
Identification of Potential Therapeutic Targets for Coronary Atherosclerosis from an Inflammatory Perspective Through Integrated Proteomics and Single-Cell Omics
Abstract
1. Introduction
2. Results
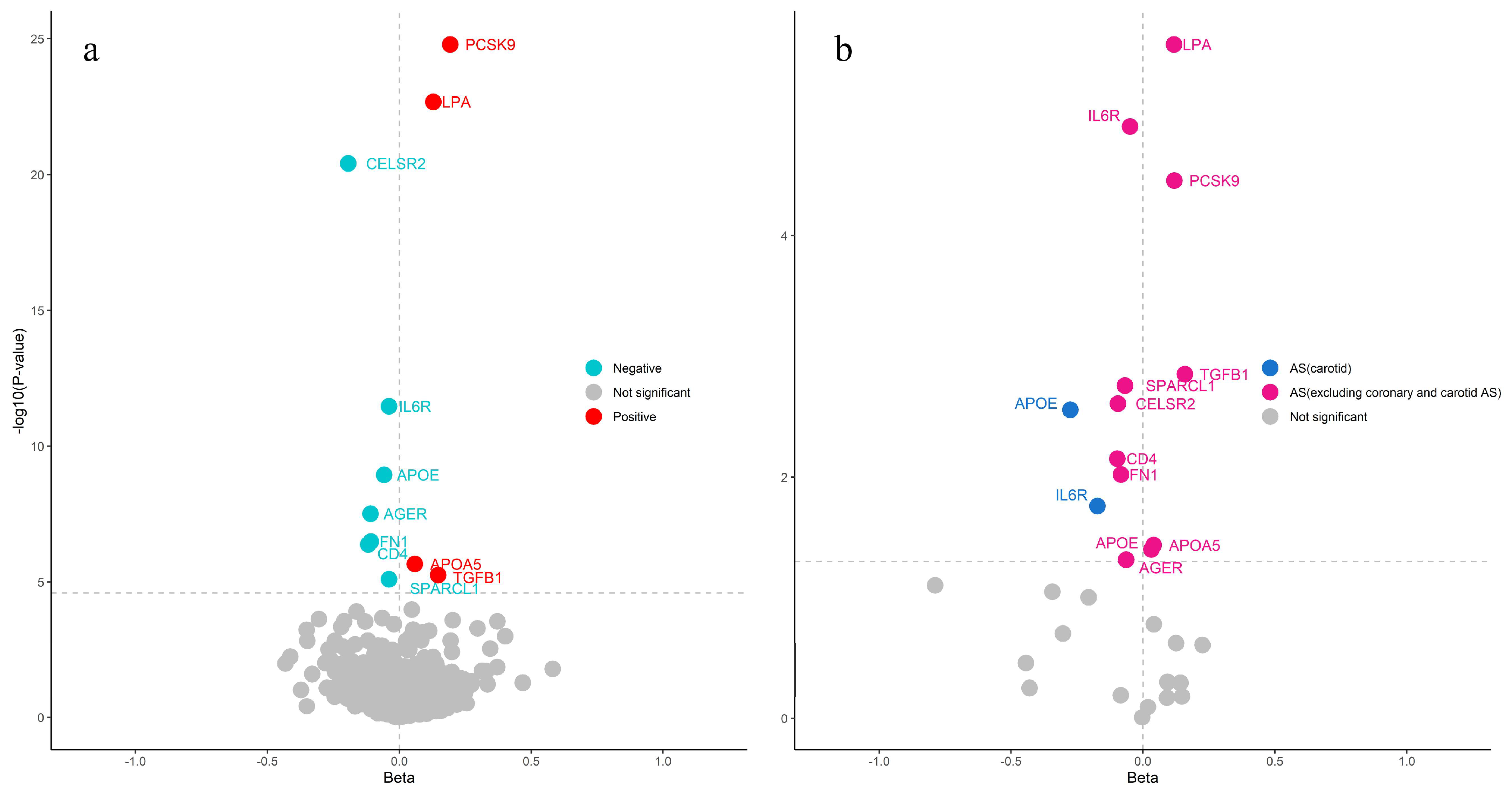
2.1. Proteome Screening for Causal Proteins in CAS
2.2. Causal Proteins for Cardiovascular Disease
2.3. Bayesian Colocalization and Reverse MR for the Causal Relationship Between Causal Proteins and CAS
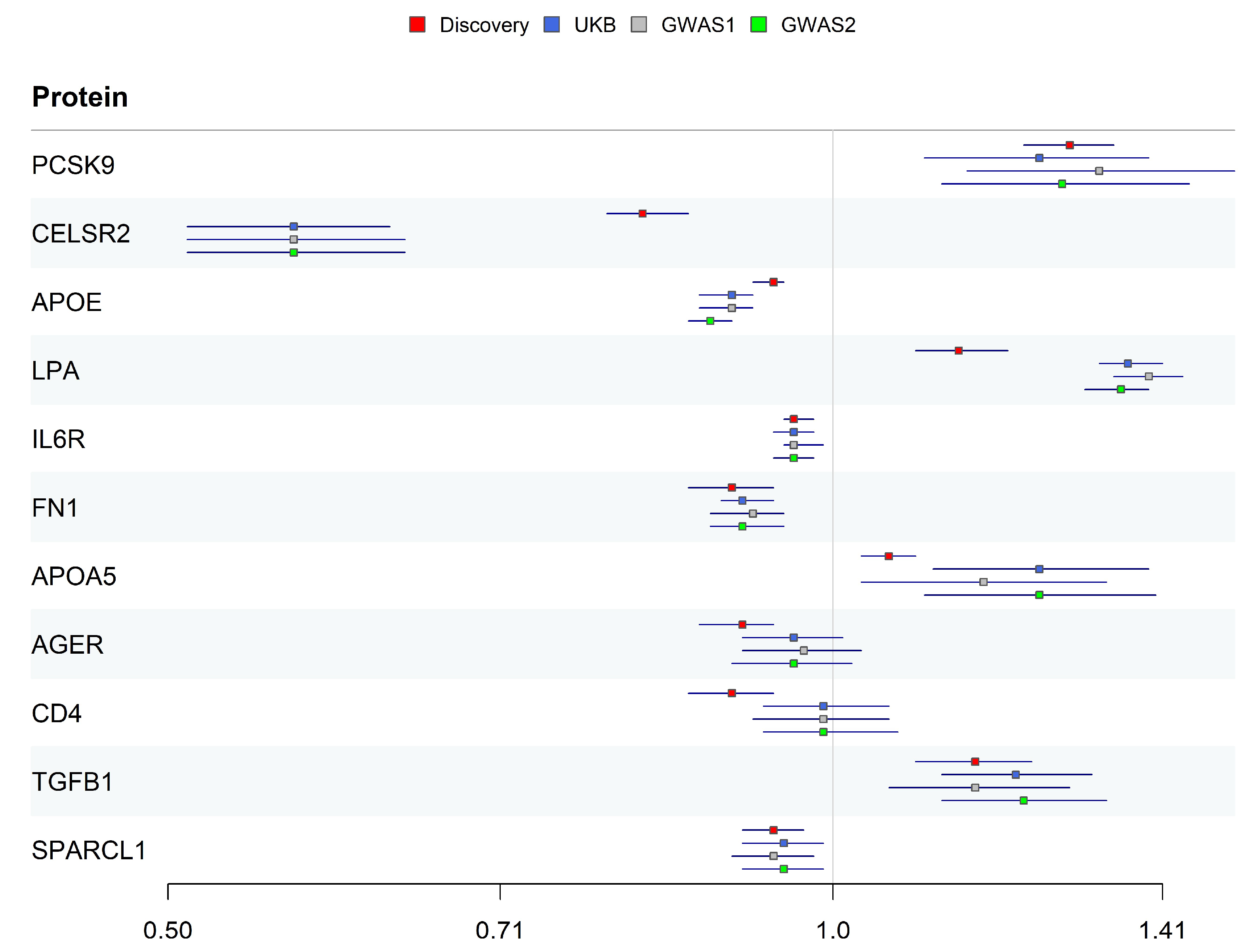
2.4. Replication Validation for Identified Proteins
2.5. Single-Cell RNA-Seq Differential Expression Analysis for Coronary Plaque Samples and Healthy Vasculature
2.6. Protein–Protein Interaction (PPI) Network, Enrichment Analysis, and Drug Repurposing
3. Discussion
4. Materials and Methods
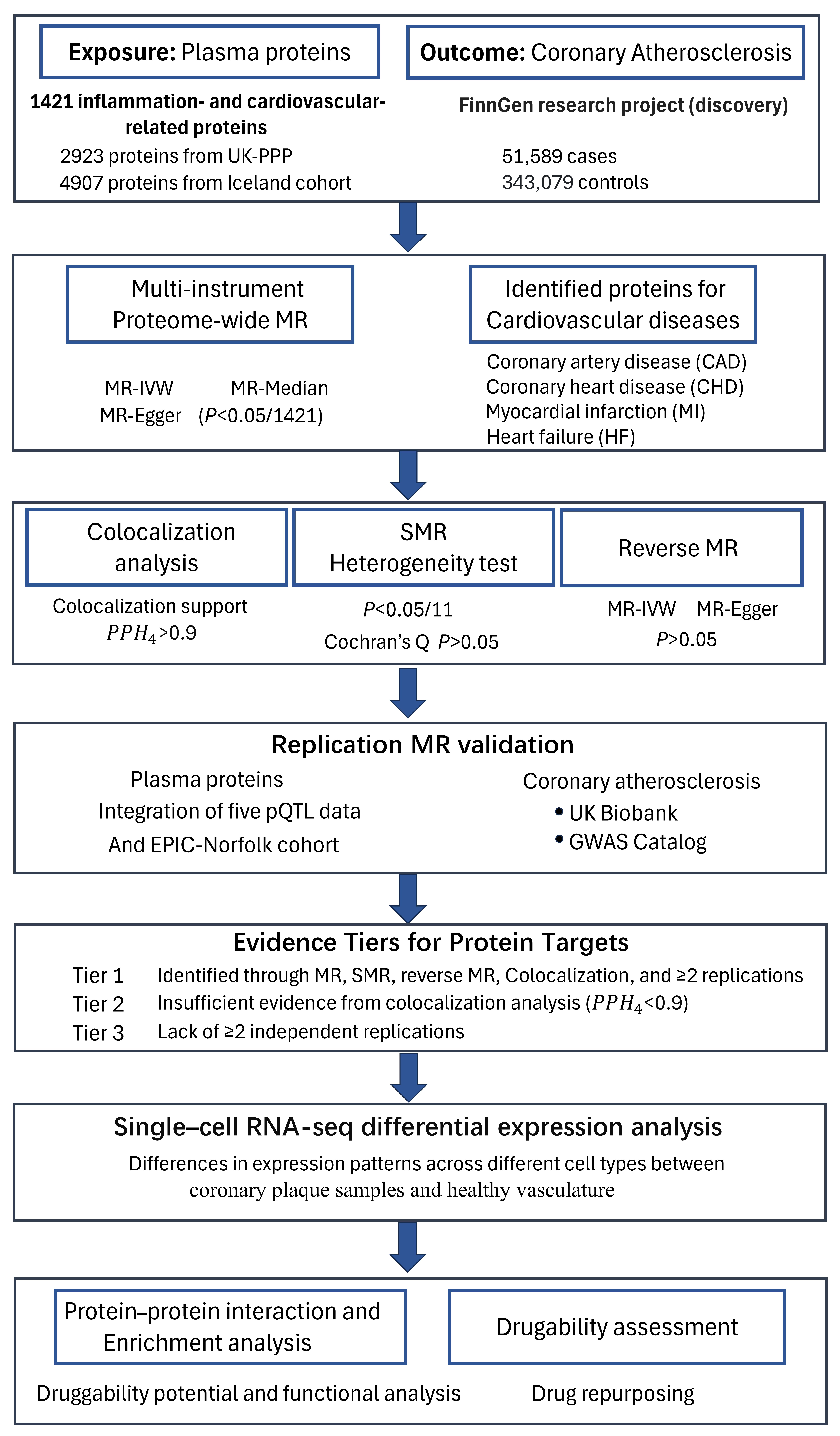
4.1. Study Design
4.2. Proteomic Data Source
4.3. GWAS Summary Statistics for Atherosclerosis and Cardiovascular Diseases
4.4. Proteome-Wide MR Analysis for Identifying Causal Proteins
4.5. Bayesian Colocalization Analysis
4.6. Single-Cell RNA-Seq Differential Expression Analysis
4.7. Protein–Protein Interaction, Enrichment Analysis, and Druggability Assessment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.A.A.; Metra, M.; Morais, J.; Atar, D. The Myth of “stable” Coronary Artery Disease. Nat. Rev. Cardiol. 2020, 17, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Theroux, P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and Atherosclerosis: Signaling Pathways and Therapeutic Intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis-No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- Ferkingstad, E.; Sulem, P.; Atlason, B.A.; Sveinbjornsson, G.; Magnusson, M.I.; Styrmisdottir, E.L.; Gunnarsdottir, K.; Helgason, A.; Oddsson, A.; Halldorsson, B.V.; et al. Large-Scale Integration of the Plasma Proteome with Genetics and Disease. Nat. Genet. 2021, 53, 1712–1721. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Cortes, A.; Koprulu, M.; Wörheide, M.A.; Oerton, E.; Cook, J.; Stewart, I.D.; Kerrison, N.D.; et al. Mapping the Proteo-Genomic Convergence of Human Diseases. Science 2021, 374, eabj1541. [Google Scholar] [CrossRef]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic Atlas of the Human Plasma Proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, E.; Glymour, M.M.; Holmes, M.V.; Kang, H.; Morrison, J.; Munafò, M.R.; Palmer, T.; Schooling, C.M.; Wallace, C.; Zhao, Q.; et al. Mendelian Randomization. Nat. Rev. Methods Primers 2022, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.; et al. Single-Cell Immune Landscape of Human Atherosclerotic Plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Hartog, J.W.L.; Voors, A.A.; Bakker, S.J.L.; Smit, A.J.; van Veldhuisen, D.J. Advanced Glycation End-Products (AGEs) and Heart Failure: Pathophysiology and Clinical Implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), Interferons, Transforming Growth Factor β, and TNF-α: Receptors, Functions, and Roles in Diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef]
- Yasuda, T.; Ishida, T.; Rader, D.J. Update on the Role of Endothelial Lipase in High-Density Lipoprotein Metabolism, Reverse Cholesterol Transport, and Atherosclerosis. Circ. J. 2010, 74, 2263–2270. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Hong, S.-C.; Yang, Q.; Yin, R.-X.; Cao, X.-L.; Chen, W.-X. Association of Variants in CELSR2-PSRC1-SORT1 with Risk of Serum Lipid Traits, Coronary Artery Disease and Ischemic Stroke. Int. J. Clin. Exp. Pathol. 2015, 8, 9543–9551. [Google Scholar]
- Samani, N.J.; Braund, P.S.; Erdmann, J.; Götz, A.; Tomaszewski, M.; Linsel-Nitschke, P.; Hajat, C.; Mangino, M.; Hengstenberg, C.; Stark, K.; et al. The Novel Genetic Variant Predisposing to Coronary Artery Disease in the Region of the PSRC1 and CELSR2 Genes on Chromosome 1 Associates with Serum Cholesterol. J. Mol. Med. 2008, 86, 1233–1241. [Google Scholar] [CrossRef]
- Wen, Q.; Weng, H.; Liu, T.; Yu, L.; Zhao, T.; Qin, J.; Li, S.; Wu, Q.; Tissir, F.; Qu, Y.; et al. Inactivating Celsr2 Promotes Motor Axon Fasciculation and Regeneration in Mouse and Human. Brain 2022, 145, 670–683. [Google Scholar] [CrossRef]
- Weerackoon, N.; Gunawardhana, K.L.; Mani, A. Wnt Signaling Cascades and Their Role in Coronary Artery Health and Disease. J. Cell. Signal. 2021, 2, 52–62. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Kumra, H.; Sabatier, L.; Hassan, A.; Sakai, T.; Mosher, D.F.; Brinckmann, J.; Reinhardt, D.P. Roles of fibronectin isoforms in neonatal vascular development and matrix integrity. PLoS Biol. 2018, 16, e2004812. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Ginsberg, M.H.; Shattil, S.J.; Deckmyn, H.; Schwartz, M.A. Matrix-Specific Suppression of Integrin Activation in Shear Stress Signaling. Mol. Biol. Cell 2006, 17, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Orem, C.; Durmuş, I.; Kilinç, K.; Baykan, M.; Gökçe, M.; Orem, A.; Topbaş, M. Plasma Fibronectin Level and Its Association with Coronary Artery Disease and Carotid Intima-Media Thickness. Coron. Artery Dis. 2003, 14, 219–224. [Google Scholar] [CrossRef]
- Ozcelik, F.; Erdogan, O.; Aktoz, M.; Ekuklu, G.; Tatli, E.; Demir, M. Diagnostic Value of Plasma Fibronectin Level in Predicting the Presence and Severity of Coronary Artery Disease. Ann. Hematol. 2009, 88, 249–253. [Google Scholar] [CrossRef]
- Rohwedder, I.; Montanez, E.; Beckmann, K.; Bengtsson, E.; Dunér, P.; Nilsson, J.; Soehnlein, O.; Fässler, R. Plasma Fibronectin Deficiency Impedes Atherosclerosis Progression and Fibrous Cap Formation. EMBO Mol. Med. 2012, 4, 564–576. [Google Scholar] [CrossRef]
- Doddapattar, P.; Gandhi, C.; Prakash, P.; Dhanesha, N.; Grumbach, I.M.; Dailey, M.E.; Lentz, S.R.; Chauhan, A.K. Fibronectin Splicing Variants Containing Extra Domain A Promote Atherosclerosis in Mice through Toll-like Receptor 4. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2391–2400. [Google Scholar] [CrossRef]
- Doddapattar, P.; Jain, M.; Dhanesha, N.; Lentz, S.R.; Chauhan, A.K. Fibronectin Containing Extra Domain A Induces Plaque Destabilization in the Innominate Artery of Aged Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 500–508. [Google Scholar] [CrossRef]
- Vavalle, J.P.; Wu, S.S.; Hughey, R.; Madamanchi, N.R.; Stouffer, G.A. Plasma Fibronectin Levels and Coronary Artery Disease. J. Thromb. Haemost. 2007, 5, 864–866. [Google Scholar] [CrossRef]
- Naschberger, E.; Liebl, A.; Schellerer, V.S.; Schütz, M.; Britzen-Laurent, N.; Kölbel, P.; Schaal, U.; Haep, L.; Regensburger, D.; Wittmann, T.; et al. Matricellular Protein SPARCL1 Regulates Tumor Microenvironment–Dependent Endothelial Cell Heterogeneity in Colorectal Carcinoma. J. Clin. Investig. 2016, 126, 4187–4204. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, Y.; Li, L. SPARCL1 Suppresses the Proliferation and Migration of Human Ovarian Cancer Cells via the MEK/ERK Signaling. Exp. Ther. Med. 2018, 16, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Cao, F.; Ding, W.; Gao, X.-J.; Chen, F.; Hu, Y.-W.; Ding, H.-Z. Prognostic Value of SPARCL1 in Patients with Colorectal Cancer. Oncol. Lett. 2018, 15, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Chen, X.; Zhang, M.; Wan, Y.; Ge, S.; Cheng, X. Sparcl1 and Atherosclerosis. J. Inflamm. Res. 2023, 16, 2121–2127. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Hu, C.; Wang, S.; Lin, L.; Qi, H.; Lin, H.; Jia, X.; Zhu, Y.; Wu, X.; Li, M.; Wang, T.; et al. Association of Serum Secreted Protein Acidic and Rich in Cysteine-like Protein 1 with Metabolic Measures and Dyslipidemia among Chinese Adults. Front. Endocrinol. 2022, 13, 1018657. [Google Scholar] [CrossRef]
- Regensburger, D.; Tenkerian, C.; Pürzer, V.; Schmid, B.; Wohlfahrt, T.; Stolzer, I.; López-Posadas, R.; Günther, C.; Waldner, M.J.; Becker, C.; et al. Matricellular Protein SPARCL1 Regulates Blood Vessel Integrity and Antagonizes Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1491–1502. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of Apolipoprotein E Genotypes with Lipid Levels and Coronary Risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef]
- Alagarsamy, J.; Jaeschke, A.; Hui, D.Y. Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9892. [Google Scholar] [CrossRef]
- Gilliland, T.C.; Liu, Y.; Mohebi, R.; Miksenas, H.; Haidermota, S.; Wong, M.; Hu, X.; Cristino, J.R.; Browne, A.; Plutzky, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Coronary Artery Disease Severity and Outcomes. J. Am. Coll. Cardiol. 2023, 81, 1780–1792. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, Z.; Yao, C.; Zhao, S. Emerging Evidences for the Opposite Role of Apolipoprotein C3 and Apolipoprotein A5 in Lipid Metabolism and Coronary Artery Disease. Lipids Health Dis. 2019, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Lv, X.; Bao, K.; Tian, X.Y.; He, L.; Shi, L.; Zhu, Y.; Ai, D. Yes-Associated Protein Targets the Transforming Growth Factor β Pathway to Mediate High-Fat/High-Sucrose Diet-Induced Arterial Stiffness. Circ. Res. 2022, 130, 6. [Google Scholar] [CrossRef]
- Zeng, Z.; Guo, R.; Wang, Z.; Yan, H.; Lv, X.; Zhao, Q.; Jiang, X.; Zhang, C.; Zhang, D.; Yang, C.; et al. Circulating Monocytes Act as a Common Trigger for the Calcification Paradox of Osteoporosis and Carotid Atherosclerosis via TGFB1-SP1 and TNFSF10-NFKB1 Axis. Front. Endocrinol. 2022, 13, 944751. [Google Scholar] [CrossRef]
- Su, W.; Li, W.; Chen, H.; Liu, H.; Huang, H.; Li, H. Advanced Glycation End Products Impair Voltage-Gated K+ Channels-Mediated Coronary Vasodilation in Diabetic Rats. PLoS ONE 2015, 10, e0142865. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Timpson, N.J.; Higgins, J.P.T.; Dimou, N.; Langenberg, C.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomisation (STROBE-MR): Explanation and Elaboration. BMJ 2021, 375, n2233. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.B.; Chiou, J.; Traylor, M.; Benner, C.; Hsu, Y.-H.; Richardson, T.G.; Surendran, P.; Mahajan, A.; Robins, C.; Vasquez-Grinnell, S.G.; et al. Plasma Proteomic Associations with Genetics and Health in the UK Biobank. Nature 2023, 622, 329–338. [Google Scholar] [CrossRef]
- Rohloff, J.C.; Gelinas, A.D.; Jarvis, T.C.; Ochsner, U.A.; Schneider, D.J.; Gold, L.; Janjic, N. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther. Nucleic Acids 2014, 3, e201. [Google Scholar] [CrossRef]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous Antibody-Based Proximity Extension Assays Provide Sensitive and Specific Detection of Low-Abundant Proteins in Human Blood. Nucleic Acids Res. 2011, 39, e102. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Haberland, V.; Baird, D.; Walker, V.; Haycock, P.C.; Hurle, M.R.; Gutteridge, A.; Erola, P.; Liu, Y.; Luo, S.; et al. Phenome-Wide Mendelian Randomization Mapping the Influence of the Plasma Proteome on Complex Diseases. Nat. Genet. 2020, 52, 1122–1131. [Google Scholar] [CrossRef]
- Koprulu, M.; Carrasco-Zanini, J.; Wheeler, E.; Lockhart, S.; Kerrison, N.D.; Wareham, N.J.; Pietzner, M.; Langenberg, C. Proteogenomic Links to Human Metabolic Diseases. Nat. Metab. 2023, 5, 516–528. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Gupta, R.; Kanai, M.; Lu, W.; Tsuo, K.; Wang, Y.; Walters, R.K.; Turley, P.; Callier, S.; Baya, N.; et al. Pan-UK Biobank GWAS Improves Discovery, Analysis of Genetic Architecture, and Resolution into Ancestry-Enriched Effects. MedRxiv 2024. [Google Scholar] [CrossRef]
- Jiang, L.; Zheng, Z.; Fang, H.; Yang, J. A Generalized Linear Mixed Model Association Tool for Biobank-Scale Data. Nat. Genet. 2021, 53, 1616–1621. [Google Scholar] [CrossRef]
- Zhou, W.; Nielsen, J.B.; Fritsche, L.G.; Dey, R.; Gabrielsen, M.E.; Wolford, B.N.; LeFaive, J.; VandeHaar, P.; Gagliano, S.A.; Gifford, A.; et al. Efficiently Controlling for Case-Control Imbalance and Sample Relatedness in Large-Scale Genetic Association Studies. Nat. Genet. 2018, 50, 1335–1341. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.-H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A Comprehensive 1000 Genomes-Based Genome-Wide Association Meta-Analysis of Coronary Artery Disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N. Engl. J. Med. 2016, 374, 1134–1144. [Google Scholar] [CrossRef]
- Palmer, T.M.; Lawlor, D.A.; Harbord, R.M.; Sheehan, N.A.; Tobias, J.H.; Timpson, N.J.; Smith, G.D.; Sterne, J.A. Using Multiple Genetic Variants as Instrumental Variables for Modifiable Risk Factors. Stat. Methods Med. Res. 2012, 21, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal Associations between Risk Factors and Common Diseases Inferred from GWAS Summary Data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef]
- Lewis, M.J.; Wang, S. Locuszoomr: An R Package for Visualizing Publication-Ready Regional Gene Locus Plots. Bioinform. Adv. 2025, 5, vbaf006. [Google Scholar] [CrossRef]
- Barcia Durán, J.G.; Das, D.; Gildea, M.; Amadori, L.; Gourvest, M.; Kaur, R.; Eberhardt, N.; Smyrnis, P.; Cilhoroz, B.; Sajja, S.; et al. Immune Checkpoint Landscape of Human Atherosclerosis and Influence of Cardiometabolic Factors. Nat. Cardiovasc. Res. 2024, 3, 1482–1502. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to Characterize Multiomics Data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | UniProt | OR (95% CI) | Pdiscovery | Preplication a | Preverse | Colocalization b PPH4 | Panel | Tier |
|---|---|---|---|---|---|---|---|---|
| PCSK9 | Q8NBP7 | 1.28 (1.22–1.34) | 1.61 × 10−25 | 5.64 × 10−5 | 0.64 | 0.999/0.999 | Cardiometabolic | 1 |
| CELSR2 | Q9HCU4 | 0.82 (0.79–0.86) | 3.88 × 10−21 | 1.70 × 10−29 | 0.04 | 0.997/0.994 | Inflammation | 1 |
| APOE | P02649 | 0.94 (0.92–0.95) | 2.70 × 10−11 | 1.44 × 10−21 | 0.95 | 0.000/0.000 | Inflammation | 2 |
| LPA | P08519 | 1.14 (1.09–1.2) | 5.51 × 10−9 | 4.11 × 10−90 | 0.45 | 0.000/0.000 | Inflammation | 2 |
| IL6R | P08887 | 0.96 (0.95–0.98) | 7.77 × 10−8 | 2.05 × 10−5 | 0.78 | 0.986/0.972 | Cardiometabolic | 1 |
| FN1 | P02751 | 0.9 (0.86–0.94) | 3.22 × 10−7 | 3.47 × 10−8 | 0.53 | 0.972/0.945 | Inflammation | 1 |
| APOA5 | Q6Q788 | 1.06 (1.03–1.09) | 2.17 × 10−6 | 1.62 × 10−4 | 0.44 | 0.000/0.000 | Cardiometabolic | 2 |
| AGER | Q15109 | 0.91 (0.87–0.94) | 2.58 × 10−6 | 0.12 | 0.64 | 0.020/0.009 | Inflammation | 3 |
| CD4 | P01730 | 0.9 (0.86–0.94) | 5.33 × 10−6 | 0.71 | 0.12 | 0.983/0.966 | Inflammation | 3 |
| TGFB1 | P01137 | 1.16 (1.09–1.23) | 5.56 × 10−6 | 2.77 × 10−6 | 0.62 | 0.450/0.290 | Inflammation | 2 |
| SPARCL1 | Q14515 | 0.94 (0.91–0.97) | 1.05 × 10−5 | 5.08 × 10−3 | 0.37 | 0.963/0.929 | Cardiometabolic | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Xie, F.; Wang, M.; Ji, J.; Song, Y.; Dai, Y.; Wang, L.; Kang, Z.; Cao, L. Identification of Potential Therapeutic Targets for Coronary Atherosclerosis from an Inflammatory Perspective Through Integrated Proteomics and Single-Cell Omics. Int. J. Mol. Sci. 2025, 26, 6201. https://doi.org/10.3390/ijms26136201
Wang H, Xie F, Wang M, Ji J, Song Y, Dai Y, Wang L, Kang Z, Cao L. Identification of Potential Therapeutic Targets for Coronary Atherosclerosis from an Inflammatory Perspective Through Integrated Proteomics and Single-Cell Omics. International Journal of Molecular Sciences. 2025; 26(13):6201. https://doi.org/10.3390/ijms26136201
Chicago/Turabian StyleWang, Hesong, Fengzhe Xie, Meng Wang, Jianxin Ji, Yongzhen Song, Yanyan Dai, Liuying Wang, Zheng Kang, and Lei Cao. 2025. "Identification of Potential Therapeutic Targets for Coronary Atherosclerosis from an Inflammatory Perspective Through Integrated Proteomics and Single-Cell Omics" International Journal of Molecular Sciences 26, no. 13: 6201. https://doi.org/10.3390/ijms26136201
APA StyleWang, H., Xie, F., Wang, M., Ji, J., Song, Y., Dai, Y., Wang, L., Kang, Z., & Cao, L. (2025). Identification of Potential Therapeutic Targets for Coronary Atherosclerosis from an Inflammatory Perspective Through Integrated Proteomics and Single-Cell Omics. International Journal of Molecular Sciences, 26(13), 6201. https://doi.org/10.3390/ijms26136201