
Immunothrombosis in Sepsis: Cellular Crosstalk, Molecular Triggers, and Therapeutic Opportunities—A Review

Abstract
1. Introduction
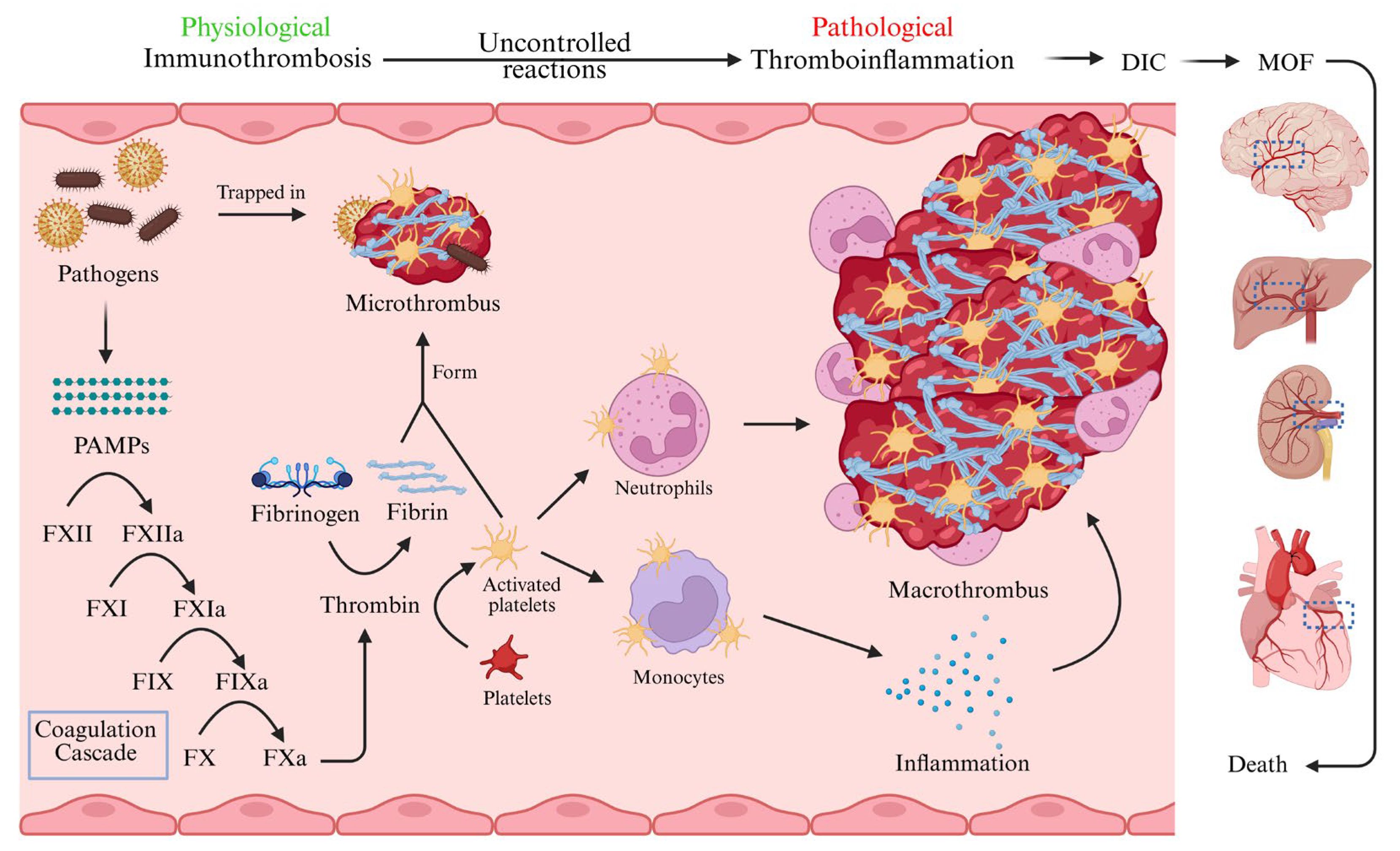
2. Pathophysiology of Sepsis-Induced Immunothrombosis
2.1. Sepsis-Induced Hyperinflammation
2.2. Sepsis-Induced Immunothrombosis
2.3. Crosstalk Between Inflammation and Coagulation
3. Cellular Activation
3.1. Cellular Activation in Sepsis-Induced Immunothrombosis
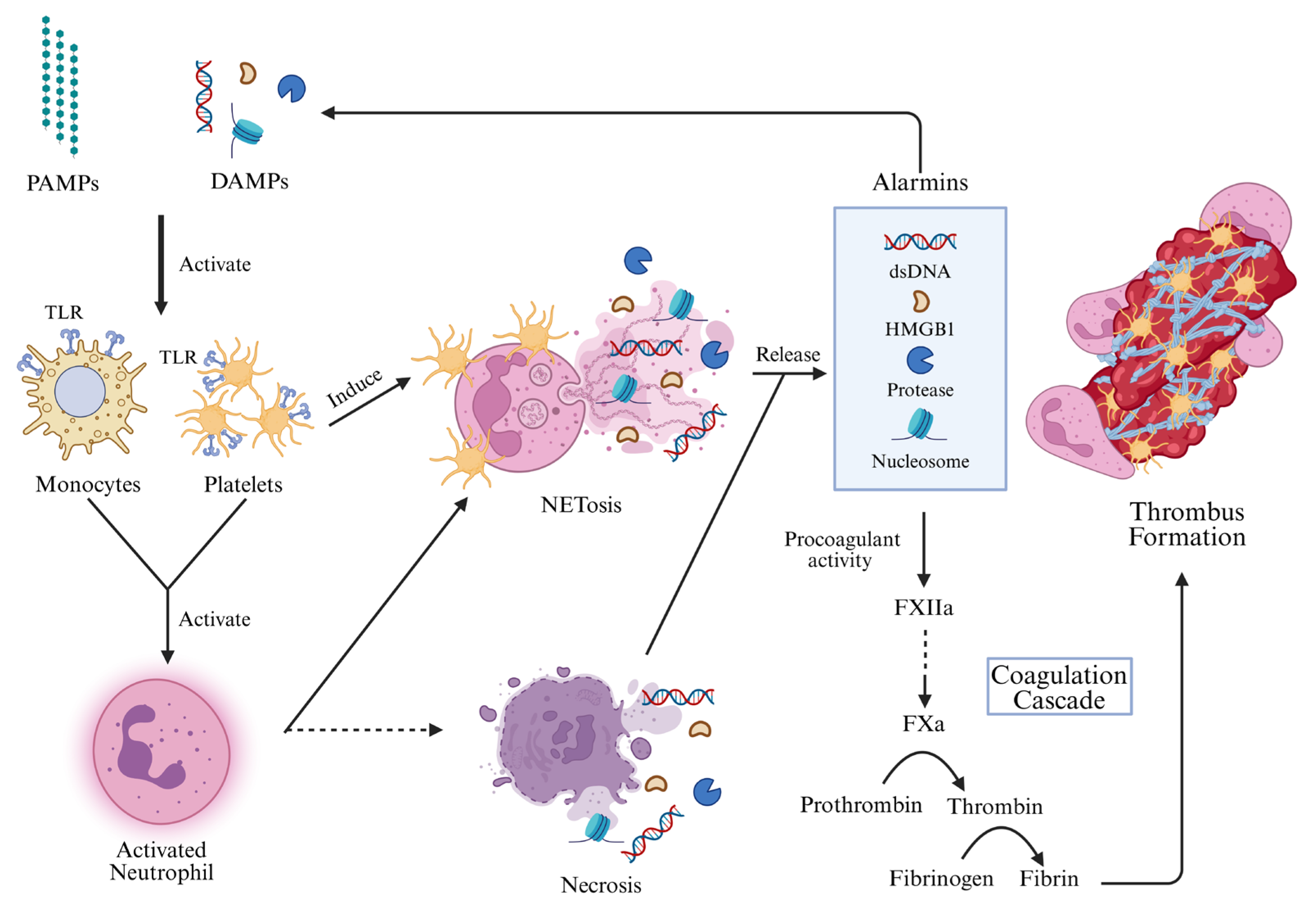
3.2. Activation of Leukocytes
3.2.1. Activation of Monocytes
3.2.2. Activation of Neutrophils
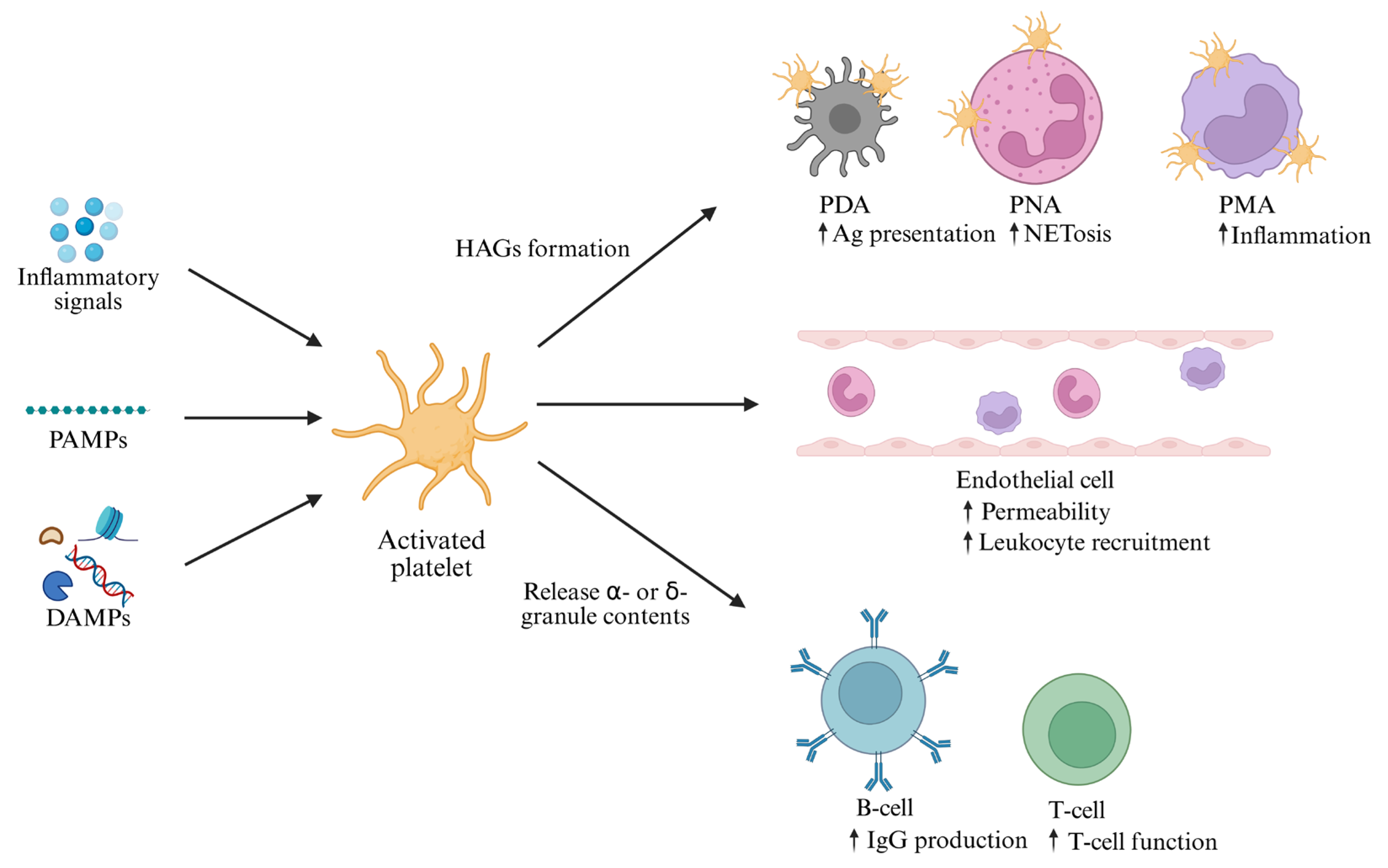
3.3. Activation of Platelets
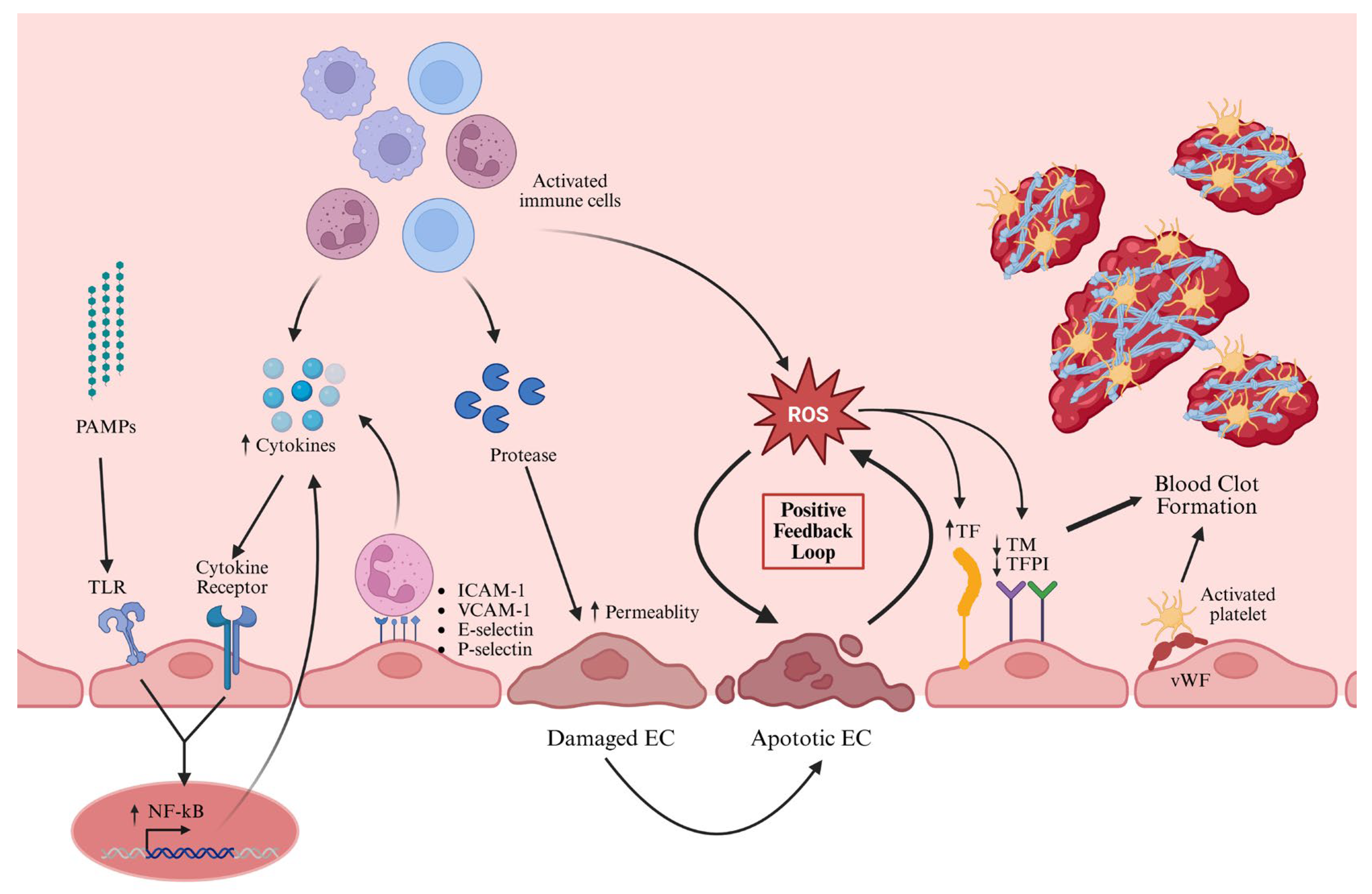
3.4. Activation of ECs
4. Cellular Signaling Pathways in Immunothrombosis
4.1. NLRP3 Inflammasome Pathway
4.2. cGAS-STING Pathway
4.3. Other Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Molecules | Source of Cells | Pathways Involved in | Roles in the Thrombosis Formation | References |
|---|---|---|---|---|
| Phosphatidylinositol 3-kinases (PI3Ks) | Platelets | PI3K/Akt pathway | Phosphorylates phosphoinositide lipids at the 3 position of the inositol ring and regulates cell growth. | [94] |
| Integrin αIIbβ3 | Platelets | PI3K/Akt pathway | Initiates intracellular signaling pathways for platelet activation and is involved in establishing cell–cell contact during platelet aggregation and thrombus formation. | [94] |
| FcγRIIA | Platelets (abundant), monocytes, neutrophils | In different pathways | Major transmembrane signaling adapter for αIIbβ3 outside-in signaling pathway in thrombosis. | [100] |
| cGAS | All cells | cGAS-STING pathway | cGAS detects aberrant abnormal DNAs, and activated cGAS initiates the synthesis of a signaling molecule known as cGAMP in which formation of 2′-3′-cGAMP from ATP and GTP. In turn it binds STING. | [91,92] |
| STING | All cells | cGAS-STING pathway | Activated STING prompts the production of interferons and proinflammatory factors, including TNF-α, IL-6, among others, through the activation of transcription factors like IRF3 and NF-κB. | [91,92] |
| NLRP3 | Macrophages/monocytes/Tohoko Hospital pediatrics 1 (THP1) | Inflammasome pathway, cGAS-STING pathway | Promotes the production of proinflammatory cytokines through activation of caspases. | [86,88] |
| Cytosolic DNA | Damaged mitochondria | cGAS-STING pathway, inflammasome pathway | Activates NLRP3 through upregulating the cGAS-STING axis. | [89] |
| NF-kB | All cells | NF-kB signaling pathway and other pathways | A crucial transcription factor involved in the expression of genes for the activation of the inflammatory response and homeostasis. | [46,101] |
| TF | Monocytes, ECs | Extrinsic coagulation pathway | Complexes with FVII/VIIa to proteolytically activate factors IX to IXa and X to Xa, resulting in thrombin generation. | [10] |
| Thrombin | Enzymatic cleavage of prothrombin | Common pathway of coagulation | Activates protease-activated receptors, which are critical for the interplay between inflammation and coagulation, boosting proinflammatory cytokine secretion and activating platelets. Converts fibrinogen to fibrin for clot formation. Involved in NETosis together with activated platelets and neutrophils. | [10] |
| NETs | Neutrophils | PAD4 pathway | Trigger a proinflammatory and procoagulant endothelial phenotype by inhibiting anticoagulation and inducing TF expression. Platelet-mediated coagulation activation. | [52,54] |
5. Molecular Mechanisms of Sepsis-Induced Immunothrombosis
6. Therapeutic Strategies for Sepsis-Induced Immunothrombosis
| Type of Drugs | Target of Drugs | Mechanism | Clinical Relevance | References |
|---|---|---|---|---|
| Low molecular weight heparins (Bemiparin, Certoparin, Dalteparin, Enoxaparin, Nadroparin, Parnaparin, Reviparin, and Tinzaparin) | Antithrombin III | Anticoagulant and anti-inflammatory | Inhibit coagulation by activating antithrombin III, which binds to and inhibits factor Xa Improve sepsis outcome | [158,159] |
| Fondaparinux | Thrombin | Anticoagulant | Reduces coagulation in COVID-19 patients | [160,161] |
| Enoxaparin | Thrombin | Anticoagulant | Reduces coagulation in COVID-19 patients | [160] |
| Ticlopidine | Platelet P2Y12 | Antagonist | Prevents platelet activation (commercially available) | [162] |
| Clopidogrel | Platelet P2Y12 | Antagonist | Prevents platelet activation (commercially available) | [162] |
| Prasugrel | Platelet P2Y12 | Antagonist | Prevents platelet activation (commercially available) | [162] |
| Ticagrelor | Platelet P2Y12 | Antagonist | Prevents platelet activation (commercially available) | [162] |
| Cangrelor | Platelet P2Y12 | Antagonist | Prevents platelet activation (commercially available) | [162] |
| DNases | NETs | Digest DNA | Prevent platelet activation and coagulation | [26] |
| Dimetil fumarate | Gasdermin D | Inhibit interaction with caspases | Prevents pyroptosis | [163] |
| Thrombomodulin | HMGB1 | Inhibit HMGB1 | Reduces inflammation | [164] |
| Recombinant human activated protein C | Factor Va and factor VIIIa | Anticoagulant | Reduces mortality in severe sepsis | [150] |
| Types of Drugs | Target of Drugs | Subject | Outcome | Study Type | References |
|---|---|---|---|---|---|
| Eptifibatide | GPIIb/IIIa receptor | Septic shock patients | Reduces endothelial injury, reduces platelet consumption and improves sequential organ failure assessment (SOFA) score | RCT | [140] |
| MCC950 | NLRP3 | Sprague Dawley rat treated by cecal ligation and puncture (CLP) | Inhibits NLRP3 activation and reduces platelet activation as well as reduces multi-organ injury | In vivo | [145] |
| Glycyrrhizin | HMBG1 | Mice (C57BL/6) and THP 1 cell | Attenuates caspase-11-dependent immune responses and coagulopathy by inhibiting HMBG1 | In vivo and in vitro | [165] |
| Ticagrelor | NETs | Patient sample and cell line | Prevents platelet activation and coagulation | Ex vivo and in vivo | [166] |
| Aspirin | Platelets, NF-kB and HMGB1 | Sepsis patients | Reduces 30-day mortality | RCT | [141] |
| Forsythiaside B (FTB) (DNase1, Cl-amidine) | NETs, PAD4 | Sprague Dawley rats treated by CLP | Alleviate coagulopathy | In vivo | [97,143] |
| P2Y12 antagonists (23 chemically synthesized compounds) | Platelets | In vitro | Inhibit platelet activation through P2Y12 antagonist activity | In vitro | [162] |
| Esaridin E | Integrin αvβ3 | CLP mice (Male C57BL/6 mice) | Improves endothelial hyperpermeability by inhibiting vWF binding to αvβ3 | In vivo | [167] |
| AntiCD14 antibody | CD14 | Baboon (Papio anubis) | Reduces activation of complement, proinflammatory cytokines and inflammatory cells | In vivo | [42] |
| Acetylsalicylic acid (ASA) | Platelets, neutrophils | Mice (C57Bl/6 mice) | Reduces platelet activation, neutrophil recruitment and NET formation | In vivo | [168] |
| Heparin | Alarmin HMGB1 | Mice (Alb-cre mice) | Inhibits alarmin HMGB1-LPS interaction and prevents lethal effect of LPS sepsis | In vivo | [158] |
| Resveratrol-loaded silver nanoparticle | Proinflammatory cytokines | Rat (Sprague Dawley rats) | Reduces proinflammatory cytokines and inhibits activation of NF-kB | In vivo | [169] |
| Histidine-rich glycoprotein | FXII | Rabbit (Male New Zealand white rabbits), endothelial cell line | Decreases thrombosis associated with the catheter Reduces sepsis-induced shock and DIC | In vitro and In vivo | [149,170,171] |
| Emelin | Plasminogen activator inhibitor-1 (PAI-1) | Mice (Male Kunming mice) | Alleviates sepsis-induced DIC | In vivo | [147] |
| Matrine | NLRP3 | THP1, J774A.1 cell line and Mice (C57BL/6 mice) | Suppresses of NLRP3 inflammasome activation through regulating protein tyrosine phosphatase non-receptor type 2 (PTPN2)/JNK/SREBP2 signaling | In vitro and In vivo | [85] |
| Amitriptyline (AMIT) | Inflammatory cytokines (TNF-α) | CLP mice (Male CF-1 outbred mice) | Reduces level of TNF-α and alleviates SIC without bleeding complication | In vivo | [148] |
| Combination of Probenecid Nanocrystals and Cefotaxime Sodium | Inflammatory cytokines and NETs | Mice (C57BL/6 mice) | Promote sepsis recovery by reducing immunothrombosis formation | In vivo | [157] |
| Combination of ulinastatin with TIENAM | Inflammatory cytokines | CLP Mice (C57BL/6 mice) | Reduce inflammation and NF-kB pathways suppressed | In vivo | [156] |
| Parthenolides | Mitochondrial mediated apoptosis | Septic rat (Sprague Dawley rats) | Improves SIC | In vivo | [172] |
| Nitrofurans, acrylamides, and indole ureas (indole derivatives) | STING | Cell line and mouse model (C57BL/6J mice) | Reduce STING-mediated inflammatory cytokine production | In vitro and in vivo | [163,173] |
7. Limitations
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Zhu, L.; Dong, H.; Li, L.; Liu, X. The Mechanisms of Sepsis Induced Coagulation Dysfunction and Its Treatment. J. Inflamm. Res. 2025, 18, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, S.M.K.; Bhat, B.V. Role of microRNAs in sepsis. Inflamm. Res. 2017, 66, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med. 2019, 7, 2050312119835043. [Google Scholar] [CrossRef]
- La Via, L.; Sangiorgio, G.; Stefani, S.; Marino, A.; Nunnari, G.; Cocuzza, S.; La Mantia, I.; Cacopardo, B.; Stracquadanio, S.; Spampinato, S.; et al. The Global Burden of Sepsis and Septic Shock. Epidemiologia 2024, 5, 456–478. [Google Scholar] [CrossRef]
- Tsantes, A.G.; Parastatidou, S.; Tsantes, E.A.; Bonova, E.; Tsante, K.A.; Mantzios, P.G.; Vaiopoulos, A.G.; Tsalas, S.; Konstantinidi, A.; Houhoula, D.; et al. Sepsis-Induced Coagulopathy: An Update on Pathophysiology, Biomarkers, and Current Guidelines. Life 2023, 13, 350. [Google Scholar] [CrossRef]
- Southeast Asia Infectious Disease Clinical Research Network. Causes and outcomes of sepsis in southeast Asia: A multinational multicentre cross-sectional study. Lancet Glob. Health 2017, 5, e157–e167. [Google Scholar] [CrossRef]
- Muratsu, A.; Oda, S.; Onishi, S.; Yoshimura, J.; Matsumoto, H.; Togami, Y.; Mitsuyama, Y.; Ito, H.; Okuzaki, D.; Ogura, H.; et al. Bacterial sepsis causes more dramatic pathogenetic changes in the Th1 pathway than does viral (COVID-19) sepsis: A prospective observational study of whole blood transcriptomes. Virol. J. 2024, 21, 190. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Sepsis-induced Coagulopathy and Disseminated Intravascular Coagulation. Anesthesiology 2020, 132, 1238–1245. [Google Scholar] [CrossRef]
- Ryan, T.A.J.; O’Neill, L.A.J. Innate immune signaling and immunothrombosis: New insights and therapeutic opportunities. Eur. J. Immunol. 2022, 52, 1024–1034. [Google Scholar] [CrossRef]
- Schrottmaier, W.C.; Assinger, A. The Concept of Thromboinflammation. Hamostaseologie 2024, 44, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levi, M.; Levy, J.H. Intracellular communication and immunothrombosis in sepsis. J. Thromb. Haemost. 2022, 20, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Massberg, S. Blood coagulation in immunothrombosis-At the frontline of intravascular immunity. Semin. Immunol. 2016, 28, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Helms, J.; Connors, J.M.; Levy, J.H. The pathophysiology, diagnosis, and management of sepsis-associated disseminated intravascular coagulation. J. Intensive Care 2023, 11, 24. [Google Scholar] [CrossRef]
- Joosten, S.C.M.; Wiersinga, W.J.; Poll, T.V. Dysregulation of Host-Pathogen Interactions in Sepsis: Host-Related Factors. Semin. Respir. Crit. Care Med. 2024, 45, 469–478. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Dartiguelongue, J.B. Systemic inflammation and sepsis. Part II: Functional consequences of the storm. Arch. Argent. Pediatr. 2021, 119, e1–e10. [Google Scholar] [CrossRef]
- Carcillo, J.A.; Shakoory, B. Cytokine Storm and Sepsis-Induced Multiple Organ Dysfunction Syndrome. Adv. Exp. Med. Biol. 2024, 1448, 441–457. [Google Scholar] [CrossRef]
- Joosten, S.C.; Wiersinga, W.J.; van der Poll, T. (Eds.) Dysregulation of Host–Pathogen Interactions in Sepsis: Host-Related Factors. In Seminars in Respiratory and Critical Care Medicine; Thieme Medical Publishers: New York, NY, USA, 2024. [Google Scholar]
- Liufu, R.; Chen, Y.; Wan, X.X.; Liu, R.T.; Jiang, W.; Wang, C.; Peng, J.M.; Weng, L.; Du, B. Sepsis-induced Coagulopathy: The Different Prognosis in Severe Pneumonia and Bacteremia Infection Patients. Clin. Appl. Thromb. Hemost. 2023, 29, 10760296231219249. [Google Scholar] [CrossRef]
- Meziani, F.; Iba, T.; Levy, J.H.; Helms, J. Sepsis-induced coagulopathy: A matter of timeline. Intensive Care Med. 2024, 50, 1404–1405. [Google Scholar] [CrossRef]
- Iba, T.; Umemura, Y.; Wada, H.; Levy, J.H. Roles of Coagulation Abnormalities and Microthrombosis in Sepsis: Pathophysiology, Diagnosis, and Treatment. Arch. Med. Res. 2021, 52, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N. Sepsis-Induced Coagulopathy: A Prelude to DIC. Anesth. Analg. 2024, 138, 695. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; Levi, M. Crosstalk between inflammation and coagulation: The lessons of sepsis. Curr. Vasc. Pharmacol. 2012, 10, 632–638. [Google Scholar] [CrossRef]
- Jong, E.; Van Gorp, E.C.M.; Levi, M.; Cate, H.T. The Crosstalk of Inflammation and Coagulation in Infectious Disease and Their Roles in Disseminated Intravascular Coagulation. In Consultative Hemostasis and Thrombosis; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 190–201. [Google Scholar]
- Arshad, A.; Ahmed, W.; Rehman, N.; Naseem, Z.; Ghos, Z. Tackling a deadly global phenomenon: Sepsis induced coagulopathy: A narrative review. J. Pak. Med. Assoc. 2024, 74, 959–966. [Google Scholar] [CrossRef]
- Schouten, M.; Wiersinga, W.J.; Levi, M.; van der Poll, T. Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol. 2008, 83, 536–545. [Google Scholar] [CrossRef]
- Iba, T.; Helms, J.; Levy, J.H. Sepsis-induced coagulopathy (SIC) in the management of sepsis. Ann. Intensive Care 2024, 14, 148. [Google Scholar] [CrossRef]
- Maneta, E.; Aivalioti, E.; Tual-Chalot, S.; Emini Veseli, B.; Gatsiou, A.; Stamatelopoulos, K.; Stellos, K. Endothelial dysfunction and immunothrombosis in sepsis. Front. Immunol. 2023, 14, 1144229. [Google Scholar] [CrossRef]
- Wu, M.; Yan, Y.; Xie, X.; Bai, J.; Ma, C.; Du, X. Effect of endothelial responses on sepsis-associated organ dysfunction. Chin. Med. J. 2024, 137, 2782–2792. [Google Scholar] [CrossRef]
- Raia, L.; Zafrani, L. Endothelial Activation and Microcirculatory Disorders in Sepsis. Front. Med. 2022, 9, 907992. [Google Scholar] [CrossRef]
- Noone, D.; Preston, R.J.S.; Rehill, A.M. The Role of Myeloid Cells in Thromboinflammatory Disease. Semin. Thromb. Hemost. 2024, 50, 998–1011. [Google Scholar] [CrossRef]
- Joffre, J.; Liles, W.C. Editorial: Endothelial activation and microcirculatory disorders in sepsis and critical illness. Front. Med. 2022, 9, 1133408. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.; Middleton, E.A. The role of platelets and megakaryocytes in sepsis and ARDS. J. Physiol. 2024, 602, 6047–6063. [Google Scholar] [CrossRef] [PubMed]
- Leick, M.; Azcutia, V.; Newton, G.; Luscinskas, F.W. Leukocyte recruitment in inflammation: Basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res. 2014, 355, 647–656. [Google Scholar] [CrossRef]
- Sorrells, M.G.; Seo, Y.; Magnen, M.; Broussard, B.; Sheybani, R.; Shah, A.M.; O’Neal, H.R.; Tse, H.T.K., Jr.; Looney, M.R.; Di Carlo, D. Biophysical Changes of Leukocyte Activation (and NETosis) in the Cellular Host Response to Sepsis. Diagnostics 2023, 13, 1435. [Google Scholar] [CrossRef]
- Belok, S.H.; Bosch, N.A.; Klings, E.S.; Walkey, A.J. Evaluation of leukopenia during sepsis as a marker of sepsis-defining organ dysfunction. PLoS ONE 2021, 16, e0252206. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef]
- Hanna, M.O.F.; Abdelhameed, A.M.; Abou-Elalla, A.A.; Hassan, R.M.; Kostandi, I. Neutrophil and monocyte receptor expression in patients with sepsis: Implications for diagnosis and prognosis of sepsis. Pathog. Dis. 2019, 77, ftz055. [Google Scholar] [CrossRef]
- Sun, X.F.; Luo, W.C.; Huang, S.Q.; Zheng, Y.J.; Xiao, L.; Zhang, Z.W.; Liu, R.H.; Zhong, Z.W.; Song, J.Q.; Nan, K.; et al. Immune-cell signatures of persistent inflammation, immunosuppression, and catabolism syndrome after sepsis. Med 2025, 6, 100569. [Google Scholar] [CrossRef]
- Arts, R.J.; Gresnigt, M.S.; Joosten, L.A.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukoc. Biol. 2017, 101, 151–164. [Google Scholar] [CrossRef]
- Keshari, R.S.; Silasi, R.; Popescu, N.I.; Regmi, G.; Chaaban, H.; Lambris, J.D.; Lupu, C.; Mollnes, T.E.; Lupu, F. CD14 inhibition improves survival and attenuates thrombo-inflammation and cardiopulmonary dysfunction in a baboon model of Escherichia coli sepsis. J. Thromb. Haemost. 2021, 19, 429–443. [Google Scholar] [CrossRef]
- Musgrave, K.M.; Scott, J.; Sendama, W.; Gardner, A.I.; Dewar, F.; Lake, C.J.; Spronk, H.M.H.; van Oerle, R.; Visser, M.; Ten Cate, H.; et al. Tissue factor expression in monocyte subsets during human immunothrombosis, endotoxemia and sepsis. Thromb. Res. 2023, 228, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Sachetto, A.T.A.; Mackman, N. Monocyte Tissue Factor Expression: Lipopolysaccharide Induction and Roles in Pathological Activation of Coagulation. Thromb. Haemost. 2023, 123, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Pekayvaz, K.; Kilani, B.; Joppich, M.; Eivers, L.; Brambs, S.; Knottenberg, V.; Akgöl, S.; Yue, K.; Li, L.; Martinez-Navarro, A.; et al. Immunothrombolytic monocyte-neutrophil axes dominate the single-cell landscape of human thrombosis and correlate with thrombus resolution. Immunity 2025, 58, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Derler, M.; Basílio, J.; Schmid, J.A. NF-κB in monocytes and macrophages-an inflammatory master regulator in multitalented immune cells. Front. Immunol. 2023, 14, 1134661. [Google Scholar] [CrossRef]
- Hou, F.F.; Mi, J.H.; Wang, Q.; Tao, Y.L.; Guo, S.B.; Ran, G.H.; Wang, J.C. Macrophage polarization in sepsis: Emerging role and clinical application prospect. Int. Immunopharmacol. 2025, 144, 113715. [Google Scholar] [CrossRef]
- Fu, G.; Deng, M.; Neal, M.D.; Billiar, T.R.; Scott, M.J. Platelet-Monocyte Aggregates: Understanding Mechanisms and Functions in Sepsis. Shock 2021, 55, 156–166. [Google Scholar] [CrossRef]
- Rolling, C.C.; Barrett, T.J.; Berger, J.S. Platelet-monocyte aggregates: Molecular mediators of thromboinflammation. Front. Cardiovasc. Med. 2023, 10, 960398. [Google Scholar] [CrossRef]
- Stiel, L.; Meziani, F.; Helms, J. Neutrophil Activation During Septic Shock. Shock 2018, 49, 371–384. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Retter, A.; Singer, M.; Annane, D. "The NET effect": Neutrophil extracellular traps-a potential key component of the dysregulated host immune response in sepsis. Crit. Care 2025, 29, 59. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Liu, Z. Impact of Neutrophil Extracellular Traps on Thrombosis Formation: New Findings and Future Perspective. Front. Cell. Infect. Microbiol. 2022, 12, 910908. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yu, Y.; Qu, M.; Qiu, Z.; Zhang, H.; Miao, C.; Guo, K. Neutrophil extracellular traps contribute to immunothrombosis formation via the STING pathway in sepsis-associated lung injury. Cell Death Discov. 2023, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Qi, H.; Kan, K.; Chen, J.; Xie, H.; Guo, X.; Zhang, L. Neutrophil Extracellular Traps Promote Hypercoagulability in Patients with Sepsis. Shock 2017, 47, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Ma, X. [Neutrophil extracellular traps and coagulation dysfunction in sepsis]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2017, 29, 752–755. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Qu, M.; Yu, Y.; Chen, Z.; Zhu, S.; Guo, K.; Chen, W.; Miao, C. Tissue Factor-Enriched Neutrophil Extracellular Traps Promote Immunothrombosis and Disease Progression in Sepsis-Induced Lung Injury. Front. Cell. Infect. Microbiol. 2021, 11, 677902. [Google Scholar] [CrossRef]
- Setarehaseman, A.; Mohammadi, A.; Maitta, R.W. Thrombocytopenia in Sepsis. Life 2025, 15, 274. [Google Scholar] [CrossRef]
- Garcia, C.; Compagnon, B.; Poëtte, M.; Gratacap, M.-P.; Lapébie, F.-X.; Voisin, S.; Minville, V.; Payrastre, B.; Vardon-Bounes, F.; Ribes, A. Platelet versus megakaryocyte: Who is the real bandleader of thromboinflammation in sepsis? Cells 2022, 11, 1507. [Google Scholar] [CrossRef]
- Rayes, J.; Bourne, J.H.; Brill, A.; Watson, S.P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res. Pract. Thromb. Haemost. 2020, 4, 23–35. [Google Scholar] [CrossRef]
- Bo, Y.; Lu, Q.; Li, B.; Sha, R.; Yu, H.; Miao, C. The role of platelets in central hubs of inflammation: A literature review. Medicine 2024, 103, e38115. [Google Scholar] [CrossRef]
- Scherlinger, M.; Richez, C.; Tsokos, G.C.; Boilard, E.; Blanco, P. The role of platelets in immune-mediated inflammatory diseases. Nat. Rev. Immunol. 2023, 23, 495–510. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Cox, D. Sepsis—It is all about the platelets. Front. Immunol. 2023, 14, 1210219. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Y.; Tao, Y.; Dang, W.; Yang, B.; Li, Y. The role of platelets in sepsis: A review. Biomol. Biomed. 2024, 24, 741–752. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Hirsch, J.; Uzun, G.; Zlamal, J.; Singh, A.; Bakchoul, T. Platelet-neutrophil interaction in COVID-19 and vaccine-induced thrombotic thrombocytopenia. Front. Immunol. 2023, 14, 1186000. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, P.T. Platelet-leukocyte interactions in COVID-19: Contributions to hypercoagulability, inflammation, and disease severity. Res. Pract. Thromb. Haemost. 2022, 6, e12709. [Google Scholar] [CrossRef]
- Zaid, Y.; Merhi, Y. Implication of Platelets in Immuno-Thrombosis and Thrombo-Inflammation. Front. Cardiovasc. Med. 2022, 9, 863846. [Google Scholar] [CrossRef]
- Williams, B.; Zou, L.; Pittet, J.F.; Chao, W. Sepsis-Induced Coagulopathy: A Comprehensive Narrative Review of Pathophysiology, Clinical Presentation, Diagnosis, and Management Strategies. Anesth. Analg. 2024, 138, 696–711. [Google Scholar] [CrossRef]
- Bitsadze, V.; Lazarchuk, A.; Vorobev, A.; Khizroeva, J.; Tretyakova, M.; Makatsariya, N.; Gashimova, N.; Grigoreva, K.; Tatarintseva, A.; Karpova, A.; et al. Systemic Inflammatory Response Syndrome, Thromboinflammation, and Septic Shock in Fetuses and Neonates. Int. J. Mol. Sci. 2025, 26, 3259. [Google Scholar] [CrossRef]
- Li, C.; Ture, S.K.; Nieves-Lopez, B.; Blick-Nitko, S.K.; Maurya, P.; Livada, A.C.; Stahl, T.J.; Kim, M.; Pietropaoli, A.P.; Morrell, C.N. Thrombocytopenia Independently Leads to Changes in Monocyte Immune Function. Circ. Res. 2024, 134, 970–986. [Google Scholar] [CrossRef]
- Péju, E.; Fouqué, G.; Charpentier, J.; Vigneron, C.; Jozwiak, M.; Cariou, A.; Mira, J.P.; Jamme, M.; Pène, F. Clinical significance of thrombocytopenia in patients with septic shock: An observational retrospective study. J. Crit. Care 2023, 76, 154293. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Peng, T.; Zhang, P.; Cheng, G. Clinical characteristics, and outcomes of severe neonatal thrombocytopenia: A retrospective cohort study in China. BMC Pediatr. 2025, 25, 275. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, M.L.; Schäfer, K. Role of Endothelial Cells in Acute and Chronic Thrombosis. Hamostaseologie 2019, 39, 128–139. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Liles, W.C.; Joffre, J. Editorial: Endothelial activation and microcirculatory disorders in sepsis and critical illness, volume II. Front. Med. 2024, 11, 1477041. [Google Scholar] [CrossRef]
- Dolmatova, E.V.; Wang, K.; Mandavilli, R.; Griendling, K.K. The effects of sepsis on endothelium and clinical implications. Cardiovasc. Res. 2021, 117, 60–73. [Google Scholar] [CrossRef]
- Tang, F.; Zhao, X.L.; Xu, L.Y.; Zhang, J.N.; Ao, H.; Peng, C. Endothelial dysfunction: Pathophysiology and therapeutic targets for sepsis-induced multiple organ dysfunction syndrome. Biomed. Pharmacother. 2024, 178, 117180. [Google Scholar] [CrossRef]
- Chen, K.; Wang, D.; Qian, M.; Weng, M.; Lu, Z.; Zhang, K.; Jin, Y. Endothelial cell dysfunction and targeted therapeutic drugs in sepsis. Heliyon 2024, 10, e33340. [Google Scholar] [CrossRef]
- Leberzammer, J.; von Hundelshausen, P. Chemokines, molecular drivers of thromboinflammation and immunothrombosis. Front. Immunol. 2023, 14, 1276353. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Ning, B.T. Signaling pathways and intervention therapies in sepsis. Signal Transduct. Target. Ther. 2021, 6, 407. [Google Scholar] [CrossRef]
- Yan, M.; Wang, Z.; Qiu, Z.; Cui, Y.; Xiang, Q. Platelet signaling in immune landscape: Comprehensive mechanism and clinical therapy. Biomark. Res. 2024, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, F.P.; Huang, Y.R.; Li, H.D.; Cao, X.Y.; You, Y.; Meng, Z.F.; Sun, K.Y.; Shen, X.Y. Matrine suppresses NLRP3 inflammasome activation via regulating PTPN2/JNK/SREBP2 pathway in sepsis. Phytomedicine 2023, 109, 154574. [Google Scholar] [CrossRef]
- Gedefaw, L.; Ullah, S.; Leung, P.H.M.; Cai, Y.; Yip, S.P.; Huang, C.L. Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells 2021, 10, 916. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.J.; Preston, R.J.S.; O’Neill, L.A.J. Immunothrombosis and the molecular control of tissue factor by pyroptosis: Prospects for new anticoagulants. Biochem. J. 2022, 479, 731–750. [Google Scholar] [CrossRef]
- Potere, N.; Abbate, A.; Kanthi, Y.; Carrier, M.; Toldo, S.; Porreca, E.; Di Nisio, M. Inflammasome Signaling, Thromboinflammation, and Venous Thromboembolism. JACC Basic Transl. Sci. 2023, 8, 1245–1261. [Google Scholar] [CrossRef]
- Luo, X.; Zhao, Y.; Luo, Y.; Lai, J.; Ji, J.; Huang, J.; Chen, Y.; Liu, Z.; Liu, J. Cytosolic mtDNA-cGAS-STING axis contributes to sepsis-induced acute kidney injury via activating the NLRP3 inflammasome. Clin. Exp. Nephrol. 2024, 28, 375–390. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Han, Y.; Qiu, L.; Wu, H.; Song, Z.; Ke, P.; Wu, X. Focus on the cGAS-STING Signaling Pathway in Sepsis and Its Inflammatory Regulatory Effects. J. Inflamm. Res. 2024, 17, 3629–3639. [Google Scholar] [CrossRef]
- Li, Q.; Wu, P.; Du, Q.; Hanif, U.; Hu, H.; Li, K. cGAS-STING, an important signaling pathway in diseases and their therapy. MedComm 2024, 5, e511. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Y.; Zeng, Z. Advances in cGAS-STING Signaling Pathway and Diseases. Front. Cell Dev. Biol. 2022, 10, 800393. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, G.F.; Canobbio, I.; Torti, M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv. Biol. Regul. 2015, 59, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Vardon Bounes, F.; Mujalli, A.; Cenac, C.; Severin, S.; Le Faouder, P.; Chicanne, G.; Gaits-Iacovoni, F.; Minville, V.; Gratacap, M.P.; Payrastre, B. The importance of blood platelet lipid signaling in thrombosis and in sepsis. Adv. Biol. Regul. 2018, 67, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Sun, S.; Chen, Y.; Tian, R.; Chen, E.; Tan, R.; Wang, X.; Liu, Z.; Liu, J.; Qu, H. Immune effects of PI3K/Akt/HIF-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit. Care 2022, 26, 29. [Google Scholar] [CrossRef]
- Li, H.; Shan, W.; Zhao, X.; Sun, W. Neutrophils: Linking Inflammation to Thrombosis and Unlocking New Treatment Horizons. Int. J. Mol. Sci. 2025, 26, 1965. [Google Scholar] [CrossRef]
- Kohli, S.; Shahzad, K.; Jouppila, A.; Holthöfer, H.; Isermann, B.; Lassila, R. Thrombosis and Inflammation-A Dynamic Interplay and the Role of Glycosaminoglycans and Activated Protein, C. Front. Cardiovasc. Med. 2022, 9, 866751. [Google Scholar] [CrossRef]
- Xu, P.; Xin, L.; Xiao, X.; Huang, Y.; Lin, C.; Liu, X.; Wei, H.; Xu, R.; Chen, Y. Neutrophils: As a Key Bridge between Inflammation and Thrombosis. Evid. Based Complement. Alternat Med. 2022, 2022, 1151910. [Google Scholar] [CrossRef]
- Patel, P.; Michael, J.V.; Naik, U.P.; McKenzie, S.E. Platelet FcγRIIA in immunity and thrombosis: Adaptive immunothrombosis. J. Thromb. Haemost. 2021, 19, 1149–1160. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Arroyo, A.B.; de Los Reyes-Garcia, A.M.; Teruel-Montoya, R.; Vicente, V.; Gonzalez-Conejero, R.; Martinez, C. microRNAs in the haemostatic system: More than witnesses of thromboembolic diseases? Thromb. Res. 2018, 166, 1–9. [Google Scholar] [CrossRef]
- Assinger, A.; Chatterjee, M.; McFadyen, J.D. Editorial: Molecular drivers of immunothrombosis. Front. Immunol. 2024, 15, 1385966. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, B.; Fejes, Z.; Pócsi, M.; Kappelmayer, J.; Nagy, B., Jr. Role of sepsis modulated circulating microRNAs. Ejifcc 2019, 30, 128–145. [Google Scholar] [PubMed]
- Erhart, F.; Widhalm, G.; Kiesel, B.; Hackl, M.; Diendorfer, A.; Preusser, M.; Rössler, K.; Thaler, J.; Pabinger, I.; Ay, C.; et al. The plasma miRNome and venous thromboembolism in high-grade glioma: miRNA Sequencing of a nested case-control cohort. J. Cell. Mol. Med. 2024, 28, e18149. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi-Jahromi, S.S.; Aslani, M. Dysregulated miRNAs network in the critical COVID-19: An important clue for uncontrolled immunothrombosis/thromboinflammation. Int. Immunopharmacol. 2022, 110, 109040. [Google Scholar] [CrossRef]
- Pieri, M.; Vayianos, P.; Nicolaidou, V.; Felekkis, K.; Papaneophytou, C. Alterations in Circulating miRNA Levels after Infection with SARS-CoV-2 Could Contribute to the Development of Cardiovascular Diseases: What We Know So Far. Int. J. Mol. Sci. 2023, 24, 2380. [Google Scholar] [CrossRef]
- Canovas-Cervera, I.; Nacher-Sendra, E.; Osca-Verdegal, R.; Dolz-Andres, E.; Beltran-Garcia, J.; Rodriguez-Gimillo, M.; Ferrando-Sanchez, C.; Carbonell, N.; Garcia-Gimenez, J.L. The Intricate Role of Non-Coding RNAs in Sepsis-Associated Disseminated Intravascular Coagulation. Int. J. Mol. Sci. 2023, 24, 2582. [Google Scholar] [CrossRef]
- Bhat, A.A.; Riadi, Y.; Afzal, M.; Bansal, P.; Kaur, H.; Deorari, M.; Ali, H.; Shahwan, M.; Almalki, W.H.; Kazmi, I.; et al. Exploring ncRNA-mediated pathways in sepsis-induced pyroptosis. Pathol. Res. Pract. 2024, 256, 155224. [Google Scholar] [CrossRef]
- Maiese, A.; Scatena, A.; Costantino, A.; Chiti, E.; Occhipinti, C.; La Russa, R.; Di Paolo, M.; Turillazzi, E.; Frati, P.; Fineschi, V. Expression of MicroRNAs in Sepsis-Related Organ Dysfunction: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 9354. [Google Scholar] [CrossRef]
- Colbert, J.F.; Ford, J.A.; Haeger, S.M.; Yang, Y.; Dailey, K.L.; Allison, K.C.; Neudecker, V.; Evans, C.M.; Richardson, V.L.; Brodsky, K.S.; et al. A model-specific role of microRNA-223 as a mediator of kidney injury during experimental sepsis. Am. J. Physiol.-Ren. Physiol. 2017, 313, F553–F559. [Google Scholar] [CrossRef]
- Saadh, M.J.; Saeed, T.N.; Alfarttoosi, K.H.; Sanghvi, G.; Roopashree, R.; Thakur, V.; Lakshmi, L.; Kubaev, A.; Taher, W.M.; Alwan, M.; et al. Exosomes and MicroRNAs: Key modulators of macrophage polarization in sepsis pathophysiology. Eur. J. Med. Res. 2025, 30, 298. [Google Scholar] [CrossRef]
- Formosa, A.; Turgeon, P.; Dos Santos, C.C. Role of miRNA dysregulation in sepsis. Mol. Med. 2022, 28, 99. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, Y.T.; Fan, J. Exosomal mediators in sepsis and inflammatory organ injury: Unraveling the role of exosomes in intercellular crosstalk and organ dysfunction. Mil. Med. Res. 2024, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, K.I.; Sauna, Z.E.; Atreya, C.D. Role of microRNAs in Hemophilia and Thrombosis in Humans. Int. J. Mol. Sci. 2020, 21, 3598. [Google Scholar] [CrossRef]
- Yapijakis, C. The Role of MicroRNAs in Thrombosis. Adv. Exp. Med. Biol. 2021, 1339, 409–414. [Google Scholar] [CrossRef]
- Zapata-Martínez, L.; Águila, S.; de Los Reyes-García, A.M.; Carrillo-Tornel, S.; Lozano, M.L.; González-Conejero, R.; Martínez, C. Inflammatory microRNAs in cardiovascular pathology: Another brick in the wall. Front. Immunol. 2023, 14, 1196104. [Google Scholar] [CrossRef]
- Schiavello, M.; Vizio, B.; Bosco, O.; Pivetta, E.; Mariano, F.; Montrucchio, G.; Lupia, E. Extracellular Vesicles: New Players in the Mechanisms of Sepsis- and COVID-19-Related Thromboinflammation. Int. J. Mol. Sci. 2023, 24, 1920. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Liu, J.; Lv, B.; Chen, F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb. Res. 2016, 137, 211–218. [Google Scholar] [CrossRef]
- Bushra; Ahmed, S.I.; Begum, S.; Maaria; Habeeb, M.S.; Jameel, T.; Khan, A.A. Molecular basis of sepsis: A New insight into the role of mitochondrial DNA as a damage-associated molecular pattern. Mitochondrion 2024, 79, 101967. [Google Scholar] [CrossRef]
- Li, C.; Sun, X.; Yang, X.; Zhang, R.; Chen, J.; Wang, X. miRNA sequencing identifies immune-associated miRNAs and highlights the role of miR-193b-5p in sepsis and septic shock progression. Sci. Rep. 2025, 15, 5323. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, Y.; Zhao, L.; Lv, X.; Du, Y. Molecular mechanisms of Sepsis attacking the immune system and solid organs. Front. Med. 2024, 11, 1429370. [Google Scholar] [CrossRef]
- Ge, J.; Deng, Q.; Zhou, R.; Hu, Y.; Zhang, X.; Zheng, Z. Identification of key biomarkers and therapeutic targets in sepsis through coagulation-related gene expression and immune pathway analysis. Front. Immunol. 2024, 15, 1470842. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, X.; Liu, Z.; Li, X.; Chen, Y.; An, N.; Hu, X. [Unveiling the molecular features and diagnosis and treatment prospects of immunothrombosis via integrated bioinformatics analysis]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2025, 41, 228–235. [Google Scholar] [PubMed]
- Qiu, X.; Nair, M.G.; Jaroszewski, L.; Godzik, A. Deciphering Abnormal Platelet Subpopulations in COVID-19, Sepsis and Systemic Lupus Erythematosus through Machine Learning and Single-Cell Transcriptomics. Int. J. Mol. Sci. 2024, 25, 5941. [Google Scholar] [CrossRef]
- Wu, X.; Yang, J.; Yu, L.; Long, D. Plasma miRNA-223 correlates with risk, inflammatory markers as well as prognosis in sepsis patients. Medicine 2018, 97, e11352. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, Y.; Lin, S.; Li, Y.; Hua, Y.; Zhou, K. Diagnostic significance of microRNAs in sepsis. PLoS ONE 2023, 18, e0279726. [Google Scholar] [CrossRef]
- Ran, X.; Zhang, J.; Wu, Y.; Du, Y.; Bao, D.; Pei, H.; Zhang, Y.; Zhou, X.; Li, R.; Tang, X.; et al. Prognostic gene landscapes and therapeutic insights in sepsis-induced coagulopathy. Thromb. Res. 2024, 237, 1–13. [Google Scholar] [CrossRef]
- Lee, R.H.; Wang, S.; Akerman, M.; Joseph, D. Role of peak D-dimer in predicting mortality and venous thromboembolism in COVID-19 patients. Sci. Prog. 2025, 108, 368504241247982. [Google Scholar] [CrossRef]
- Bahk, J.; Rehman, A.; Ho, K.S.; Narasimhan, B.; Baloch, H.; Zhang, J.; Yip, R.; Lookstein, R.; Steiger, D.J. Predictors of pulmonary embolism in hospitalized patients with COVID-19. Thromb. J. 2023, 21, 73. [Google Scholar] [CrossRef]
- Padilla, S.; Andreo, M.; Marco, P.; Marco-Rico, A.; Ledesma, C.; Fernández-González, M.; García-Abellán, J.; Mascarell, P.; Botella, Á.; Gutiérrez, F.; et al. Enhanced prediction of thrombotic events in hospitalized COVID-19 patients with soluble thrombomodulin. PLoS ONE 2025, 20, e0319666. [Google Scholar] [CrossRef]
- Vaz, C.O.; Hounkpe, B.W.; Oliveira, J.D.; Mazetto, B.; Cardoso Jacintho, B.; Aparecida Locachevic, G.; Henrique De Oliveira Soares, K.; Carlos Silva Mariolano, J.; Castilho de Mesquita, G.; Colombera Peres, K.; et al. MicroRNA 205-5p and COVID-19 adverse outcomes: Potential molecular biomarker and regulator of the immune response. Exp. Biol. Med. 2023, 248, 1024–1033. [Google Scholar] [CrossRef]
- Curtiaud, A.; Iba, T.; Angles-Cano, E.; Meziani, F.; Helms, J. Biomarkers of sepsis-induced coagulopathy: Diagnostic insights and potential therapeutic implications. Ann. Intensive Care 2025, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Ehrenberg, A.; Toska, L.M.; Metz, L.M.; Klier, M.; Krueger, I.; Reusswig, F.; Elvers, M. Molecular Drivers of Platelet Activation: Unraveling Novel Targets for Anti-Thrombotic and Anti-Thrombo-Inflammatory Therapy. Int. J. Mol. Sci. 2020, 21, 7906. [Google Scholar] [CrossRef] [PubMed]
- Vagionas, D.; Papadakis, D.D.; Politou, M.; Koutsoukou, A.; Vasileiadis, I. Thromboinflammation in Sepsis and Heparin: A Review of Literature and Pathophysiology. Vivo 2022, 36, 2542–2557. [Google Scholar] [CrossRef] [PubMed]
- Man, C.; An, Y.; Wang, G.X.; Mao, E.Q.; Ma, L. Recent Advances in Pathogenesis and Anticoagulation Treatment of Sepsis-Induced Coagulopathy. J. Inflamm. Res. 2025, 18, 737–750. [Google Scholar] [CrossRef]
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Semak, V.; Lauková, L.; Fischer, M.B.; Weber, V. Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. Int. J. Mol. Sci. 2022, 23, 1823. [Google Scholar] [CrossRef]
- Murao, S.; Yamakawa, K. A Systematic Summary of Systematic Reviews on Anticoagulant Therapy in Sepsis. J. Clin. Med. 2019, 8, 1869. [Google Scholar] [CrossRef]
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E.; et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2562–2570. [Google Scholar] [CrossRef]
- Berthelsen, R.E.; Ostrowski, S.R.; Bestle, M.H.; Johansson, P.I. Co-administration of iloprost and eptifibatide in septic shock (CO-ILEPSS)-a randomised, controlled, double-blind investigator-initiated trial investigating safety and efficacy. Crit. Care 2019, 23, 301. [Google Scholar] [CrossRef]
- Eisen, D.P.; Leder, K.; Woods, R.L.; Lockery, J.E.; McGuinness, S.L.; Wolfe, R.; Pilcher, D.; Moore, E.M.; Shastry, A.; Nelson, M.R.; et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): A randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir. Med. 2021, 9, 186–195. [Google Scholar] [CrossRef]
- Derhaschnig, U.; Pachinger, C.; Schweeger-Exeli, I.; Marsik, C.; Jilma, B. Blockade of GPIIb/IIIa by eptifibatide and tirofiban does not alter tissue factor induced thrombin generation in human endotoxemia. Thromb. Haemost. 2003, 90, 1054–1060. [Google Scholar] [CrossRef]
- He, W.; Xi, Q.; Cui, H.; Zhang, P.; Huang, R.; Wang, T.; Wang, D. Forsythiaside B ameliorates coagulopathies in a rat model of sepsis through inhibition of the formation of PAD4-dependent neutrophil extracellular traps. Front. Pharmacol. 2022, 13, 1022985. [Google Scholar] [CrossRef] [PubMed]
- Alsabani, M.; Abrams, S.T.; Cheng, Z.; Morton, B.; Lane, S.; Alosaimi, S.; Yu, W.; Wang, G.; Toh, C.H. Reduction of NETosis by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br. J. Anaesth. 2022, 128, 283–293. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, D.C.; Baik, C.H.; Travis, O.K.; White, D.L.; Young, C.M.; Austin Pierce, W.; Shields, C.A.; Poudel, B.; Williams, J.M. NLRP3 inflammasome activation in platelets in response to sepsis. Physiol. Rep. 2019, 7, e14073. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, Y.; Wang, Y.; Proulle, V.; Andreasen, P.A.; Hong, W.; Chen, Z.; Huang, M.; Xu, P. Embelin ameliorated sepsis-induced disseminated intravascular coagulation intensities by simultaneously suppressing inflammation and thrombosis. Biomed. Pharmacother. 2020, 130, 110528. [Google Scholar] [CrossRef]
- Xia, B.T.; Beckmann, N.; Winer, L.K.; Kim, Y.; Goetzman, H.S.; Veile, R.E.; Gulbins, E.; Goodman, M.D.; Nomellini, V.; Caldwell, C.C. Amitriptyline Treatment Mitigates Sepsis-Induced Tumor Necrosis Factor Expression and Coagulopathy. Shock 2019, 51, 356–363. [Google Scholar] [CrossRef]
- Wake, H.; Mori, S.; Liu, K.; Morioka, Y.; Teshigawara, K.; Sakaguchi, M.; Kuroda, K.; Gao, Y.; Takahashi, H.; Ohtsuka, A.; et al. Histidine-Rich Glycoprotein Prevents Septic Lethality through Regulation of Immunothrombosis and Inflammation. EBioMedicine 2016, 9, 180–194. [Google Scholar] [CrossRef]
- Umemura, Y.; Nishida, T.; Yamakawa, K.; Ogura, H.; Oda, J.; Fujimi, S. Anticoagulant therapies against sepsis-induced disseminated intravascular coagulation. Acute Med. Surg. 2023, 10, e884. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, R.; Sun, H.; Liang, Y.; Ma, X. Efficacy and safety of heparin for sepsis-induced disseminated intravascular coagulation (HepSIC): Study protocol for a multicenter randomized controlled trial. Trials 2024, 25, 4. [Google Scholar] [CrossRef]
- Alkan, S.; Şener, A.; Doğan, E.; Yüksel, C.; Yüksel, B. Prophylactic Anticoagulant Treatment Might Have an Anti-inflammatory Effect and Reduce Mortality Rates in Hospitalized COVID-19 Patients? Oman Med. J. 2022, 37, e394. [Google Scholar] [CrossRef]
- Sholzberg, M.; Tang, G.H.; Rahhal, H.; AlHamzah, M.; Kreuziger, L.B.; Áinle, F.N.; Alomran, F.; Alayed, K.; Alsheef, M.; AlSumait, F.; et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. Bmj 2021, 375, n2400. [Google Scholar] [CrossRef] [PubMed]
- Sholzberg, M.; Tang, G.H.; Negri, E.; Rahhal, H.; Kreuziger, L.B.; Pompilio, C.E.; James, P.; Fralick, M.; AlHamzah, M.; Alomran, F.; et al. Coagulopathy of hospitalised COVID-19: A Pragmatic Randomised Controlled Trial of Therapeutic Anticoagulation versus Standard Care as a Rapid Response to the COVID-19 Pandemic (RAPID COVID COAG-RAPID Trial): A structured summary of a study protocol for a randomised controlled trial. Trials 2021, 22, 202. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Liu, J.; Li, A. Effect of Anticoagulant Versus Non-Anticoagulant Therapy on Mortality of Sepsis-Induced Disseminated Intravascular Coagulation: A Systematic Review and Meta-Analysis. Clin. Appl. Thromb. Hemost. 2023, 29, 10760296231157766. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Lin, C.; Lin, X.; Hu, S.; Deng, X.; Xie, L.; Ye, H.; Zhou, F.; Wu, S. Combining ulinastatin with TIENAM improves the outcome of sepsis induced by cecal ligation and puncture in mice by reducing inflammation and regulating immune responses. Int. Immunopharmacol. 2024, 141, 112927. [Google Scholar] [CrossRef]
- Meng, Z.; Huang, H.; Guo, J.; Wang, D.; Tao, X.; Dai, Q.; Bai, Y.; Ma, C.; Huang, L.; Fu, Y.; et al. Promote Sepsis Recovery through the Inhibition of Immunothrombosis via a Combination of Probenecid Nanocrystals and Cefotaxime Sodium. ACS Appl. Mater. Interfaces 2025, 17, 21013–21032. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.; Li, Z.; He, Z.; Yang, X.; Cheng, X.; Peng, Y.; Xue, Q.; Bai, Y.; Zhang, R.; et al. Heparin prevents caspase-11-dependent septic lethality independent of anticoagulant properties. Immunity 2021, 54, 454–467. [Google Scholar] [CrossRef]
- Hao, C.; Sun, M.; Wang, H.; Zhang, L.; Wang, W. Low molecular weight heparins and their clinical applications. Prog. Mol. Biol. Transl. Sci. 2019, 163, 21–39. [Google Scholar] [CrossRef]
- Cardillo, G.; Viggiano, G.V.; Russo, V.; Mangiacapra, S.; Cavalli, A.; Castaldo, G.; Agrusta, F.; Bellizzi, A.; Amitrano, M.; Iannuzzo, M.; et al. Antithrombotic and Anti-Inflammatory Effects of Fondaparinux and Enoxaparin in Hospitalized COVID-19 Patients: The FONDENOXAVID Study. J. Blood Med. 2021, 12, 69–75. [Google Scholar] [CrossRef]
- Keshari, R.S.; Silasi, R.; Popescu, N.I.; Georgescu, C.; Chaaban, H.; Lupu, C.; McCarty, O.J.T.; Esmon, C.T.; Lupu, F. Fondaparinux pentasaccharide reduces sepsis coagulopathy and promotes survival in the baboon model of Escherichia coli sepsis. J. Thromb. Haemost. 2020, 18, 180–190. [Google Scholar] [CrossRef]
- Al-Najjar, B.O.; Saqallah, F.G.; Abbas, M.A.; Al-Hijazeen, S.Z.; Sibai, O.A. P2Y(12) antagonists: Approved drugs, potential naturally isolated and synthesised compounds, and related in-silico studies. Eur. J. Med. Chem. 2022, 227, 113924. [Google Scholar] [CrossRef]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, X.; Wang, X.; Liang, F.; Tang, Y. Glycyrrhizin attenuates caspase-11-dependent immune responses and coagulopathy by targeting high mobility group box 1. Int. Immunopharmacol. 2022, 107, 108713. [Google Scholar] [CrossRef] [PubMed]
- Mitsios, A.; Chrysanthopoulou, A.; Arampatzioglou, A.; Angelidou, I.; Vidali, V.; Ritis, K.; Skendros, P.; Stakos, D. Ticagrelor Exerts Immune-Modulatory Effect by Attenuating Neutrophil Extracellular Traps. Int. J. Mol. Sci. 2020, 21, 3625. [Google Scholar] [CrossRef]
- Liu, Y.S.; Chen, W.L.; Zeng, Y.W.; Li, Z.H.; Zheng, H.L.; Pan, N.; Zhao, L.Y.; Wang, S.; Chen, S.H.; Jiang, M.H.; et al. Isaridin E Protects against Sepsis by Inhibiting Von Willebrand Factor-Induced Endothelial Hyperpermeability and Platelet-Endothelium Interaction. Mar. Drugs 2024, 22, 283. [Google Scholar] [CrossRef]
- Carestia, A.; Davis, R.P.; Grosjean, H.; Lau, M.W.; Jenne, C.N. Acetylsalicylic acid inhibits intravascular coagulation during Staphylococcus aureus-induced sepsis in mice. Blood 2020, 135, 1281–1286. [Google Scholar] [CrossRef]
- Üstündağ, H.; Kalindemirtaş, F.D.; Doğanay, S.; Demir, Ö.; Kurt, N.; Tahir Huyut, M.; Özgeriş, B.; Kariper, İ.A. Enhanced efficacy of resveratrol-loaded silver nanoparticle in attenuating sepsis-induced acute liver injury: Modulation of inflammation, oxidative stress, and SIRT1 activation. Shock 2023, 60, 688–697. [Google Scholar] [CrossRef]
- Malik, R.A.; Liao, P.; Zhou, J.; Hussain, R.; Fredenburgh, J.C.; Hettrick, L.; Revenko, A.S.; Weitz, J.I. Histidine-rich glycoprotein attenuates catheter thrombosis. Blood Adv. 2023, 7, 5651–5660. [Google Scholar] [CrossRef]
- Pradana, A.N.K.; Akahoshi, T.; Guo, J.; Mizuta, Y.; Matsunaga, S.; Narahara, S.; Murata, M.; Yamaura, K. Changes of Histidine-Rich Glycoprotein Levels in Critically Ill Septic Patients. Shock 2024, 62, 351–356. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, X.; Li, Y.; Wu, Y.; Du, Y.; Yang, H.; Liu, Z.; Pei, H.; Li, R.; Luo, H.; et al. Parthenolide improves sepsis-induced coagulopathy by inhibiting mitochondrial-mediated apoptosis in vascular endothelial cells through BRD4/BCL-xL pathway. J. Transl. Med. 2025, 23, 80. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, G.; Wang, Y.; Peng, Y.; Xu, L.; Jiang, T.; Ma, J.; Dong, W.; Chen, C.P. Development of indole derivatives as inhibitors targeting STING-dependent inflammation. Bioorganic Med. Chem. 2025, 126, 118216. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aklilu, A.; Lai, M.S.-L.; Jiang, Z.; Yip, S.P.; Huang, C.-L. Immunothrombosis in Sepsis: Cellular Crosstalk, Molecular Triggers, and Therapeutic Opportunities—A Review. Int. J. Mol. Sci. 2025, 26, 6114. https://doi.org/10.3390/ijms26136114
Aklilu A, Lai MS-L, Jiang Z, Yip SP, Huang C-L. Immunothrombosis in Sepsis: Cellular Crosstalk, Molecular Triggers, and Therapeutic Opportunities—A Review. International Journal of Molecular Sciences. 2025; 26(13):6114. https://doi.org/10.3390/ijms26136114
Chicago/Turabian StyleAklilu, Addis, Michael Siu-Lun Lai, Zhiwei Jiang, Shea Ping Yip, and Chien-Ling Huang. 2025. "Immunothrombosis in Sepsis: Cellular Crosstalk, Molecular Triggers, and Therapeutic Opportunities—A Review" International Journal of Molecular Sciences 26, no. 13: 6114. https://doi.org/10.3390/ijms26136114
APA StyleAklilu, A., Lai, M. S.-L., Jiang, Z., Yip, S. P., & Huang, C.-L. (2025). Immunothrombosis in Sepsis: Cellular Crosstalk, Molecular Triggers, and Therapeutic Opportunities—A Review. International Journal of Molecular Sciences, 26(13), 6114. https://doi.org/10.3390/ijms26136114