
The Perioperative Biochemical and Clinical Considerations of Pheochromocytoma Management

 and
and
Abstract
1. Introduction
2. Genetic and Molecular Insights
2.1. Cluster 1 (Pseudohypoxia)
2.2. Cluster 2 (Kinase Signaling)
2.3. Cluster 3 (Wnt Signaling)
3. Biochemical Considerations
Biochemical Markers and Diagnostic Testing
4. Clinical Considerations
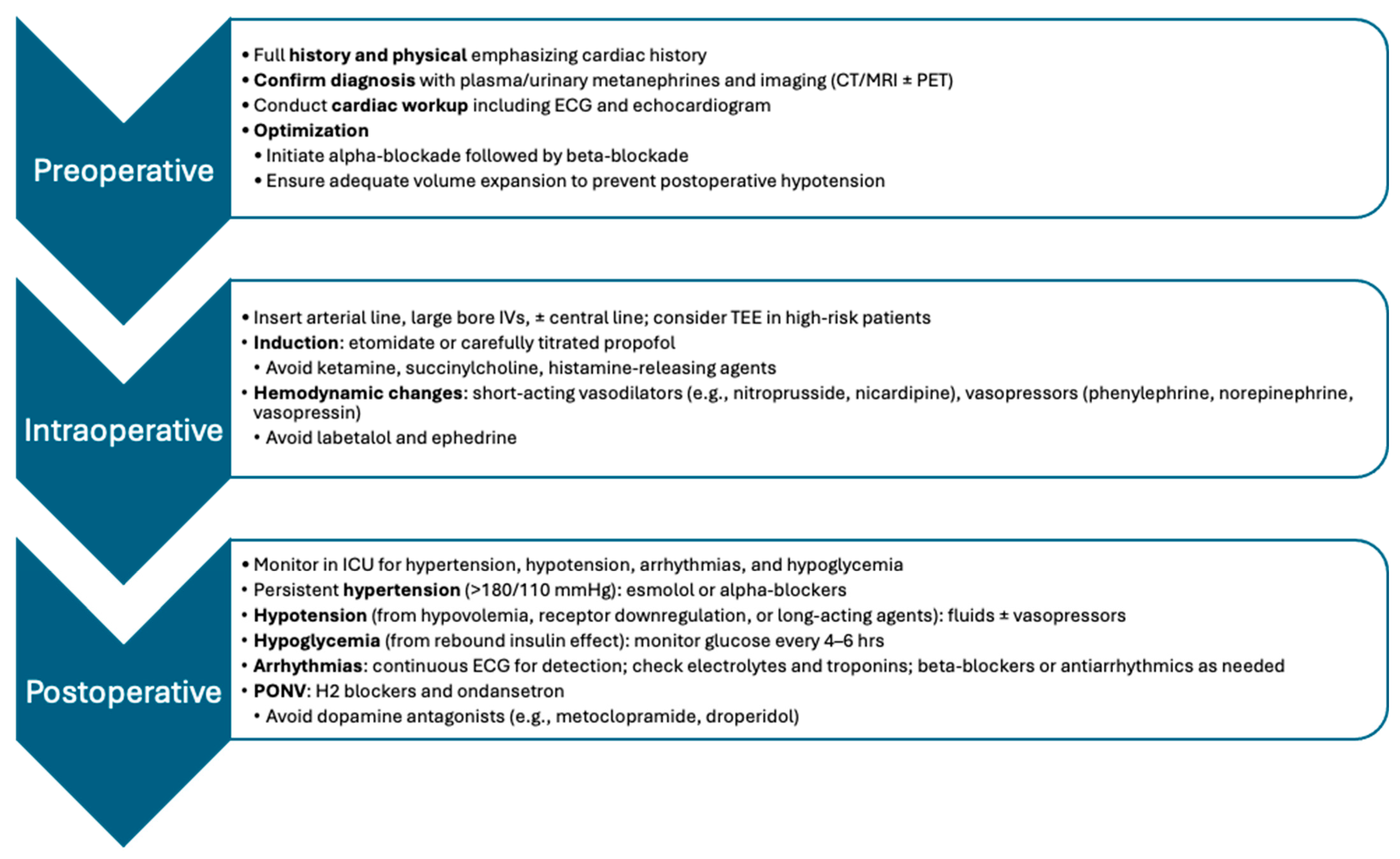
4.1. Preoperative Assessment and Optimization
4.2. Intraoperative Management
4.3. Complications
4.4. Postoperative Care
4.5. Targeted Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Iodine-131 MIBG | I-311-Metaiodbenzylguanidine Therapy |
| Car-T | Chimeric Antigen Receptor T-Cell Therapy |
| CD40 | Cluster of Differentiation 40 |
| CT | Computed Tomography |
| COX4I2 | Cytochrome C Oxidase Subunit 4 Isoform 2 |
| DNA | Deoxyribonucleic Acid |
| ECG | Electrocardiogram |
| HDAC | Histone Deacetylase |
| HIF | Hypoxia-Inducible Factor |
| HIF-2α | Hypoxia-Inducible Factor-2 Alpha |
| IL13 alpha2 | Interleukin 13 Receptor Alpha 2 |
| MRI | Magnetic Resonance Imaging |
| mTOR | Mammalian Target of Rapamycin |
| MBTA Therapy | Mannan-BAM, TLR Ligands, and Anti-CD40 Antibody Therapy |
| Mannan-BAM | Mannan-Conjugated to Biocompatible Anchor for Cell Membrane |
| MIBG | Metaiodobenzylguanidine |
| MEN2 | Multiple Endocrine Neoplasia Type 2 |
| MAX | MYC-Associated Factor X |
| NF1 | Neurofibromatosis Type 1 |
| MGMT | O(6)-Methylguanine-DNA Methyltransferase |
| PVs | Pathogenic Variants |
| PRRT | Peptide Receptor Radionuclide Therapy |
| PARP | Poly (ADP-Ribose) Polymerase |
| RET | Rearranged During Transfection |
| RNA | Ribonucleic acid |
| SDH | Succinate Dehydrogenase |
| SDHA, SDHB, SDHC, SDHD respectively | Succinate Dehydrogenase A, B, C, D |
| TLR | Toll-Like Receptor |
| TMEM127 | Transmembrane Protein 127 |
| TKIs | Tyrosine Kinase Inhibitors |
| VHL | Von Hippel–Lindau Disease |
| Wnt | Wingless and Int-1 Genes |
References
- Maneu, V.; Borges, R.; Gandía, L.; García, A.G. Forty years of the adrenal chromaffin cell through ISCCB meetings around the world. Pflugers Arch. Eur. J. Physiol. 2023, 475, 667–690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prichard, B.N.; Owens, C.W.; Smith, C.C.; Walden, R.J. Heart and catecholamines. Acta Cardiol. 1991, 46, 309–322. [Google Scholar] [PubMed]
- Szatko, A.; Gli nicki, P.; Gietka-Czernel, M. Pheochromocytoma/paraganglioma-associated cardiomyopathy. Front. Endocrinol. 2023, 14, 1204851. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strosberg, J.R. Update on the management of unusual neuroendocrine tumors: Pheochromocytoma and paraganglioma, medullary thyroid cancer and adrenocortical carcinoma. Semin. Oncol. 2013, 40, 120–133. [Google Scholar] [CrossRef]
- Guerrero, M.A.; Schreinemakers, J.M.; Vriens, M.R.; Suh, I.; Hwang, J.; Shen, W.T.; Gosnell, J.; Clark, O.H.; Duh, Q.-Y. Clinical spectrum of pheochromocytoma. J. Am. Coll. Surg. 2009, 209, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Aygun, N.; Uludag, M. Pheochromocytoma and paraganglioma: From epidemiology to clinical findings. SiSli Etfal Hastan Tip. Bulteni/Med. Bull. Sisli Hosp. 2020, 54, 159–168. [Google Scholar] [CrossRef]
- Conzo, G.; Pasquali, D.; Colantuoni, V.; Circelli, L.; Tartaglia, E.; Gambardella, C.; Napolitano, S.; Mauriello, C.; Avenia, N.; Santini, L.; et al. Current concepts of pheochromocytoma. Int. J. Surg. 2014, 12, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, F.-A.; Charalampopoulos, A. Pheochromocytoma. Endocr. Regul. 2019, 53, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Assie, G.; Baudin, E.; Eisenhofer, G.; de la Fouchardiere, C.; Haak, H.; de Krijger, R.; Porpiglia, F.; Terzolo, M.; Berruti, A.; et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1476–1490, Erratum in Ann. Oncol. 2023, 34, 631. [Google Scholar] [CrossRef] [PubMed]
- Oshmyansky, A.R.; Mahammedi, A.; Dackiw, A.; Ball, D.W.; Schulick, R.D.; Zeiger, M.A.; Siegelman, S.S. Serendipity in the diagnosis of pheochromocytoma. J. Comput. Assist. Tomogr. 2013, 37, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, H.; Cerullo, I.; Bartlett, E.K.; Roses, R.E.; Cohen, D.L.; Kelz, R.R.; Karakousis, G.C.; Fraker, D.L. Clinicopathologic characteristics of incidentally identified pheochromocytoma. Ann. Surg. Oncol. 2015, 22, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Pheochromocytoma and Extra-adrenal Paraganglioma: Updates. Arch. Pathol. Lab. Med. 2008, 132, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Erlic, Z.; Neumann, H.P. Diagnosing patients with hereditary paraganglial tumours. Lancet Oncol. 2009, 10, 741. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Wachtel, H.; Fishbein, L. Genetics of pheochromocytoma and paraganglioma. Curr. Opin. Endocrinol. Diabetes Obes 2021, 28, 283–290. [Google Scholar] [CrossRef]
- Jochmanová, I.; Zelinka, T.; Widimský, J.; Pacak, K. HIF Signaling pathway in pheochromocytoma and other neuroendocrine tumors. Physiol. Res. 2014, 63, S251–S262. [Google Scholar] [CrossRef]
- McMahon, J.J.F.-H.K.Q.; Findeis, S.K. Von Hippel-Lindau Disease. J. Pediatr. Genet. 2016, 5, 116–123. [Google Scholar] [CrossRef]
- Taïeb, D.; Nölting, S.; Perrier, N.D.; Fassnacht, M.; Carrasquillo, J.A.; Grossman, A.B.; Clifton-Bligh, R.; Wanna, G.B.; Schwam, Z.G.; Amar, L.; et al. Management of phaeochromocytoma and paraganglioma in patients with germline SDHB pathogenic variants: An international expert Consensus statement. Nat. Rev. Endocrinol. 2024, 20, 168–184. [Google Scholar] [CrossRef]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993, 2, 851–856. [Google Scholar] [CrossRef]
- Fishbein, L. Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2016, 30, 135–150. [Google Scholar] [CrossRef]
- Imai, T.; Uchino, S.; Okamoto, T.; Suzuki, S.; Kosugi, S.; Kikumori, T.; Sakurai, A. High penetrance of pheochromocytoma in multiple endocrine neoplasia 2 caused by germ line RET codon 634 mutation in Japanese patients. Eur. J. Endocrinol. 2013, 168, 683–687. [Google Scholar] [CrossRef]
- Siqueira, D.R.; Ceolin, L.; Ferreira, C.V.; Romitti, M.; Maia, S.C.; Maciel, L.M.Z.; Maia, A.L. Role of RET genetic variants in MEN2-associated pheochromocytoma. Eur. J. Endocrinol. 2014, 170, 821–828. [Google Scholar] [CrossRef]
- Wells, S.A.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised american thyroid association guidelines for the management of medullary thyroid carcinoma. Thyroid® 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Aylsworth, A.; Carey, J.C.; Korf, B.; Marks, J.; Pyeritz, R.E.; Rubenstein, A.; Viskochil, D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997, 278, 51–57. [Google Scholar] [CrossRef]
- Vlenterie, M.; Flucke, U.; Hofbauer, L.C.; Timmers, H.J.; Gastmeier, J.; Aust, D.E.; van der Graaf, W.T.; Wesseling, P.; Eisenhofer, G.; Lenders, J.W. Pheochromocytoma and gastrointestinal stromal tumors in patients with neurofibromatosis type I. Am. J. Med. 2013, 126, 174–180. [Google Scholar] [CrossRef]
- Shinall, M.C.; Solorzano, C.C. Pheochromocytoma in Neurofibromatosis Type 1: When Should it Be Suspected? Endocr. Pract. 2014, 20, 792–796. [Google Scholar] [CrossRef]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. 21 May 2008 [Updated 21 September 2023]. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1548/ (accessed on 15 May 2025).
- Petr, E.J.; Else, T. Pheochromocytoma and Paraganglioma in Neurofibromatosis type 1: Frequent surgeries and cardiovascular crises indicate the need for screening. Clin. Diabetes Endocrinol. 2018, 4, 15. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Toledo, S.P.A.; Lourenço, D.M., Jr.; Sekiya, T.; Lucon, A.M.; Baena, M.E.S.; Castro, C.C.; Bortolotto, L.A.; Zerbini, M.C.; Siqueira, S.A.C.; Toledo, R.A.; et al. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J. Clin. Endocrinol. Metab. 2015, 100, E308–E318. [Google Scholar] [CrossRef]
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef]
- Cascón, A.; Robledo, M. MAX and MYC: A Heritable Breakup. Cancer Res. 2012, 72, 3119–3124. [Google Scholar] [CrossRef]
- Comino-Mendez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef]
- Park, M.J.; Park, S.S.; Lee, B.H.; Jang, J.S.; Kim, W.W.; Kim, S.J.; Lee, Y.-M.; Lee, K.E.; Sung, T.-Y.; Seong, M.-W.; et al. Phenotype of pheochromocytoma and paraganglioma by germline mutation: A Korean multicenter study. Endocr. Relat. Cancer 2025, 32, e240269. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Nölting, S.; Bechmann, N.; Taieb, D.; Beuschlein, F.; Fassnacht, M.; Kroiss, M.; Eisenhofer, G.; Grossman, A.; Pacak, K. Personalized Management of Pheochromocytoma and Paraganglioma. Endocr. Rev. 2022, 43, 199–239. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Li, F.; Zhang, Y.; Gao, X.; Wang, Y.; Zhou, H. The connection between tricarboxylic acid cycle enzyme mutations and pseudohypoxic signaling in pheochromocytoma and paraganglioma. Front. Endocrinol. 2023, 14, 1274239. [Google Scholar] [CrossRef]
- Monteagudo, M.; Calsina, B.; Salazar-Hidalgo, M.E.; Martínez-Montes, Á.M.; Piñeiro-Yáñez, E.; Caleiras, E.; Martín, M.C.; Rodríguez-Perales, S.; Letón, R.; Gil, E.; et al. MAML3-fusions modulate vascular and immune tumour microenvironment and confer high metastatic risk in pheochromocytoma and paraganglioma. Best Pr. Res. Clin. Endocrinol. Metab. 2024, 38, 101931. [Google Scholar] [CrossRef]
- Jordan, D.K.; Patil, S.R.; Divelbiss, J.E.; Vemuganti, S.; Headley, C.; Waziri, M.H.; Gurll, N.J. Cytogenetic abnormalities in tumors of patients with von Hippel-Lindau disease. Cancer Genet. Cytogenet. 1989, 42, 227–241. [Google Scholar] [CrossRef]
- Comino-Méndez, I.; de Cubas, A.A.; Bernal, C.; Álvarez-Escolá, C.; Sánchez-Malo, C.; Ramírez-Tortosa, C.L.; Pedrinaci, S.; Rapizzi, E.; Ercolino, T.; Bernini, G.; et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum. Mol. Genet. 2013, 22, 2169–2176. [Google Scholar] [CrossRef]
- Buffet, A.; Morin, A.; Castro-Vega, L.-J.; Habarou, F.; Lussey-Lepoutre, C.; Letouzé, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.-P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar] [CrossRef]
- Luo, Z.; Yan, X.; Liu, Y.; Nan, F.; Lei, Y.; Ren, Y.; Li, L. Prognostic significance of Ki-67 in assessing the risk of progression, relapse or metastasis in pheochromocytomas and paragangliomas. Ann. Med. 2025, 57, 2478312. [Google Scholar] [CrossRef]
- Celada, L.; Cubiella, T.; San-Juan-Guardado, J.; Martínez, A.S.J.; Valdés, N.; Jiménez-Fonseca, P.; Díaz, I.; Enguita, J.M.; Astudillo, A.; Álvarez-González, E.; et al. Differential HIF2α Protein Expression in Human Carotid Body and Adrenal Medulla under Physiologic and Tumorigenic Conditions. Cancers 2022, 14, 2986. [Google Scholar] [CrossRef]
- Tsang, V.H.M.; Dwight, T.; Benn, D.E.; Meyer-Rochow, G.Y.; Gill, A.J.; Sywak, M.; Sidhu, S.; Veivers, D.; Sue, C.M.; Robinson, B.G.; et al. Overexpression of miR-210 is associated with SDH-related pheochromocytomas, paragangliomas, and gastrointestinal stromal tumours. Endocr. Relat. Cancer 2014, 21, 415–426. [Google Scholar] [CrossRef]
- Ruff, S.M.; Ayabe, R.I.; Malekzadeh, P.; Good, M.L.; Wach, M.M.; Gonzales, M.K.; Tirosh, A.; Nilubol, N.; Pacak, K.; Kebebew, E.; et al. MicroRNA-210 May Be a Preoperative Biomarker of Malignant Pheochromocytomas and Paragangliomas. J. Surg. Res. 2019, 243, 1–7. [Google Scholar] [CrossRef]
- Calsina, B.; Castro-Vega, L.J.; Torres-Pérez, R.; Inglada-Pérez, L.; Currás-Freixes, M.; Roldán-Romero, J.M.; Mancikova, V.; Letón, R.; Remacha, L.; Santos, M.; et al. Integrative multi-omics analysis identifies a prognostic miRNA signature and a targetable miR-21-3p/TSC2/mTOR axis in metastatic pheochromocytoma/paraganglioma. Theranostics 2019, 9, 4946–4958. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Guan, X.; Zhao, J.; Shen, L.; Liu, J. Exosomal double-stranded DNA as a biomarker for the diagnosis and preoperative assessment of pheochromocytoma and paraganglioma. Mol. Cancer 2018, 17, 128. [Google Scholar] [CrossRef]
- Khatami, F.; Reis, L.O.; Ebrahimi, M.; Nasiri, S.; Tavangar, S.M.; Pishkuhi, M.A.; Shafiee, G.; Heshmat, R.; Aghamir, S.M.K. The role of methylation quantification of circulating tumor DNA (ctDNA) as a diagnostic biomarker of Pheochromocytomas (PCCs) and Paragangliomas (PGLs). J. Diabetes Metab. Disord. 2024, 23, 2065–2072. [Google Scholar] [CrossRef]
- Khalil, B.; Rosani, A.; Warrington, S.J. Physiology, Catecholamines. In StatPearls [Internet]; U.S. National Library of Medicine: Bethesda, MD, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507716/ (accessed on 11 December 2024).
- Sawka, A.M.; Jaeschke, R.; Singh, R.J.; Young, W.F. A Comparison of biochemical tests for pheochromocytoma: Measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J. Clin. Endocrinol. Metab. 2003, 88, 553–558. [Google Scholar] [CrossRef]
- Sawka, A.M.; Prebtani, A.P.; Thabane, L.; Gafni, A.; Levine, M.; YoungJr, W.F. A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr. Disord. 2004, 4, 2. [Google Scholar] [CrossRef]
- Pacak, K.; Eisenhofer, G.; Ilias, I. Diagnosis of pheochromocytoma with special emphasis on MEN2 syndrome. Hormones 2009, 8, 111–116. [Google Scholar] [CrossRef]
- Timmers, H.J.L.M.; Taïeb, D.; Pacak, K.; Lenders, J.W.M. Imaging of Pheochromocytomas and Paragangliomas. Endocr. Rev. 2024, 45, 414–434. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Evans, D.B.; Lee, J.E.; Perrier, N.D. Pheochromocytoma: Advances in genetics, diagnosis, localization, and treatment. Hematol. Clin. N. Am. 2007, 21, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Herring, J.; Enquist, E.; Keiser, H.R.; Linehan, W.M. von Recklinghausen’s disease and pheochromocytomas. J. Urol. 1999, 162, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Dluhy, R.G. Pheochromocytoma—Death of an Axiom. N. Engl. J. Med. 2002, 346, 1486–1488. [Google Scholar] [CrossRef] [PubMed]
- Gruber, L.M.; Erickson, D.; Babovic-Vuksanovic, D.; Thompson, G.B.; Young, W.F.; Bancos, I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin. Endocrinol. 2016, 86, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.-P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.M.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Young, W.F., Jr.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 381, 552. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Roizen, M.F.; Horrigan, R.W.; Koike, M.; Eger, I.E., 2nd; Mulroy, M.F.; Frazer, B.; Eger, B.T. A prospective randomized trial of four anesthetic techniques for resection of pheochromocytoma. Anesthesiology 1982, 57, A43. [Google Scholar] [CrossRef]
- Buitenwerf, E.; Osinga, T.E.; Timmers, H.J.L.M.; Lenders, J.W.M.; Feelders, R.A.; Eekhoff, E.M.W.; Haak, H.R.; Corssmit, E.P.M.; Bisschop, P.H.L.T.; Valk, G.D.; et al. Efficacy of α-blockers on hemodynamic control during pheochromocytoma resection: A Randomized controlled trial. J. Clin. Endocrinol. Metab. 2019, 105, 2381–2391. [Google Scholar] [CrossRef]
- García, M.I.D.O.; Palasí, R.; Gómez, R.C.; Marco, J.L.P.; Merino-Torres, J.F. Surgical and Pharmacological Management of Functioning Pheochromocytoma and Paraganglioma. In Paraganglioma: A Multidisciplinary Approach [Internet]; Mariani-Costantini, R., Ed.; Codon Publications: Brisbane, AU, USA, 2019; Chapter 4. [Google Scholar] [CrossRef]
- Butz, J.J.; Weingarten, T.N.; Cavalcante, A.N.; Bancos, I.; Young, W.F.; McKenzie, T.J.; Schroeder, D.R.; Martin, D.P.; Sprung, J. Perioperative hemodynamics and outcomes of patients on metyrosine undergoing resection of pheochromocytoma or paraganglioma. Int. J. Surg. 2017, 46, 1–6. [Google Scholar] [CrossRef]
- Zelinka, T.; Petrák, O.; Turková, H.; Holaj, R.; Štrauch, B.; Kršek, M.; Vránková, A.B.; Musil, Z.; Dušková, J.; Kubinyi, J.; et al. High incidence of cardiovascular complications in pheochromocytoma. Horm. Metab. Res. 2012, 44, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Y-Hassan, S.; Falhammar, H. Cardiovascular Manifestations and Complications of Pheochromocytomas and Paragangliomas. J. Clin. Med. 2020, 9, 2435. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Araujo-Castro, M.; Pascual-Corrales, E.; Chavez, L.N.; Lorca, A.M.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Álvaro, J.L.; Ojeda, C.M.; López, S.R.; Durbán, R.B.; et al. Protocol for presurgical and anesthetic management of pheochromocytomas and sympathetic paragangliomas: A multidisciplinary approach. J. Endocrinol. Investig. 2021, 44, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Bruynzeel, H.; Feelders, R.A.; Groenland, T.H.N.; Meiracker, A.H.v.D.; van Eijck, C.H.J.; Lange, J.F.; de Herder, W.W.; Kazemier, G. Risk factors for hemodynamic instability during surgery for pheochromocytoma. J. Clin. Endocrinol. Metab. 2010, 95, 678–685. [Google Scholar] [CrossRef]
- Kiernan, C.M.; Du, L.; Chen, X.; Broome, J.T.; Shi, C.; Peters, M.F.; Solorzano, C.C. Predictors of hemodynamic instability during surgery for pheochromocytoma. Ann. Surg. Oncol. 2014, 21, 3865–3871. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Araki, S.; Kijima, T.; Waseda, Y.; Komai, Y.; Nakanishi, Y.; Uehara, S.; Yasuda, Y.; Yoshida, S.; Yokoyama, M.; Ishioka, J.; et al. Incidence and predictive factors of hypoglycemia after pheochromocytoma resection. Int. J. Urol. 2018, 26, 273–277. [Google Scholar] [CrossRef]
- Kamath, A.S.; Singh, K. Perioperative Management of Pheochromocytoma. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK589634/ (accessed on 6 July 2023).
- Pacak, K. Preoperative Management of the pheochromocytoma patient. J. Clin. Endocrinol. Metab. 2007, 92, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, J.; Dodd, S.; Martin, Y.N. Perioperative Management of Pheochromocytoma. J. Cardiothorac. Vasc. Anesth. 2017, 31, 1427–1439. [Google Scholar] [CrossRef]
- Atallah, F.; Bastide-Heulin, T.; Soulieé, M.; Crouzil, F.; Galiana, A.; Samii, K.; Virenque, C. Haemodynamic changes during retroperitoneoscopic adrenalectomy for phaeochromocytoma. Br. J. Anaesth. 2001, 86, 731–733. [Google Scholar] [CrossRef]
- Fagundes, G.F.C.; Almeida, M.Q. Perioperative Management of Pheochromocytomas and Sympathetic Paragangliomas. J. Endocr. Soc. 2022, 6, bvac004. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hines, R.L.; Marschall, K.E. Endocrine disease. In Stoelting’s Anesthesia and Co-Existing Disease, 7th ed.; Hines, R.L., Marschall, K.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 392–395. [Google Scholar]
- Jha, A.; Taïeb, D.; Carrasquillo, J.A.; Pryma, D.A.; Patel, M.; Millo, C.; de Herder, W.W.; Del Rivero, J.; Crona, J.; Shulkin, B.L.; et al. High-Specific-Activity-131I-MIBG versus 177Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2021, 27, 2989–2995. [Google Scholar] [CrossRef]
- Takenaka, J.; Watanabe, S.; Abe, T.; Hirata, K.; Uchiyama, Y.; Kimura, R.; Shinohara, N.; Kudo, K. Prognostic value of [18F]FDG-PET prior to [131I]MIBG treatment for pheochromocytoma and paraganglioma (PPGL). Ann. Nucl. Med. 2023, 37, 10–17. [Google Scholar] [CrossRef]
- Hadoux, J.; Favier, J.; Scoazec, J.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Déandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef]
- Sukrithan, V.; Perez, K.; Pandit-Taskar, N.; Jimenez, C. Management of metastatic pheochromocytomas and paragangliomas: When and what. Curr. Probl. Cancer 2024, 51, 101116. [Google Scholar] [CrossRef]
- Del Rivero, J.; Perez, K.; Geyer, S.M.; Khalil, M.F.; Vijendran, A.; Kordaris-Corkill, A.; Shergill, A.; Spencer, K.R.; Soares, H.P.; Lopez, C.D.; et al. Alliance A021804: A prospective, multi-institutional phase II trial evaluating temozolomide versus temozolomide and olaparib for advanced pheochromocytoma and paraganglioma. J. Clin. Oncol. 2023, 41, TPS4191. [Google Scholar] [CrossRef]
- Mak, I.Y.F.; Hayes, A.R.; Khoo, B.; Grossman, A. Peptide Receptor Radionuclide Therapy as a Novel Treatment for Metastatic and Invasive Phaeochromocytoma and Paraganglioma. Neuroendocrinology 2019, 109, 287–298. [Google Scholar] [CrossRef]
- Kolasinska-Ćwikła, A.; Pęczkowska, M.; Ćwikła, J.B.; Michałowska, I.; Pałucki, J.M.; Bodei, L.; Lewczuk-Myślicka, A.; Januszewicz, A. A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. J. Clin. Med. 2019, 8, 952. [Google Scholar] [CrossRef]
- Jaiswal, S.K.; Sarathi, V.; Memon, S.S.; Garg, R.; Malhotra, G.; Verma, P.; Shah, R.; Sehemby, M.K.; Patil, V.A.; Jadhav, S.; et al. 177Lu-DOTATATE therapy in metastatic/inoperable pheochromocytoma-paraganglioma. Endocr. Connect. 2020, 9, 864–873. [Google Scholar] [CrossRef]
- Fischer, A.; Kloos, S.; Maccio, U.; Friemel, J.; Remde, H.; Fassnacht, M.; Pamporaki, C.; Eisenhofer, G.; Timmers, H.J.L.M.; Robledo, M.; et al. Metastatic Pheochromocytoma and Paraganglioma: Somatostatin Receptor 2 Expression, Genetics, and Therapeutic Responses. J. Clin. Endocrinol. Metab. 2023, 108, 2676–2685. [Google Scholar] [CrossRef]
- Gubbi, S.; Al-Jundi, M.; Del Rivero, J.; Jha, A.; Knue, M.; Zou, J.; Turkbey, B.; Carrasquillo, J.A.; Lin, E.; Pacak, K.; et al. Case Report: Primary Hypothyroidism Associated With Lutetium 177-DOTATATE Therapy for Metastatic Paraganglioma. Front. Endocrinol. 2021, 11, 587065. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 2009, 41, 697–702. [Google Scholar] [CrossRef]
- Deschler-Baier, B.; Konda, B.; Massarelli, E.; Hu, M.I.; Wirth, L.J.; Xu, X.; Wright, J.; Clifton-Bligh, R.J. Clinical Activity of Selpercatinib in RET-mutant Pheochromocytoma. J. Clin. Endocrinol. Metab. 2025, 110, e600–e606. [Google Scholar] [CrossRef]
- Belzutifan/MK-6482 for the Treatment of Advanced Pheochromocytoma/Paraganglioma (PPGL), Pancreatic Neuroendocrine Tumor (pNET), Von Hippel-Lindau (VHL) Disease-Associated Tumors, Advanced Gastrointestinal Stromal Tumor (wt GIST), or Solid Tumors With HIF-2α Related Genetic Alterations (MK-6482-015). 2021. Available online: https://clinicaltrials.gov/study/NCT04924075 (accessed on 15 May 2025).
- FDA Approves Merck’s WELIREG (Belzutifan) for the Treatment of Adults and Pediatric Patients 12 Years and Older with Locally Advanced, Unresectable, or Metastatic Pheochromocytoma or Paraganglioma (PPGL). 2025. Available online: https://www.merck.com/news/fda-approves-mercks-welireg-belzutifan-for-the-treatment-of-adults-and-pediatric-patients-12-years-and-older-with-locally-advanced-unresectable-or-metastatic-pheochromocytoma-or-par/ (accessed on 18 June 2025).
- Comandone, A.; Vana, F.; Comandone, T.; Tucci, M. Antiangiogenic Therapy in Clear Cell Renal Carcinoma (CCRC): Pharmacological Basis and Clinical Results. Cancers 2021, 13, 5896. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baudin, E.; Goichot, B.; Berruti, A.; Hadoux, J.; Moalla, S.; Laboureau, S.; Nölting, S.; de la Fouchardière, C.; Kienitz, T.; Deutschbein, T.; et al. Sunitinib for metastatic progressive phaeochromocytomas and paragangliomas: Results from FIRSTMAPPP, an academic, multicentre, international, randomised, placebo-controlled, double-blind, phase 2 trial. Lancet 2024, 403, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Habra, M.A.; Campbell, M.T.; Tamsen, G.; Cruz-Goldberg, D.; Long, J.; Bassett, R.; Dantzer, R.; Balderrama-Brondani, V.; Varghese, J.; et al. Cabozantinib in patients with unresectable and progressive metastatic phaeochromocytoma or paraganglioma (the Natalie Trial): A single-arm, phase 2 trial. Lancet Oncol. 2024, 25, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Jasim, S.; Suman, V.J.; Jimenez, C.; Harris, P.; Sideras, K.; Burton, J.K.; Worden, F.P.; Auchus, R.J.; Bible, K.C. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine 2017, 57, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Nölting, S.; Grossman, A.; Pacak, K. Metastatic Phaeochromocytoma: Spinning Towards More Promising Treatment Options. Exp. Clin. Endocrinol. Diabetes 2019, 127, 117–128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eid, M.; Foukal, J.; Sochorová, D.; Tuček, Š.; Starý, K.; Kala, Z.; Mayer, J.; Němeček, R.; Trna, J.; Kunovský, L. Management of pheochromocytomas and paragangliomas: Review of current diagnosis and treatment options. Cancer Med. 2023, 12, 13942–13957. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ullrich, M.; Richter, S.; Liers, J.; Drukewitz, S.; Friedemann, M.; Kotzerke, J.; Ziegler, C.G.; Nölting, S.; Kopka, K.; Pietzsch, J. Epigenetic drugs in somatostatin type 2 receptor radionuclide theranostics and radiation transcriptomics in mouse pheochromocytoma models. Theranostics 2023, 13, 278–294. [Google Scholar] [CrossRef]
- Kaplinsky, A.; Halperin, R.; Shlomai, G.; Tirosh, A. Role of epigenetic regulation on catecholamine synthesis in pheochromocytoma and paraganglioma. Cancer 2024, 130, 3289–3296. [Google Scholar] [CrossRef]
- Pollard, J.H.; Menda, Y.; Zamba, K.; Madsen, M.; O’Dorisio, M.S.; O’Dorisio, T.; Bushnell, D. Potential for Increasing Uptake of Radiolabeled 68Ga-DOTATOC and 123I-MIBG in Patients with Midgut Neuroendocrine Tumors Using a Histone Deacetylase Inhibitor Vorinostat. Cancer Biotherapy Radiopharm. 2021, 36, 632–641. [Google Scholar] [CrossRef]
- Martiniova, L.; Perera, S.M.; Brouwers, F.M.; Alesci, S.; Abu-Asab, M.; Marvelle, A.F.; Kiesewetter, D.O.; Thomasson, D.; Morris, J.C.; Kvetnansky, R.; et al. Increased uptake of [¹²³I]meta-iodobenzylguanidine, [¹⁸F]fluorodopamine, and [³H]norepinephrine in mouse pheochromocytoma cells and tumors after treatment with the histone deacetylase inhibitors. Endocr Relat Cancer 2011, 18, 143–157. [Google Scholar] [CrossRef]
- Manta, A.; Kazanas, S.; Karamaroudis, S.; Gogas, H.; Ziogas, D.C. Histone deacetylase inhibitors as a novel therapeutic approach for pheochromocytomas and paragangliomas. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2022, 30, 211–219. [Google Scholar] [CrossRef]
- Uher, O.; Vanova, K.H.; Lencova, R.; Frejlachova, A.; Wang, H.; Zhuang, Z.; Zenka, J.; Pacak, K. Identification of Immune Cell Infiltration in Murine Pheochromocytoma during Combined Mannan-BAM, TLR Ligand, and Anti-CD40 Antibody-Based Immunotherapy. Cancers 2021, 13, 1030412. [Google Scholar] [CrossRef]
- Senders, Z.J.; Martin, R.C.G. Intratumoral Immunotherapy and Tumor Ablation: A Local Approach with Broad Potential. Cancers 2022, 14, 1754. [Google Scholar] [CrossRef]
- Lookian, P.P.; Zhao, D.; Medina, R.; Wang, H.; Zenka, J.; Gilbert, M.R.; Pacak, K.; Zhuang, Z. Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3455. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jimenez, C.; Armaiz-Pena, G.; Dahia, P.L.M.; Lu, Y.; Toledo, R.A.; Varghese, J.; Habra, M.A. Endocrine and Neuroendocrine Tumors Special Issue—Checkpoint Inhibitors for Adrenocortical Carcinoma and Metastatic Pheochromocytoma and Paraganglioma: Do They Work? Cancers 2022, 14, 467. [Google Scholar] [CrossRef]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef]
- Colby, C. Another Car T-Cell Clinical Trial Open to Neuroendocrine Cancer Patients. 2024. Available online: https://netrf.org/2024/08/21/another-car-t-cell-clinical-trial-open-to-neuroendocrine-cancer-patients/#:~:text=CAR%20T-cell%20therapies%20work,Adrenocortical%20Carcinoma (accessed on 18 June 2025).
- Fang, L.; Tian, W.; Zhang, C.; Wang, X.; Li, W.; Zhang, Q.; Zhang, Y.; Zheng, J. Oncolytic adenovirus-mediated expression of CCL5 and IL12 facilitates CA9-targeting CAR-T therapy against renal cell carcinoma. Pharmacol. Res. 2023, 189, 106701. [Google Scholar] [CrossRef]
- Ronca, R.; Supuran, C.T. Carbonic anhydrase IX: An atypical target for innovative therapies in cancer. Biochim. et Biophys. Acta (BBA) Rev. Cancer 2024, 1879, 189120. [Google Scholar] [CrossRef]

{kind=link}
| Non-Selective Alpha-Blockers | Selective Alpha-Blockers | |
|---|---|---|
| Drug | Phenoxybenzamine | Doxazosin Prazosin |
| Mechanism | Irreversible blockade of α1 and α2 receptors | Reversible blockade of α1 only |
| Pharmacodynamics | Slower onset Longer half-life | Faster onset Shorter half-life |
| Hemodynamics | Reflex tachycardia (due to α2 blockade) Postoperative hypotension (due to longer half-life) Better intraoperative blood pressure control | Less reflex tachycardia and postoperative hypotension |
| Accessibility | More expensive Lower availability | Less expensive Greater availability |
| Indications | Large, secreting tumors Longer optimization | Shorter surgeries Faster optimization |
| Drug Class | Safe | Avoid |
|---|---|---|
| Antiemetics | H2 Blockers, Ondansetron | Metoclopramide, Droperidol, Amisulpride |
| Induction Agents | Propofol, Etomidate, Fentanyl | Ketamine |
| Neuromuscular Blockers | Vecuronium, Cisatracurium, Rocuronium | Succinylcholine, Pancuronium |
| Inhaled Agents | Enflurane, Isoflurane, Sevoflurane, Nitrous Oxide | Desflurane, Halothane |
| Anti-hypertensives and Beta-Blockers | Dihydropyridine Ca2+ Channel Blockers (Nicardipine, Clevidipine), Sodium Nitroprusside, Nitroglycerine, Diltiazem, Magnesium Sulfate, Phentolamine, Esmolol | Labetalol, Propranolol, Sotalol |
| Vasoactive Agents | Norepinephrine, Vasopressin | Ephedrine |
| Opioids | Hydromorphone, Fentanyl, Remifentanil | Morphine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gombert, A.J.; Nerantzinis, A.M.; Li, J.; Wang, W.; Yeung, I.Y.; Costa, A.; Bergese, S.D. The Perioperative Biochemical and Clinical Considerations of Pheochromocytoma Management. Int. J. Mol. Sci. 2025, 26, 6080. https://doi.org/10.3390/ijms26136080
Gombert AJ, Nerantzinis AM, Li J, Wang W, Yeung IY, Costa A, Bergese SD. The Perioperative Biochemical and Clinical Considerations of Pheochromocytoma Management. International Journal of Molecular Sciences. 2025; 26(13):6080. https://doi.org/10.3390/ijms26136080
Chicago/Turabian StyleGombert, Alexa J., Alexandra M. Nerantzinis, Jennifer Li, Weidong Wang, Isaac Y. Yeung, Ana Costa, and Sergio D. Bergese. 2025. "The Perioperative Biochemical and Clinical Considerations of Pheochromocytoma Management" International Journal of Molecular Sciences 26, no. 13: 6080. https://doi.org/10.3390/ijms26136080
APA StyleGombert, A. J., Nerantzinis, A. M., Li, J., Wang, W., Yeung, I. Y., Costa, A., & Bergese, S. D. (2025). The Perioperative Biochemical and Clinical Considerations of Pheochromocytoma Management. International Journal of Molecular Sciences, 26(13), 6080. https://doi.org/10.3390/ijms26136080