in Primary Open-Angle Glaucoma and in Pseudoexfoliative Glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Mitochondrial Damage in the Trabecular Meshwork of Patients with Glaucoma. Arch. Ophthalmol. 2010, 128, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Pulliero, A.; Izzotti, A. The Dysfunction of the Trabecular Meshwork during Glaucoma Course. J. Cell Physiol. 2015, 230, 510–525. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial Abnormalities in Patients with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Leung, K.W.; Zhang, Y.-H.; Duan, S.; Zhong, X.-F.; Jiang, R.-Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial Complex I Defect Induces ROS Release and Degeneration in Trabecular Meshwork Cells of POAG Patients: Protection by Antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O’Malley, B.; Spiegelman, B.M. Activation of PPARgamma Coactivator-1 through Transcription Factor Docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef]
- Martínez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The Hitchhiker’s Guide to PGC-1α Isoform Structure and Biological Functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional Integration of Mitochondrial Biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial Biogenesis: Pharmacological Approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear Control of Respiratory Gene Expression in Mammalian Cells. J. Cell Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the Human Mitochondrial Transcription Factor A Gene by Nuclear Respiratory Factors: A Potential Regulatory Link between Nuclear and Mitochondrial Gene Expression in Organelle Biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Hock, M.B.; Kralli, A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spåhr, H.; Bonekamp, N.A.; Kühl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V.; et al. Cross-Strand Binding of TFAM to a Single mtDNA Molecule Forms the Mitochondrial Nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef]
- Ngo, H.B.; Kaiser, J.T.; Chan, D.C. The Mitochondrial Transcription and Packaging Factor Tfam Imposes a U-Turn on Mitochondrial DNA. Nat. Struct. Mol. Biol. 2011, 18, 1290–1296. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct Structural Features of TFAM Drive Mitochondrial DNA Packaging versus Transcriptional Activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef]
- Xu, W.; Boyd, R.M.; Tree, M.O.; Samkari, F.; Zhao, L. Mitochondrial Transcription Factor A Promotes DNA Strand Cleavage at Abasic Sites. Proc. Natl. Acad. Sci. USA 2019, 116, 17792–17799. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The Molecular Mechanism of the Catalase Reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.P.; Chung, M.L.; Anderson, P.J.; Johnson, M.; Epstein, D.L. Hydrogen Peroxide Removal by the Calf Aqueous Outflow Pathway. Investig. Ophthalmol. Vis. Sci. 1988, 29, 976–981. [Google Scholar]
- Gong, B.; Shi, Y.; Qu, C.; Ye, Z.; Yin, Y.; Tan, C.; Shuai, P.; Li, J.; Guo, X.; Cheng, Y.; et al. Association of Catalase Polymorphisms with Primary Open-Angle Glaucoma in a Chinese Population. Ophthalmic Genet. 2018, 39, 35–40. [Google Scholar] [CrossRef]
- Graybeal, K.; Sanchez, L.; Zhang, C.; Stiles, L.; Zheng, J.J. Characterizing the Metabolic Profile of Dexamethasone Treated Human Trabecular Meshwork Cells. Exp. Eye Res. 2022, 214, 108888. [Google Scholar] [CrossRef]
- Watanabe, M.; Sato, T.; Tsugeno, Y.; Umetsu, A.; Suzuki, S.; Furuhashi, M.; Ida, Y.; Hikage, F.; Ohguro, H. Human Trabecular Meshwork (HTM) Cells Treated with TGF-Β2 or Dexamethasone Respond to Compression Stress in Different Manners. Biomedicines 2022, 10, 1338. [Google Scholar] [CrossRef]
- Kennedy, S.; Williams, C.; Tsaturian, E.; Morgan, J.T. Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells. Curr. Issues Mol. Biol. 2024, 46, 9867–9880. [Google Scholar] [CrossRef]
- Nacarelli, T.; Azar, A.; Sell, C. Inhibition of mTOR Prevents ROS Production Initiated by Ethidium Bromide-Induced Mitochondrial DNA Depletion. Front. Endocrinol. 2014, 5, 122. [Google Scholar] [CrossRef]
- Desjardins, P.; Frost, E.; Morais, R. Ethidium Bromide-Induced Loss of Mitochondrial DNA from Primary Chicken Embryo Fibroblasts. Mol. Cell Biol. 1985, 5, 1163–1169. [Google Scholar] [CrossRef]
- Nass, M.M. Differential Effects of Ethidium Bromide on Mitochondrial and Nuclear DNA Synthesis in Vivo in Cultured Mammalian Cells. Exp. Cell Res. 1972, 72, 211–222. [Google Scholar] [CrossRef]
- Zylber, E.; Vesco, C.; Penman, S. Selective Inhibition of the Synthesis of Mitochondria-Associated RNA by Ethidium Bromide. J. Mol. Biol. 1969, 44, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zylber, E.; Penman, S. Mitochondrial-Associated 4 S RNA Synthesis Inhibition by Ethidium Bromide. J. Mol. Biol. 1969, 46, 201–204. [Google Scholar] [CrossRef]
- Nass, M.M. Abnormal DNA Patterns in Animal Mitochondria: Ethidium Bromide-Induced Breakdown of Closed Circular DNA and Conditions Leading to Oligomer Accumulation. Proc. Natl. Acad. Sci. USA 1970, 67, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Khozhukhar, N.; Spadafora, D.; Rodriguez, Y.; Alexeyev, M. Elimination of Mitochondrial DNA from Mammalian Cells. Curr. Protoc. Cell Biol. 2018, 78, 20.11.1–20.11.14. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, R.D. The Effect of Ethidium Bromide on Mitochondrial DNA Synthesis and Mitochondrial DNA Structure in HeLa Cells. J. Cell Biol. 1971, 51, 116–122. [Google Scholar] [CrossRef]
- Kavli, B.; Slupphaug, G.; Mol, C.D.; Arvai, A.S.; Peterson, S.B.; Tainer, J.A.; Krokan, H.E. Excision of Cytosine and Thymine from DNA by Mutants of Human Uracil-DNA Glycosylase. EMBO J. 1996, 15, 3442–3447. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Persistent Damage Induces Mitochondrial DNA Degradation. DNA Repair 2013, 12, 488–499. [Google Scholar] [CrossRef]
- Sharma, R.; Grover, A. Myocilin-Associated Glaucoma: A Historical Perspective and Recent Research Progress. Mol. Vis. 2021, 27, 480–493. [Google Scholar]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.-K.; Zhang, G.; Zhang, B.-T. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Front. Cell Dev. Biol. 2020, 8, 593269. [Google Scholar] [CrossRef]
- Browne, J.G.; Ho, S.L.; Kane, R.; Oliver, N.; Clark, A.F.; O’Brien, C.J.; Crean, J.K. Connective Tissue Growth Factor Is Increased in Pseudoexfoliation Glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3660–3666. [Google Scholar] [CrossRef]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bösl, M.; Bosserhoff, A.; Köstler, J.; Wagner, R.; Tamm, E.R.; et al. Connective Tissue Growth Factor Causes Glaucoma by Modifying the Actin Cytoskeleton of the Trabecular Meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Brodskaya, M.W. Fibronectin Detection in Drainage Outflow System of Human Eyes in Ageing and Progression of Open-Angle Glaucoma. Mech. Ageing Dev. 1989, 47, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-F.; Tane, N.; Roy, S. Fibronectin Overexpression Inhibits Trabecular Meshwork Cell Monolayer Permeability. Mol. Vis. 2004, 10, 750–757. [Google Scholar] [PubMed]
- Bradley, J.M.; Vranka, J.; Colvis, C.M.; Conger, D.M.; Alexander, J.P.; Fisk, A.S.; Samples, J.R.; Acott, T.S. Effect of Matrix Metalloproteinases Activity on Outflow in Perfused Human Organ Culture. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2649–2658. [Google Scholar]
- Yang, Y.-F.; Sun, Y.Y.; Acott, T.S.; Keller, K.E. Effects of Induction and Inhibition of Matrix Cross-Linking on Remodeling of the Aqueous Outflow Resistance by Ocular Trabecular Meshwork Cells. Sci. Rep. 2016, 6, 30505. [Google Scholar] [CrossRef]
- Dan, J.; Belyea, D.; Gertner, G.; Leshem, I.; Lusky, M.; Miskin, R. Plasminogen Activator Inhibitor-1 in the Aqueous Humor of Patients with and without Glaucoma. Arch. Ophthalmol. 2005, 123, 220–224. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Welge-Lussen, U.; Lütjen-Drecoll, E. The Effect of TGF-Beta2 on Human Trabecular Meshwork Extracellular Proteolytic System. Exp. Eye Res. 2003, 77, 757–765. [Google Scholar] [CrossRef]
- Wang, W.-H.; McNatt, L.G.; Pang, I.-H.; Millar, J.C.; Hellberg, P.E.; Hellberg, M.H.; Steely, H.T.; Rubin, J.S.; Fingert, J.H.; Sheffield, V.C.; et al. Increased Expression of the WNT Antagonist sFRP-1 in Glaucoma Elevates Intraocular Pressure. J. Clin. Investig. 2008, 118, 1056–1064. [Google Scholar] [CrossRef]
- Raghunathan, V.K.; Morgan, J.T.; Dreier, B.; Reilly, C.M.; Thomasy, S.M.; Wood, J.A.; Ly, I.; Tuyen, B.C.; Hughbanks, M.; Murphy, C.J.; et al. Role of Substratum Stiffness in Modulating Genes Associated with Extracellular Matrix and Mechanotransducers YAP and TAZ. Investig. Ophthalmol. Vis. Sci. 2013, 54, 378–386. [Google Scholar] [CrossRef]
- Raghunathan, V.K.; Morgan, J.T.; Chang, Y.-R.; Weber, D.; Phinney, B.; Murphy, C.J.; Russell, P. Transforming Growth Factor Beta 3 Modifies Mechanics and Composition of Extracellular Matrix Deposited by Human Trabecular Meshwork Cells. ACS Biomater. Sci. Eng. 2015, 1, 110–118. [Google Scholar] [CrossRef]
- Raghunathan, V.K.; Morgan, J.T.; Park, S.A.; Weber, D.; Phinney, B.S.; Murphy, C.J.; Russell, P. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4447–4459. [Google Scholar] [CrossRef]
- Morgan, J.T.; Raghunathan, V.K.; Chang, Y.-R.; Murphy, C.J.; Russell, P. Wnt Inhibition Induces Persistent Increases in Intrinsic Stiffness of Human Trabecular Meshwork Cells. Exp. Eye Res. 2015, 132, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-M.; Park, J.-H.; Park, I.-H.; Lee, H.-M. Doxycycline Inhibits TGF-Β1-Induced Extracellular Matrix Production in Nasal Polyp-Derived Fibroblasts. Int. Forum Allergy Rhinol. 2016, 6, 256–263. [Google Scholar] [CrossRef]
- Fujita, H.; Sakamoto, N.; Ishimatsu, Y.; Kakugawa, T.; Hara, S.; Hara, A.; Amenomori, M.; Ishimoto, H.; Nagata, T.; Mukae, H.; et al. Effects of Doxycycline on Production of Growth Factors and Matrix Metalloproteinases in Pulmonary Fibrosis. Respiration 2011, 81, 420–430. [Google Scholar] [CrossRef]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between Mitochondria and Other Organelles: A Brand-New Perspective on Mitochondria in Cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear Sensing of Breaks in Mitochondrial DNA Enhances Immune Surveillance. Nature 2021, 591, 477–481. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Wang, P.; Wang, G. Non-Coding RNA Regulated Cross-Talk Between Mitochondria and Other Cellular Compartments. Front. Cell Dev. Biol. 2021, 9, 688523. [Google Scholar] [CrossRef] [PubMed]
- Baleriola, J.; García-Feijoo, J.; Martínez-de-la-Casa, J.M.; Fernández-Cruz, A.; de la Rosa, E.J.; Fernández-Durango, R. Apoptosis in the Trabecular Meshwork of Glaucomatous Patients. Mol. Vis. 2008, 14, 1513–1516. [Google Scholar]
- Alvarado, J.; Murphy, C.; Juster, R. Trabecular Meshwork Cellularity in Primary Open-Angle Glaucoma and Nonglaucomatous Normals. Ophthalmology 1984, 91, 564–579. [Google Scholar] [CrossRef]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular Senescence in the Glaucomatous Outflow Pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as Playmakers of Apoptosis, Autophagy and Senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Ruchko, M.; Gorodnya, O.; LeDoux, S.P.; Alexeyev, M.F.; Al-Mehdi, A.-B.; Gillespie, M.N. Mitochondrial DNA Damage Triggers Mitochondrial Dysfunction and Apoptosis in Oxidant-Challenged Lung Endothelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L530–L535. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef]
- Zhang, L.; Qi, J.; Zhang, X.; Zhao, X.; An, P.; Luo, Y.; Luo, J. The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells. Int. J. Mol. Sci. 2022, 23, 6667. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Frye, A.M.; Phuong, T.T.T.; Yarishkin, O.; Jo, A.O.; Xu, Y.; Lakk, M.; Iuso, A.; Redmon, S.N.; Ambati, B.; et al. TRPV4 Regulates Calcium Homeostasis, Cytoskeletal Remodeling, Conventional Outflow and Intraocular Pressure in the Mammalian Eye. Sci. Rep. 2016, 6, 30583. [Google Scholar] [CrossRef] [PubMed]
- Guarino, B.D.; Paruchuri, S.; Thodeti, C.K. The Role of TRPV4 Channels in Ocular Function and Pathologies. Exp. Eye Res. 2020, 201, 108257. [Google Scholar] [CrossRef]
- Lapajne, L.; Rudzitis, C.N.; Cullimore, B.; Ryskamp, D.; Lakk, M.; Redmon, S.N.; Yarishkin, O.; Krizaj, D. TRPV4: Cell Type-Specific Activation, Regulation and Function in the Vertebrate Eye. Curr. Top. Membr. 2022, 89, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.D.; Chen, Y.-L.; Kasetti, R.B.; Maddineni, P.; Mayhew, W.; Millar, J.C.; Ellis, D.Z.; Sonkusare, S.K.; Zode, G.S. Impaired TRPV4-eNOS Signaling in Trabecular Meshwork Elevates Intraocular Pressure in Glaucoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2022461118. [Google Scholar] [CrossRef]
- Luo, N.; Conwell, M.D.; Chen, X.; Kettenhofen, C.I.; Westlake, C.J.; Cantor, L.B.; Wells, C.D.; Weinreb, R.N.; Corson, T.W.; Spandau, D.F.; et al. Primary Cilia Signaling Mediates Intraocular Pressure Sensation. Proc. Natl. Acad. Sci. USA 2014, 111, 12871–12876. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wm, S.; Md, C.; Cm, R. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and Fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Zaykov, V.; Chaqour, B. The CCN2/CTGF Interactome: An Approach to Understanding the Versatility of CCN2/CTGF Molecular Activities. J. Cell Commun. Signal 2021, 15, 567–580. [Google Scholar] [CrossRef]
- Stecher, V.J.; Kaplan, J.E.; Connolly, K.; Mielens, Z.; Saelens, J.K. Fibronectin in Acute and Chronic Inflammation. Arthritis Rheum. 1986, 29, 394–399. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Bajaj, R.; Wang, L.; Yan, Y.; Ritzenthaler, J.D.; Gewirtz, A.T.; Roman, J.; Merlin, D.; Sitaraman, S.V. Epithelial-Derived Fibronectin Expression, Signaling, and Function in Intestinal Inflammation. J. Biol. Chem. 2007, 282, 32965–32973. [Google Scholar] [CrossRef]
- Brenmoehl, J.; Falk, W.; Göke, M.; Schölmerich, J.; Rogler, G. Inflammation Modulates Fibronectin Isoform Expression in Colonic Lamina Propria Fibroblasts (CLPF). Int. J. Color. Dis. 2008, 23, 947–955. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Lambert, A.J.; Boysen, H.M.; Buckingham, J.A.; Yang, T.; Podlutsky, A.; Austad, S.N.; Kunz, T.H.; Buffenstein, R.; Brand, M.D. Low Rates of Hydrogen Peroxide Production by Isolated Heart Mitochondria Associate with Long Maximum Lifespan in Vertebrate Homeotherms. Aging Cell 2007, 6, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Cree, L.M.; Patel, S.K.; Pyle, A.; Lynn, S.; Turnbull, D.M.; Chinnery, P.F.; Walker, M. Age-Related Decline in Mitochondrial DNA Copy Number in Isolated Human Pancreatic Islets. Diabetologia 2008, 51, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in Skeletal Muscle Mitochondrial Function with Aging in Humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Kaaman, M.; Sparks, L.M.; van Harmelen, V.; Smith, S.R.; Sjölin, E.; Dahlman, I.; Arner, P. Strong Association between Mitochondrial DNA Copy Number and Lipogenesis in Human White Adipose Tissue. Diabetologia 2007, 50, 2526–2533. [Google Scholar] [CrossRef]
- Laderman, K.A.; Penny, J.R.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-Dependent Functional Alterations of Mitochondrial DNA (mtDNA) from Human Fibroblasts Transferred into mtDNA-Less Cells. J. Biol. Chem. 1996, 271, 15891–15897. [Google Scholar] [CrossRef]
- Quan, C.; Cho, M.K.; Perry, D.; Quan, T. Age-Associated Reduction of Cell Spreading Induces Mitochondrial DNA Common Deletion by Oxidative Stress in Human Skin Dermal Fibroblasts: Implication for Human Skin Connective Tissue Aging. J. Biomed. Sci. 2015, 22, 62. [Google Scholar] [CrossRef]
- Solanky, D.; Fields, J.A.; Iudicello, J.E.; Ellis, R.J.; Franklin, D.; Clifford, D.B.; Gelman, B.B.; Marra, C.M.; Morgello, S.; Rubin, L.H.; et al. Higher Buccal Mitochondrial DNA and Mitochondrial Common Deletion Number Are Associated with Markers of Neurodegeneration and Inflammation in Cerebrospinal Fluid. J. Neurovirol 2022, 28, 281–290. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Arnheim, N. Detection of a Specific Mitochondrial DNA Deletion in Tissues of Older Humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef]
- Cooper, J.M.; Mann, V.M.; Schapira, A.H. Analyses of Mitochondrial Respiratory Chain Function and Mitochondrial DNA Deletion in Human Skeletal Muscle: Effect of Ageing. J. Neurol. Sci. 1992, 113, 91–98. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High Levels of Mitochondrial DNA Deletions in Substantia Nigra Neurons in Aging and Parkinson Disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A Pattern of Accumulation of a Somatic Deletion of Mitochondrial DNA in Aging Human Tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; Pang, C.Y.; You, B.J.; Lee, H.C. Tandem Duplications and Large-Scale Deletions of Mitochondrial DNA Are Early Molecular Events of Human Aging Process. Ann. N. Y. Acad. Sci. 1996, 786, 82–101. [Google Scholar] [CrossRef]
- Zhang, C.; Linnane, A.W.; Nagley, P. Occurrence of a Particular Base Substitution (3243 A to G) in Mitochondrial DNA of Tissues of Ageing Humans. Biochem. Biophys. Res. Commun. 1993, 195, 1104–1110. [Google Scholar] [CrossRef]
- Liu, X.; Huang, C.-Y.; Cai, S.; Polansky, J.R.; Kaufman, P.L.; Brandt, C.R. Transformation of Human Trabecular Meshwork Cells with SV40 TAg Alters Promoter Utilization. Curr. Eye Res. 2002, 25, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Filla, M.S.; Liu, X.; Nguyen, T.D.; Polansky, J.R.; Brandt, C.R.; Kaufman, P.L.; Peters, D.M. In Vitro Localization of TIGR/MYOC in Trabecular Meshwork Extracellular Matrix and Binding to Fibronectin. Investig. Ophthalmol. Vis. Sci. 2002, 43, 151–161. [Google Scholar]
- Doucette, L.P.; Footz, T.; Walter, M.A. FOXC1 Regulates Expression of Prostaglandin Receptors Leading to an Attenuated Response to Latanoprost. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2548–2554. [Google Scholar] [CrossRef]
- Peotter, J.L.; Phillips, J.; Tong, T.; Dimeo, K.; Gonzalez, J.M.; Peters, D.M. Involvement of Tiam1, RhoG and ELMO2/ILK in Rac1-Mediated Phagocytosis in Human Trabecular Meshwork Cells. Exp. Cell Res. 2016, 347, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Filla, M.S.; Faralli, J.A.; Desikan, H.; Peotter, J.L.; Wannow, A.C.; Peters, D.M. Activation of Avβ3 Integrin Alters Fibronectin Fibril Formation in Human Trabecular Meshwork Cells in a ROCK-Independent Manner. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3897–3913. [Google Scholar] [CrossRef]
- Bioart. Available online: https://bioart.niaid.nih.gov/bioart/411 (accessed on 8 May 2025).
- Bioart. Available online: https://bioart.niaid.nih.gov/bioart/171 (accessed on 8 May 2025).
- Bioart. Available online: https://bioart.niaid.nih.gov/bioart/198 (accessed on 8 May 2025).
- Bioart. Available online: https://bioart.niaid.nih.gov/bioart/543 (accessed on 8 May 2025).
- Chatzispyrou, I.A.; Held, N.M.; Mouchiroud, L.; Auwerx, J.; Houtkooper, R.H. Tetracycline Antibiotics Impair Mitochondrial Function and Its Experimental Use Confounds Research. Cancer Res. 2015, 75, 4446–4449. [Google Scholar] [CrossRef]
- Clark-Walker, G.D.; Linnane, A.W. In Vivo Differentiation of Yeast Cytoplasmic and Mitochondrial Protein Synthesis with Antibiotics. Biochem. Biophys. Res. Commun. 1966, 25, 8–13. [Google Scholar] [CrossRef]
- Wüst, R.C.I.; Coolen, B.F.; Held, N.M.; Daal, M.R.R.; Alizadeh Tazehkandi, V.; Baks-Te Bulte, L.; Wiersma, M.; Kuster, D.W.D.; Brundel, B.J.J.M.; van Weeghel, M.; et al. The Antibiotic Doxycycline Impairs Cardiac Mitochondrial and Contractile Function. Int. J. Mol. Sci. 2021, 22, 4100. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Musrap, N.; Cretu, D.; Karagiannis, G.S.; Batruch, I.; Smith, C.; Drabovich, A.P.; Trudel, D.; van der Kwast, T.; Morrissey, C.; et al. Proteomic Profiling of Androgen-Independent Prostate Cancer Cell Lines Reveals a Role for Protein S during the Development of High Grade and Castration-Resistant Prostate Cancer. J. Biol. Chem. 2012, 287, 34019–34031. [Google Scholar] [CrossRef] [PubMed]
- Fuchshofer, R.; Yu, A.H.L.; Welge-Lüssen, U.; Tamm, E.R. Bone Morphogenetic Protein-7 Is an Antagonist of Transforming Growth Factor-Beta2 in Human Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 715–726. [Google Scholar] [CrossRef]
- Han, H.; Kampik, D.; Grehn, F.; Schlunck, G. TGF-Β2-Induced Invadosomes in Human Trabecular Meshwork Cells. PLoS ONE 2013, 8, e70595. [Google Scholar] [CrossRef]
- Luna, C.; Parker, M.; Challa, P.; Gonzalez, P. Long-Term Decrease of Intraocular Pressure in Rats by Viral Delivery of miR-146a. Transl. Vis. Sci. Technol. 2021, 10, 14. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.G.; Patton, J.T.; Anders, R.A.; Yolken, R.H.; Schwarz, K.B. Rotavirus Infects Human Biliary Epithelial Cells and Stimulates Secretion of Cytokines IL-6 and IL-8 via MAPK Pathway. Biomed. Res. Int. 2015, 2015, 697238. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson’s Disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence | Reference |
|---|---|---|---|
| TBP | TGTATCCACAGTGAATCTTGGTTG | GGTTCGTGGCTCTCTTATCCTC | [116] |
| CTGF | CACAAGGGCCTCTTCTGTGA | TCTCTTCCAGGTCAGCTTCG | [117] |
| FN1 | AATCCAAGCGGAGAGAGTCA | CATCCTCAGGGCTCGAGTAG | [118] |
| PAI1 | AATGTGTCATTTCCGGCTGCTGTG | ACATCCATCTTTGTGCCCTACCCT | [119] |
| SFRP1 | CTCAACAAGAACTGCCACGC | CTCGTTGTCACAGGGAGGAC | Primer-Blast [120] |
| GAPDH | ACAGTCAGCCGCATCTTCTT | ACGACCAAATCCGTTGACTC | [121] |
| PGC1A | AAACAGCAGCAGAGACAAATGC | TTGGTTTGGCTTGTAAGTGTTGTG | [122] |
| TFAM | TGTTCACAATGGATAGGCAC | TCTGGGTTTTCCAAAGCAAG | [122] |
| SOD2 | CTGGACAAACCTCAGCCCT | CTGATTTGGACAAGCAGCAA | [122] |
| CATA | TGGAAAGAAGACTCCCATCG | CCAGAAGTCCCAGACCATGT | [122] |
| BGLOB | GGTGAGTCTATGGGACGCTT | GATCCTGAGACTTCCACACTGA | Primer-Blast [120] |
| TMDQ | CCATCTTTGCAGGCACACTCATC | ATCCACCTCAACTGCCTGCTATG | [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, S.P.; Tsaturian, E.; Zhao, L.; Morgan, J.T. Methods for Mitochondrial DNA Damage and Depletion in Immortalized Trabecular Meshwork Cells. Int. J. Mol. Sci. 2025, 26, 6255. https://doi.org/10.3390/ijms26136255
Kennedy SP, Tsaturian E, Zhao L, Morgan JT. Methods for Mitochondrial DNA Damage and Depletion in Immortalized Trabecular Meshwork Cells. International Journal of Molecular Sciences. 2025; 26(13):6255. https://doi.org/10.3390/ijms26136255
Chicago/Turabian StyleKennedy, Shane P., Emily Tsaturian, Linlin Zhao, and Joshua T. Morgan. 2025. "Methods for Mitochondrial DNA Damage and Depletion in Immortalized Trabecular Meshwork Cells" International Journal of Molecular Sciences 26, no. 13: 6255. https://doi.org/10.3390/ijms26136255
APA StyleKennedy, S. P., Tsaturian, E., Zhao, L., & Morgan, J. T. (2025). Methods for Mitochondrial DNA Damage and Depletion in Immortalized Trabecular Meshwork Cells. International Journal of Molecular Sciences, 26(13), 6255. https://doi.org/10.3390/ijms26136255