
Exploring the Role of Peripheral Macrophages in Glioma Progression: The Metabolic Significance of Cyclooxygenase-2 (COX-2)

 ,
,  , , , and
, , , and
Abstract

1. Introduction
2. Results
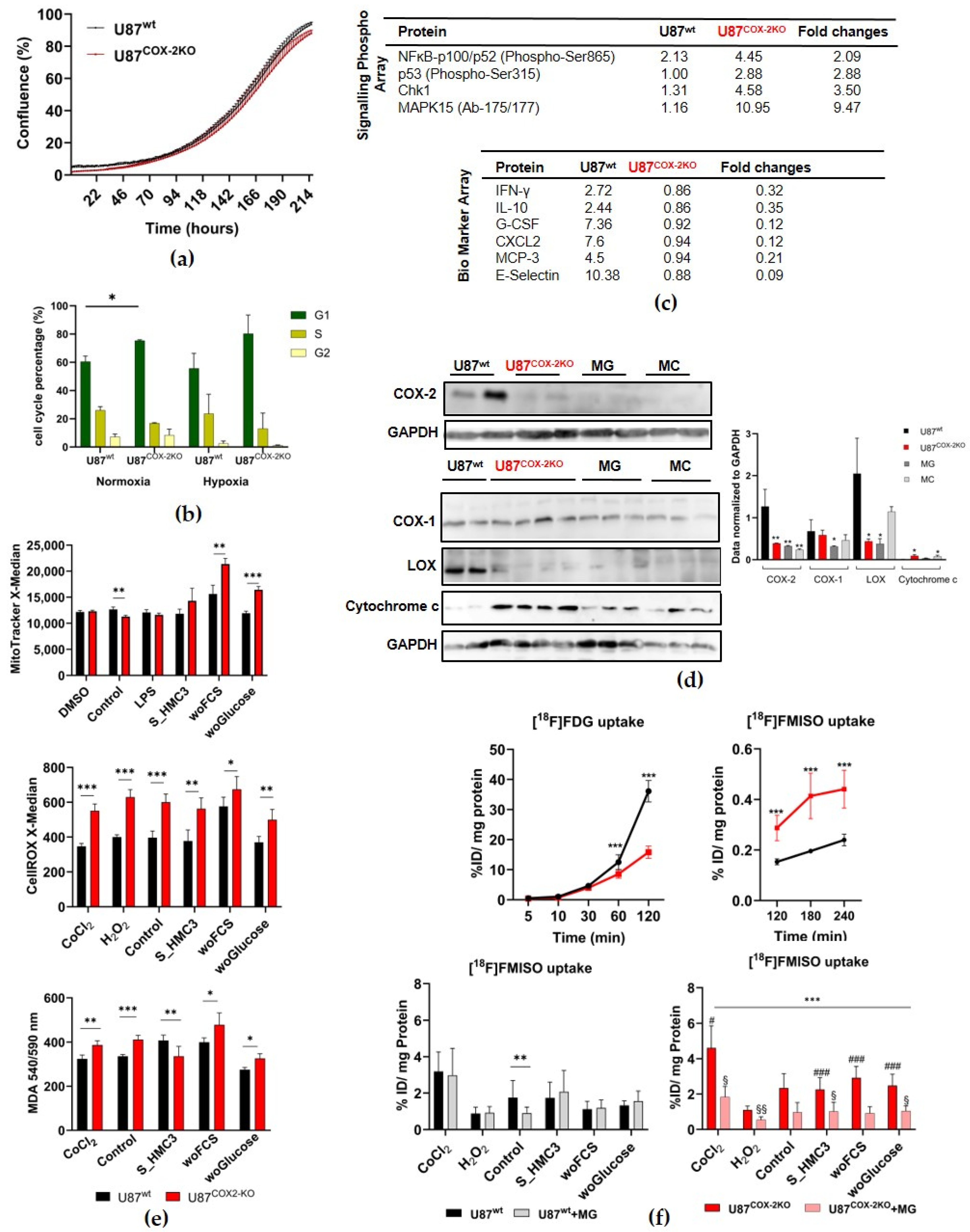
2.1. In Vitro Experiments in Adherent Cell Culture
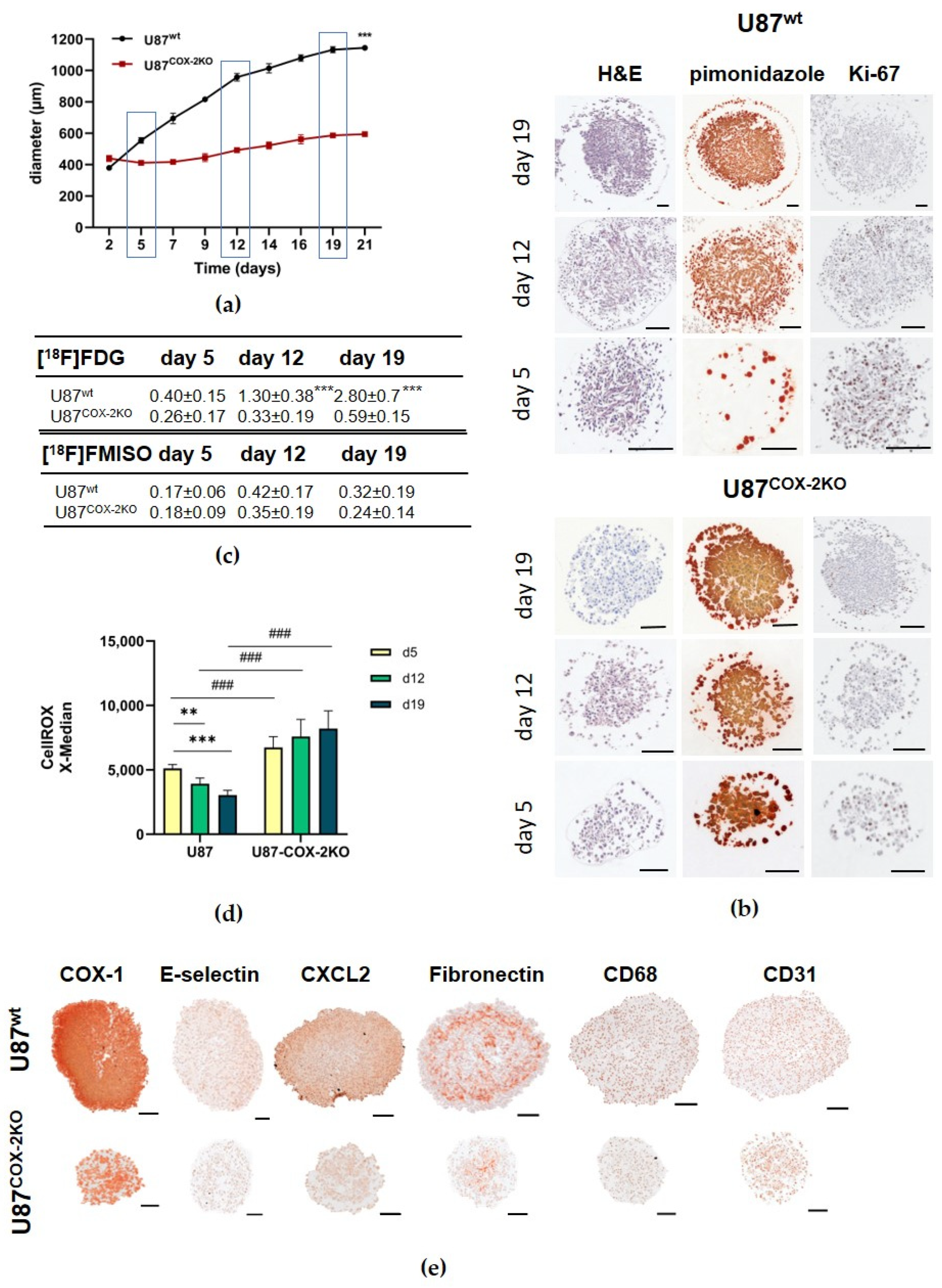
2.2. In Vitro Experiments in Spheroids
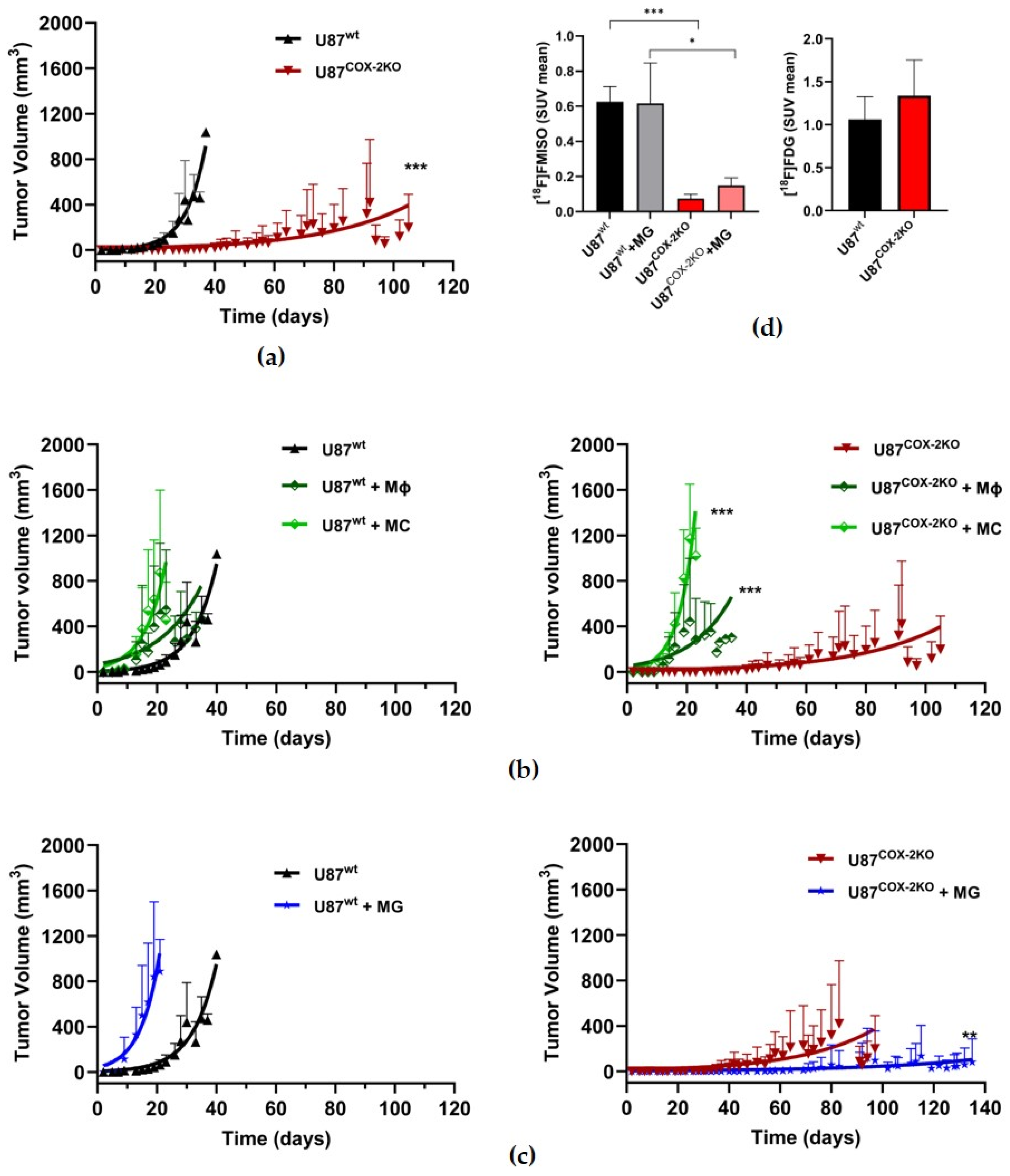
2.3. In Vivo Effects of COX-2 Knockout in Subcutaneous Xenograft Models
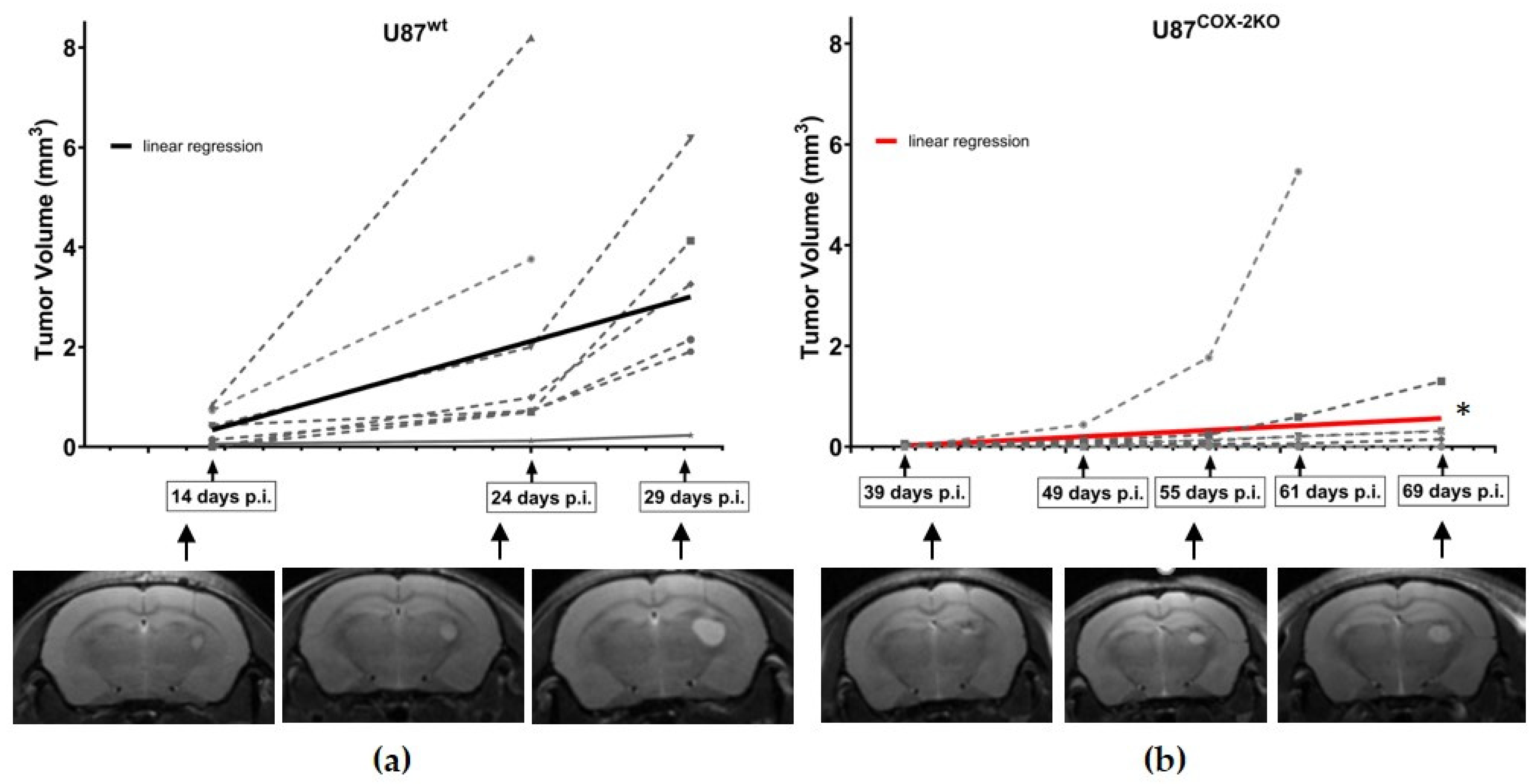
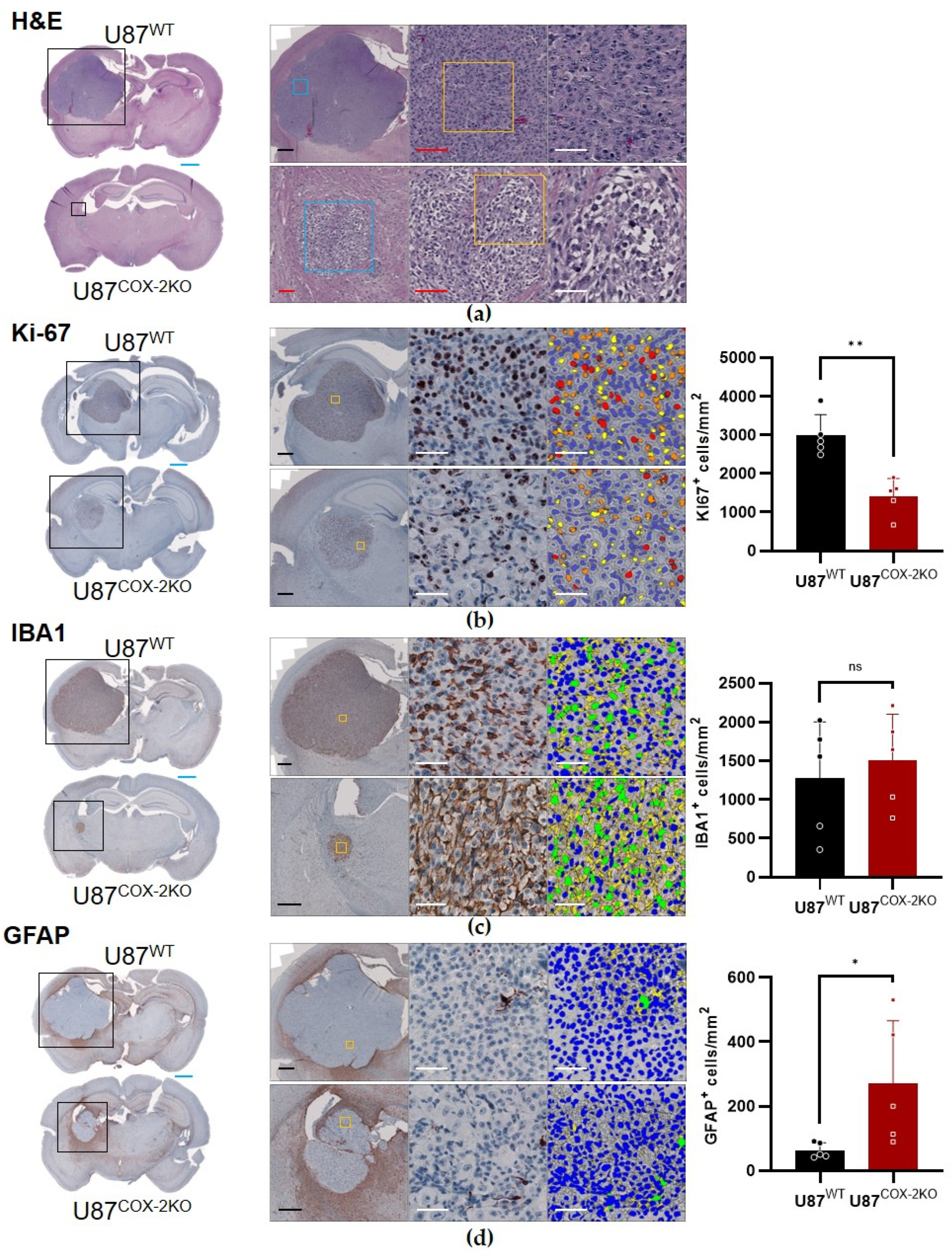
2.4. In Vivo Effects of COX-2 Knockout in Orthotopic Xenograft Models
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions
4.2. Knockdown of COX-2 with CRISPR/Cas9
4.3. Antibody Array
4.4. Growth Rate Analysis
4.5. RFP Control and Cell Cycle Analysis by FACS
4.6. Western Blot Analysis
4.7. Histological Analysis of U87wt and U87COX−2KO Spheroids
4.8. Spheroid Histology and Immunohistochemistry
4.9. Oxidative Stress and Metabolic Assay
4.10. Radiotracer Uptake Assay
4.11. Subcutaneous Glioblastoma Xenografts
4.12. Small Animal PET/CT Imaging of Subcutaneous Glioblastoma Xenografts
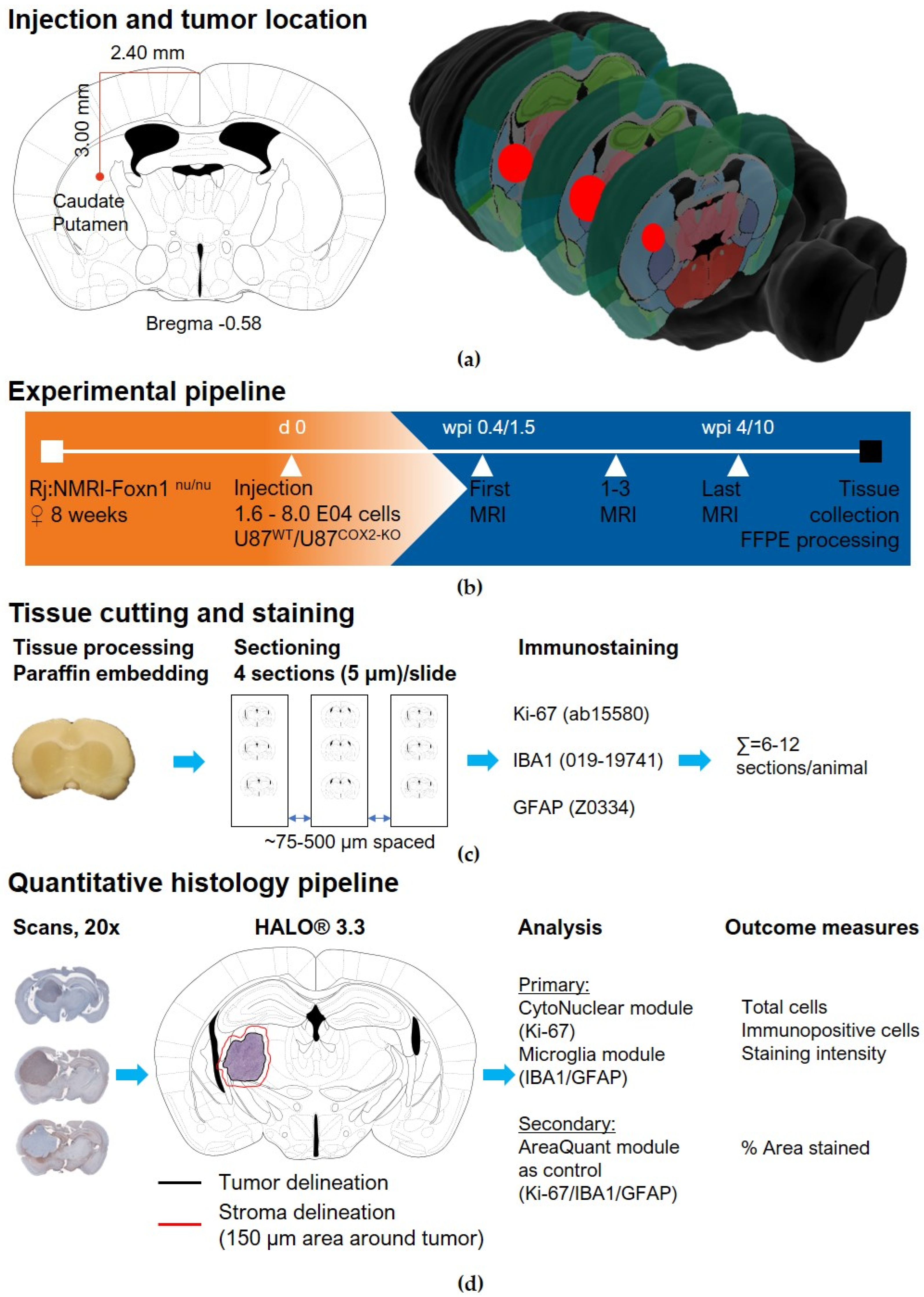
4.13. Orthotopic Glioblastoma Xenograft Tumor Model
4.14. MRI Imaging
4.15. Immunohistochemistry of U87wt and U87COX−2KO Brain Tumors
4.16. Data Acquisition and Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitusova, K.; Peltek, O.O.; Karpov, T.E.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Overcoming the Blood-Brain Barrier for the Therapy of Malignant Brain Tumor: Current Status and Prospects of Drug Delivery Approaches. J. Nanobiotechnol. 2022, 20, 412. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and Microglia: The Cerberus of Glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Chen, H.C.; Chang, W.C.; Chuang, J.Y.; Chang, K.Y.; Liou, J.P.; Hsu, T.I. The Complex Role of Eicosanoids in the Brain: Implications for Brain Tumor Development and Therapeutic Opportunities. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188957. [Google Scholar] [CrossRef]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in Glioblastoma Multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef]
- Ferreira, M.T.; Miyake, J.A.; Gomes, R.N.; Feitoza, F.; Stevannato, P.B.; da Cunha, A.S.; Serachi, F.O.; Panagopoulos, A.T.; Colquhoun, A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 4297. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ayroldi, E.; Giusti, I.; Dolo, V.; Cifone, M.G.; Cinque, B.; Palumbo, P. Cyclooxygenase-2 Upregulated by Temozolomide in Glioblastoma Cells Is Shuttled In Extracellular Vesicles Modifying Recipient Cell Phenotype. Front. Oncol. 2022, 12, 933746. [Google Scholar] [CrossRef]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in Glioblastoma Are Hypoxic, Express Extracellular Matrix Proteases, and Are Formed by an Actively Migrating Cell Population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the Hypoxia-Inducible-Factor Pathway in Glioma Growth and Angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, P.; Ji, C. Cell Death Conversion under Hypoxic Condition in Tumor Development and Therapy. Int. J. Mol. Sci. 2015, 16, 25536–25551. [Google Scholar] [CrossRef]
- Rodrigues, P.; Bangali, H.; Hammoud, A.; Mustafa, Y.F.; Al-Hetty, H.R.A.K.; Alkhafaji, A.T.; Deorari, M.M.; Al-Taee, M.M.; Zabibah, R.S.; Alsalamy, A. COX 2-Inhibitors; a Thorough and Updated Survey into Combinational Therapies in Cancers. Med. Oncol. 2024, 41, 41. [Google Scholar] [CrossRef]
- Haase-Kohn, C.; Wolf, S.; Herwig, N.; Mosch, B.; Pietzsch, J. Metastatic Potential of B16-F10 Melanoma Cells Is Enhanced by Extracellular S100A4 Derived from RAW264.7 Macrophages. Biochem. Biophys. Res. Commun. 2014, 446, 143–148. [Google Scholar] [CrossRef]
- Dean, P.T.; Hooks, S.B. Pleiotropic Effects of the COX-2/PGE2 Axis in the Glioblastoma Tumor Microenvironment. Front. Oncol. 2022, 12, 1116014. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ciafarone, A.; Topi, S.; Cifone, M.G.; Cinque, B.; Palumbo, P. Involvement of Cyclooxygenase-2 in Establishing an Immunosuppressive Microenvironment in Tumorspheres Derived from TMZ-Resistant Glioblastoma Cell Lines and Primary Cultures. Cells 2024, 13, 258. [Google Scholar] [CrossRef]
- Knudsen, A.M.; Ewald, J.D.; Pedersen, V.; Haupt-Jorgensen, M.; Hansen, E.V.R.; Kristensen, B.W. Characterisation of the tumour microenvironment in primary and recurrent glioblastomas. Neuropathol Appl Neurobiol. 2024, 50, 5. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Frederico, S.C.; Zhang, X.; Hu, B.; Kohanbash, G. Pre-Clinical Models for Evaluating Glioma Targeted Immunotherapies. Front. Immunol. 2022, 13, 1092399. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, T.H.; Chen, C.P.; Xiang, J.J.; Zhao, X.-B.; Gui, R.-Y.; Liao, X.-H. Targeting COX-2 Potently Inhibits Proliferation of Cancer Cells in Vivo but Not in Vitro in Cutaneous Squamous Cell Carcinoma. Transl. Cancer Res. 2021, 10, 2219–2228. [Google Scholar] [CrossRef]
- Lv, X.; Chen, Z.; Li, S.; Xie, H. Knockdown of Cyclooxygenase-2 Leads to Growth Inhibition and Cell Cycle Arrest in Hepatocellular Carcinoma Cells. Onco Targets Ther. 2019, 12, 4341–4349. [Google Scholar] [CrossRef]
- Haase-Kohn, C.; Laube, M.; Donat, C.K.; Belter, B.; Pietzsch, J. CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells 2022, 11, 749. [Google Scholar] [CrossRef]
- Kniess, T.; Laube, M.; Bergmann, R.; Sehn, F.; Graf, F.; Steinbach, J.; Wuest, F.; Pietzsch, J. Radiosynthesis of a ¹⁸F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Bio. Med. Chem. 2012, 11, 3410–3421. [Google Scholar] [CrossRef]
- Ullrich, M.; Richter, S.; Seifert, V.; Hauser, S.; Calsina, B.; Martínez-Montes, A.M.; Ter Laak, M.; Ziegler, C.; Timmers, H.; Eisenhofer, G.; et al. Targeting Cyclooxygenase-2 in Pheochromocytoma and Paraganglioma: Focus on Genetic Background. Cancer 2019, 11, 743. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Gugu, M.K.; Cifone, M.G.; Cinque, B.; Palumbo, P. Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. Int. J. Mol. Sci. 2022, 23, 1545. [Google Scholar] [CrossRef]
- Palumbo, P.; Lombardi, F.; Augello, F.R.; Giusti, I.; Dolo, V.; Leocata, P.; Cifone, M.G.; Cinque, B. Biological Effects of Selective COX-2 Inhibitor NS398 on Human Glioblastoma Cell Lines. Cancer Cell Int. 2020, 20, 167. [Google Scholar] [CrossRef]
- Yu, T.; Lao, X.; Zheng, H. Influencing COX-2 Activity by COX Related Pathways in Inflammation and Cancer. Mini Rev. Med. Chem. 2016, 16, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Nakanishi, H. Phosphatidylserine-Containing Liposomes: Potential Pharmacological Interventions against Inflammatory and Immune Diseases through the Production of Prostaglandin E(2) after Uptake by Myeloid Derived Phagocytes. Arch. Immunol. Et Ther. Exp. 2011, 59, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, A.; Paczek, L.; Burdzinska, A. The Role of COX-2 and PGE2 in the Regulation of Immunomodulation and Other Functions of Mesenchymal Stromal Cells. Biomedicines 2023, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Pavlacky, J.; Polak, J. Technical Feasibility and Physiological Relevance of Hypoxic Cell Culture Models. Front. Endocrinol. 2020, 11, 57. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef]
- Rana, R.; Huirem, R.S.; Kant, R.; Chauhan, K.; Sharma, S.; Yashavarddhan, M.H.; Chhabra, S.S.; Acharya, R.; Kalra, S.K.; Gupta, A.; et al. Cytochrome C as a Potential Clinical Marker for Diagnosis and Treatment of Glioma. Front. Oncol. 2022, 12, 960787. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-Specific Regulation of Cytochrome c by Post-Translational Modifications: Respiration, the Mitochondrial Membrane Potential, ROS, and Apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c Release from Mitochondria Proceeds by a Two-Step Process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Colecchia, D.; Strambi, A.; Sanzone, S.; Iavarone, C.; Rossi, M.; Dall’Armi, C.; Piccioni, F.; Verrotti di Pianella, A.; Chiariello, M. MAPK15/ERK8 Stimulates Autophagy by Interacting with LC3 and GABARAP Proteins. Autophagy 2012, 8, 1724–1740. [Google Scholar] [CrossRef]
- Dutta, S.; Mahalanobish, S.; Saha, S.; Mandal, M.; Begam, S.; Sadhukhan, P.; Ghosh, S.; Brahmachari, G.; Sil, P.C. Biological Evaluation of the Novel 3,3′-((4-Nitrophenyl)Methylene)Bis(4-Hydroxy-2H-Chromen-2-One) Derivative as Potential Anticancer Agents via the Selective Induction of Reactive Oxygen Species-Mediated Apoptosis. Cell Signal 2023, 111, 110876. [Google Scholar] [CrossRef]
- Kern, M.A.; Haugg, A.M.; Koch, A.F.; Schilling, T.; Breuhahn, K.; Walczak, H.; Fleischer, B.; Trautwein, C.; Michalski, C.; Schulze-Bergkamen, H.; et al. Cyclooxygenase-2 Inhibition Induces Apoptosis Signaling via Death Receptors and Mitochondria in Hepatocellular Carcinoma. Cancer Res. 2006, 66, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.; Olesch, C.; Hanaka, H.; Rådmark, O.; Weigert, A.; Brüne, B. Necrosis in DU145 Prostate Cancer Spheroids Induces COX-2/mPGES-1-Derived PGE2 to Promote Tumor Growth and to Inhibit T Cell Activation. Int. J. Cancer 2013, 133, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhong, K.; Wang, Z.; Zhang, Z.; Tang, X.; Tong, A.; Zhou, L. Tumor-Associated Microglia and Macrophages in Glioblastoma: From Basic Insights to Therapeutic Opportunities. Front. Immunol. 2022, 13, 964898. [Google Scholar] [CrossRef]
- Acker, G.; Zollfrank, J.; Jelgersma, C.; Nieminen-Kelha, M.; Kremenetskaia, I.; Mueller, S.; Ghori, A.; Vajkoczy, P.; Brandenburg, S. The CXCR2/CXCL2 Signalling Pathway—An Alternative Therapeutic Approach in High-Grade Glioma. Eur. J. Cancer 2020, 126, 106–115. [Google Scholar] [CrossRef]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident Microglia Rather than Peripheral Macrophages Promote Vascularization in Brain Tumors and Are Source of Alternative Pro-Angiogenic Factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Ercolano, G.; De Cicco, P.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Carriero, R.; Kunderfranco, P.; Ianaro, A. Knockdown of PTGS2 by CRISPR/CAS9 System Designates a New Potential Gene Target for Melanoma Treatment. Front. Pharmacol. 2019, 10, 1456. [Google Scholar] [CrossRef]
- Leite, D.M.; Zvar Baskovic, B.; Civita, P.; Neto, C.; Gumbleton, M.; Pilkington, G.J. A Human Co-Culture Cell Model Incorporating Microglia Supports Glioblastoma Growth and Migration, and Confers Resistance to Cytotoxics. FASEB J. 2020, 34, 1710–1727. [Google Scholar] [CrossRef]
- Kolliopoulos, C.; Ali, M.M.; Castillejo-Lopez, C.; Heldin, C.H.; Heldin, P. CD44 Depletion in Glioblastoma Cells Suppresses Growth and Stemness and Induces Senescence. Cancers 2022, 14, 3747. [Google Scholar] [CrossRef]
- Dugandzija, T.; Drljaca, J.; Bulajic, D.; Isakovic, A.; Stilinovic, N.; Sekulic, S.; Capo, I. Hallmarks of Tumor-Associated Microglia Response to Experimental U87 Human Glioblastoma Xenograft. Tissue Cell 2021, 72, 101557. [Google Scholar] [CrossRef]
- Simone, L.; Capobianco, D.L.; Di Palma, F.; Binda, E.; Legnani, F.G.; Vescovi, A.L.; Svelto, M.; Pisani, F. GFAP Serves as a Structural Element of Tunneling Nanotubes between Glioblastoma Cells and Could Play a Role in the Intercellular Transfer of Mitochondria. Front. Cell Dev. Biol. 2023, 11, 1221671. [Google Scholar] [CrossRef]
- Tang, G.; Yue, Z.; Talloczy, Z.; Hagemann, T.; Cho, W.; Messing, A.; Sulzer, D.L.; Goldman, J.E. Autophagy Induced by Alexander Disease-Mutant GFAP Accumulation Is Regulated by P38/MAPK and mTOR Signaling Pathways. Hum. Mol. Genet. 2008, 17, 1540–1555. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Huang, B.; Qiu, S.; Li, X.; He, L.; Peng, Y. Tumor-Associated Macrophages Induce Vasculogenic Mimicry of Glioblastoma Multiforme through Cyclooxygenase-2 Activation. Oncotarget 2016, 7, 83976–83986. [Google Scholar] [CrossRef] [PubMed]
- Tjiu, J.W.; Chen, J.S.; Shun, C.T.; Lin, S.J.; Liao, Y.H.; Chu, C.Y.; Tsai, T.F.; Chiu, H.C.; Dai, Y.S.; Inoue, H.; et al. Tumor-Associated Macrophage-Induced Invasion and Angiogenesis of Human Basal Cell Carcinoma Cells by Cyclooxygenase-2 Induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef]
- Zheng, T.; Chen, K.; Zhang, X.; Feng, H.; Shi, Y.; Liu, L.; Zhang, J.; Chen, Y. Knockdown of TXNDC9 Induces Apoptosis and Autophagy in Glioma and Mediates Cell Differentiation by P53 Activation. Aging 2020, 12, 18649–18659. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Erö, C.; Gewaltig, M.O.; Keller, D.; Markram, H. A Cell Atlas for the Mouse Brain. Front. Neuroinform. 2019, 28, 13. [Google Scholar] [CrossRef]
- Rodarie, D.; Verasztó, C.; Roussel, J.; Reimann, M.; Keller, D.; Ramaswamy, S.; Markram, H.; Gewaltig, M.O. A method to estimate the cellular composition of the mouse brain from heterogeneous datasets. PLoS Comput. Biol. 2022, 18, 12. [Google Scholar] [CrossRef]
- Roussel, Y.; Verasztó, C.; Rodarie, D.; Damart, T.; Reimann, M.; Ramaswamy, S.; Markram, H.; Keller, D. Mapping of morpho-electric features to molecular identity of cortical inhibitory neurons. PLoS Comput. Biol. 2023, 19, e1010058. [Google Scholar] [CrossRef]
- Rabaan, A.A.; AlSaihati, H.; Bukhamsin, R.; Bakhrebah, M.A.; Nassar, M.S.; Alsaleh, A.A.; Alhashem, Y.N.; Bukhamseen, A.Y.; Al-Ruhimy, K.; Alotaibi, M.; et al. Application of CRISPR/Cas9 Technology in Cancer Treatment: A Future Direction. Curr. Oncol. 2023, 30, 1954–1976. [Google Scholar] [CrossRef]
- Mosch, B.; Mueller, K.; Steinbach, J.; Pietzsch, J. Influence of irradiation on metabolism and metastatic potential of B16-F10 melanoma cells. Int. J. Radiat. Biol. 2009, 85, 1002–1012. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, Q.; Li, W.; Lu, Y.; Ni, Y.; Ma, L.; Fu, Y. SOX5 induces lung adenocarcinoma angiogenesis by inducing the expression of VEGF through STAT3 signaling. Onco Targets Ther. 2018, 11, 5733–5741. [Google Scholar] [CrossRef]
- Rix, J.; Uckermann, O.; Kirsche, K.; Schackert, G.; Koch, E.; Kirsch, M.; Galli, R. Correlation of biomechanics and cancer cell phenotype by combined Brillouin and Raman spectroscopy of U87-MG glioblastoma cells. J. R. Soc. Interface 2022, 19, 209. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Mean ± SD | Median | |
|---|---|---|---|
| U87wt | 44 | 2.0 ± 1.0 | 1.8 |
| U87wt + MC | 18 | 2.3 ± 1.1 | 1.7 |
| U87wt + Mϕ | 23 | 2.1 ± 1.1 | 1.8 |
| U87wt + MG | 13 | 3.1 ± 0.4 | 3.0 |
| U87COX-2KO | 38 | 0.9 ± 0.4 | 1.0 |
| U87COX-2KO+MC | 18 | 2.0 ± 0.7 | 1.8 |
| U87COX-2KO +Mϕ | 23 | 1.8 ± 0.7 | 1.8 |
| U87COX-2KO + MG | 23 | 0.4 ± 0.6 | 0.0 |
| Parameters | p Value |
|---|---|
| U87wt vs U87COX-2KO | <0.001 |
| U87wt vs U87wt + MC | 0.57 |
| U87wt vs U87wt + Mϕ | 0.98 |
| U87wt vs U87wt + MG | 0.3 |
| U87COX-2KO vs U87COX-2KO + MC | <0.001 |
| U87COX-2KO vs U87COX-2KO + Mϕ | <0.001 |
| U87COX-2KO vs U87COX-2KO + MG | <0.002 |
| U87wt + MC vs U87COX-2KO + MC | 0.78 |
| U87wt + Mϕ vs U87COX-2KO + Mϕ | 0.67 |
| U87wt + MG vs U87COX-2KO + MG | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietzsch, J.; Toussaint, M.; Donat, C.K.; Doctor, A.; Meister, S.; Wodtke, J.; Laube, M.; Hofheinz, F.; Rix, J.; Deuther-Conrad, W.; et al. Exploring the Role of Peripheral Macrophages in Glioma Progression: The Metabolic Significance of Cyclooxygenase-2 (COX-2). Int. J. Mol. Sci. 2025, 26, 6198. https://doi.org/10.3390/ijms26136198
Pietzsch J, Toussaint M, Donat CK, Doctor A, Meister S, Wodtke J, Laube M, Hofheinz F, Rix J, Deuther-Conrad W, et al. Exploring the Role of Peripheral Macrophages in Glioma Progression: The Metabolic Significance of Cyclooxygenase-2 (COX-2). International Journal of Molecular Sciences. 2025; 26(13):6198. https://doi.org/10.3390/ijms26136198
Chicago/Turabian StylePietzsch, Jens, Magali Toussaint, Cornelius Kurt Donat, Alina Doctor, Sebastian Meister, Johanna Wodtke, Markus Laube, Frank Hofheinz, Jan Rix, Winnie Deuther-Conrad, and et al. 2025. "Exploring the Role of Peripheral Macrophages in Glioma Progression: The Metabolic Significance of Cyclooxygenase-2 (COX-2)" International Journal of Molecular Sciences 26, no. 13: 6198. https://doi.org/10.3390/ijms26136198
APA StylePietzsch, J., Toussaint, M., Donat, C. K., Doctor, A., Meister, S., Wodtke, J., Laube, M., Hofheinz, F., Rix, J., Deuther-Conrad, W., & Haase-Kohn, C. (2025). Exploring the Role of Peripheral Macrophages in Glioma Progression: The Metabolic Significance of Cyclooxygenase-2 (COX-2). International Journal of Molecular Sciences, 26(13), 6198. https://doi.org/10.3390/ijms26136198