
Cortisol-Induced Chromatin Remodeling and Gene Expression in Skeletal Muscle of Rainbow Trout: Integrative ATAC-Seq and RNA-Seq Analysis

 , , and
, , and
Abstract

1. Introduction
2. Results
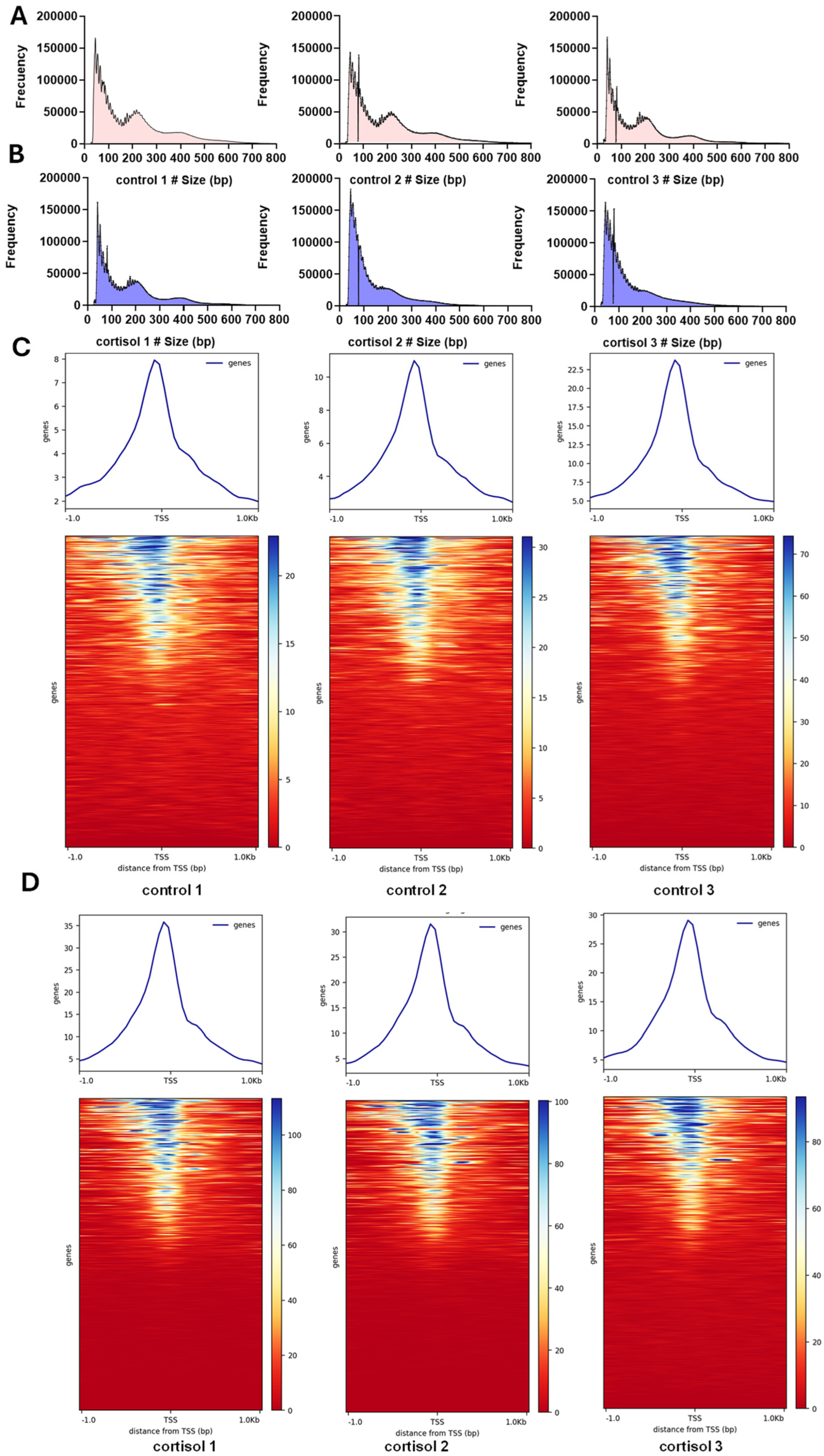
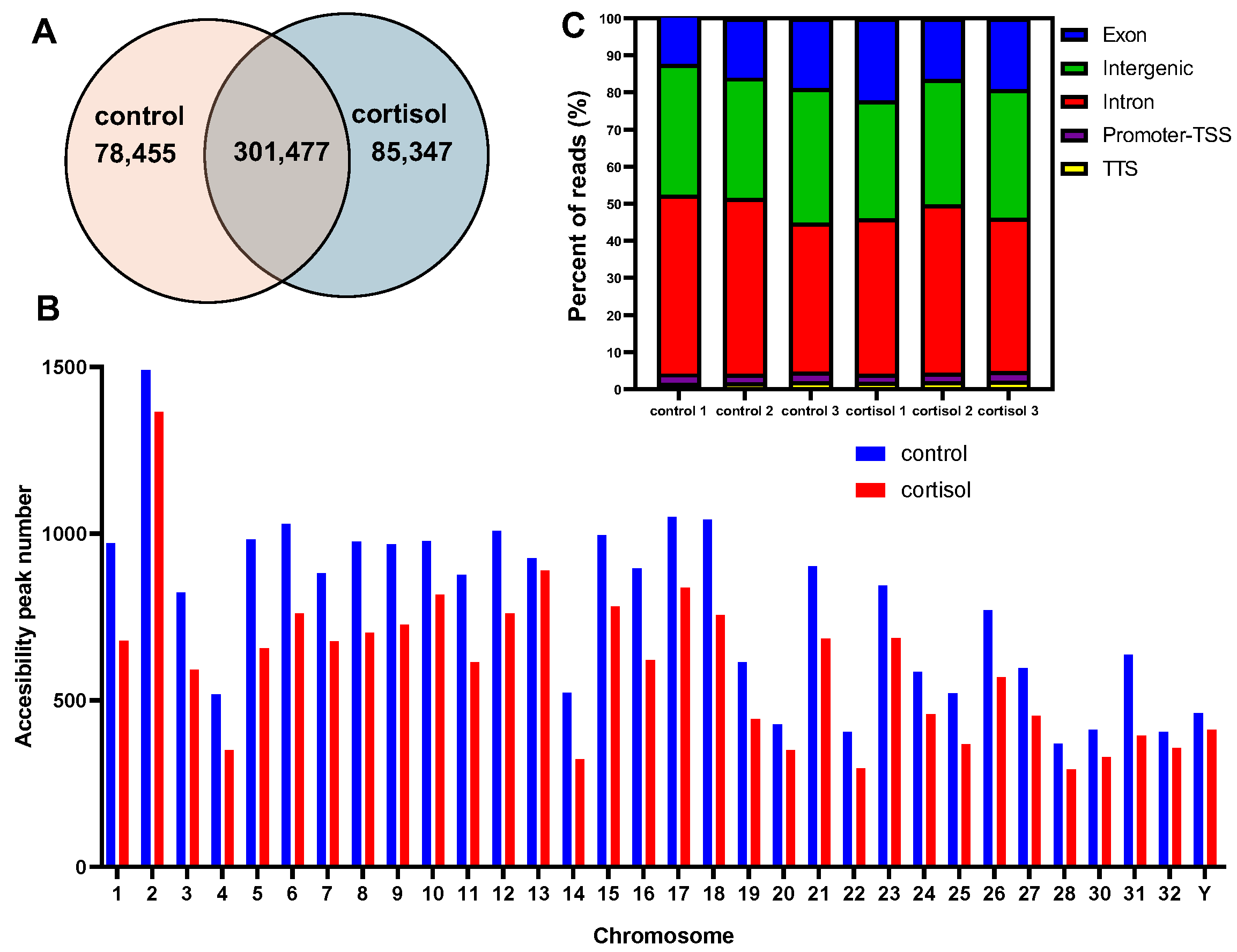
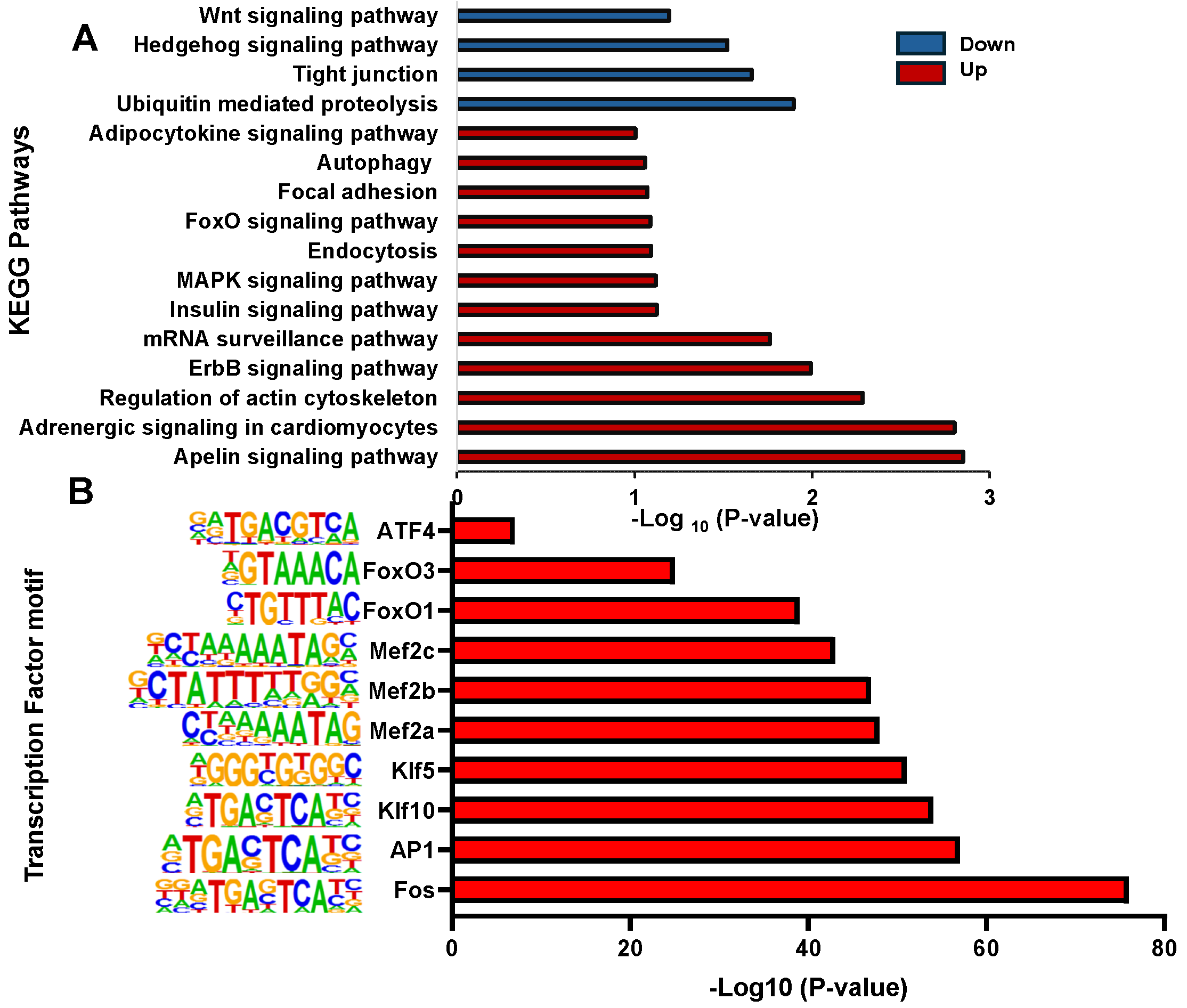
2.1. Analysis of Chromatin Accessibility Induced by Cortisol in Rainbow Trout Skeletal Muscle
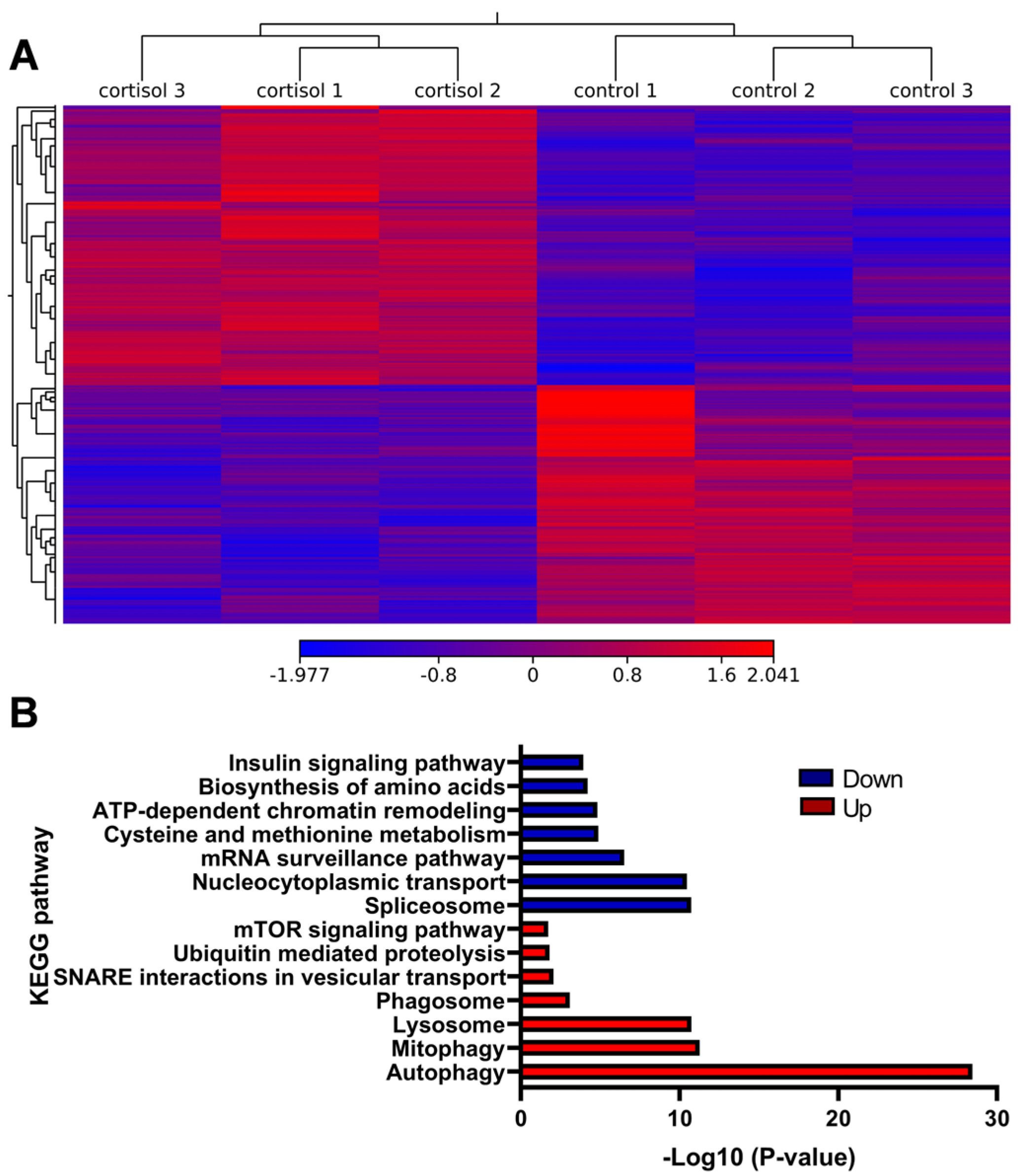
2.2. Analysis of Gene Expression Induced by Cortisol in Rainbow Trout Skeletal Muscle
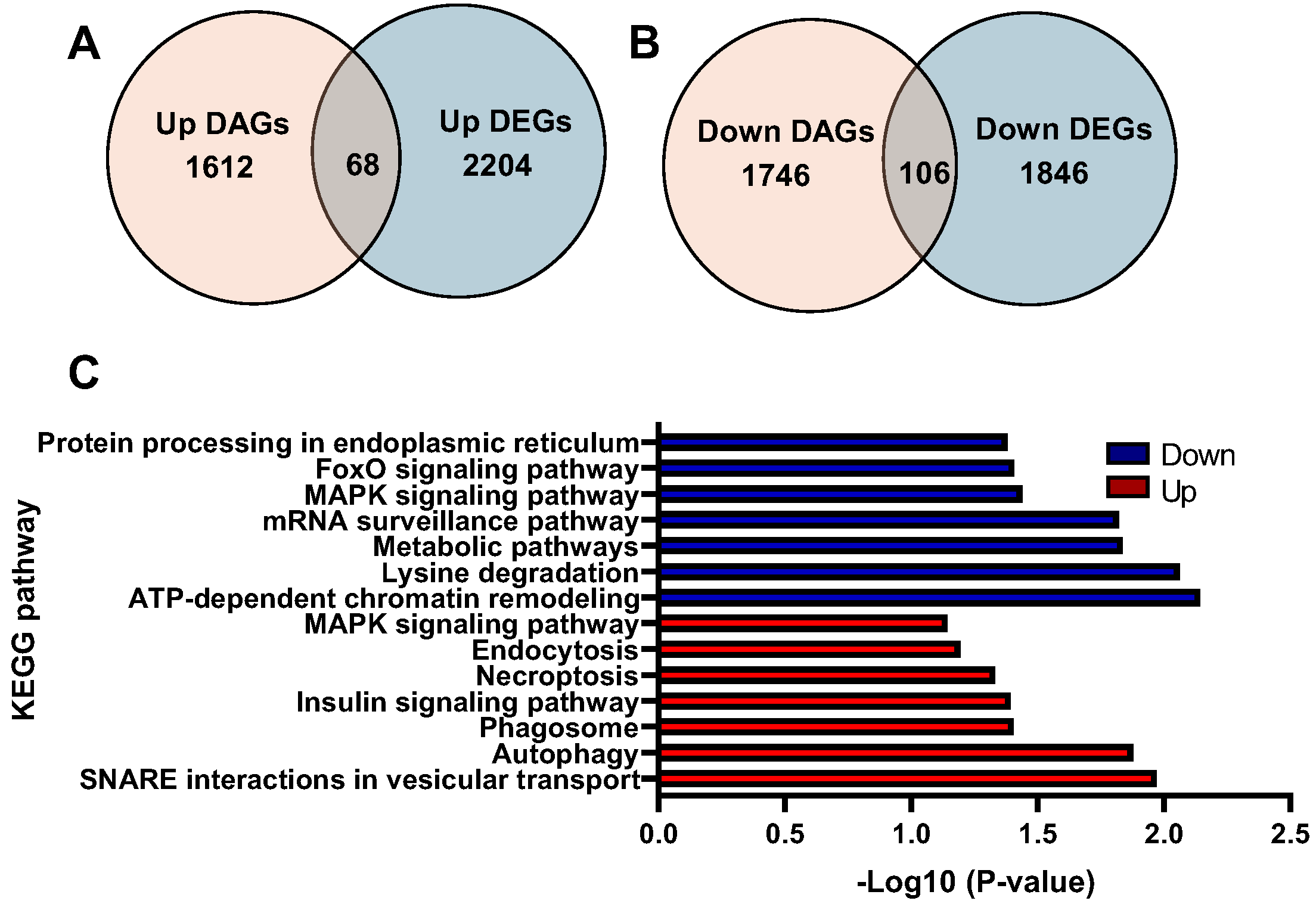
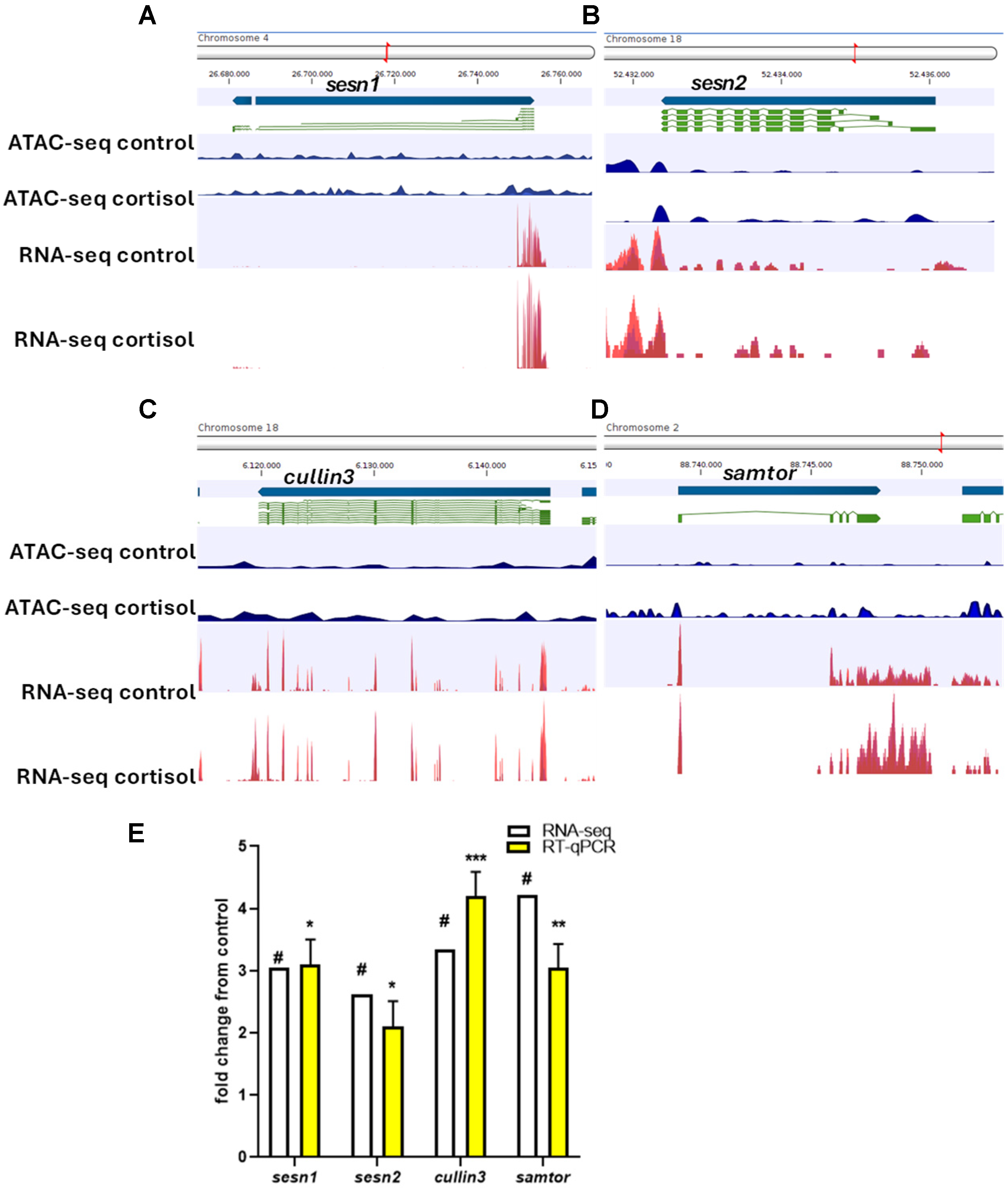
2.3. Integrative Analysis of ATAC-Seq/RNA-Seq and Validation
3. Discussion
4. Materials and Methods
4.1. Experimental Protocol
4.2. ATAC-Seq Library Construction and Sequencing
4.3. ATAC-Seq Analysis
4.4. RNA-Seq Library Construction, Sequencing, and Analysis
4.5. RNA-Seq Analysis
4.6. Real-Time PCR Validation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GR | Glucocorticoid receptor |
| MR | Mineralocorticoid receptor |
| DAGs | Differentially accessed genes |
| DARs | Differentially accessed regions |
| DEGs | Differentially expressed genes |
| qPCR | Real-time PCR |
| GO | Gene ontology |
| RQN | RNA quality number |
References
- Food & Agriculture Organization. Food and Agriculture Organization of the United Nations State of World Fisheries and Aquaculture 2024: Blue Transformation in Action; FAO: Rome, Italy, 2024; ISBN 978-92-5-138763-4. [Google Scholar]
- Ceballos-Concha, A.; Asche, F.; Cárdenas-Retamal, R. Salmon Aquaculture in Chile: Production Growth and Socioeconomic Impacts. Rev. Aquac. 2025, 17, e12993. [Google Scholar] [CrossRef]
- D’Agaro, E.; Gibertoni, P.; Esposito, S. Recent Trends and Economic Aspects in the Rainbow Trout (Oncorhynchus mykiss) Sector. Appl. Sci. 2022, 12, 8773. [Google Scholar] [CrossRef]
- Wind, T.; Schumann, M.; Hofer, S.; Schulz, C.; Brinker, A. Life Cycle Assessment of Rainbow Trout Farming in the Temperate Climate Zone Based on the Typical Farm Concept. J. Clean. Prod. 2022, 380, 134851. [Google Scholar] [CrossRef]
- Food & Agriculture Organization. The State of World Fisheries and Aquaculture 2022: Towards Blue Transformation; FAO: Rome, Italy, 2022; ISBN 978-92-5-136364-5. [Google Scholar]
- Gaffney, L.P.; Lavery, J.M. Research Before Policy: Identifying Gaps in Salmonid Welfare Research that Require Further Study to Inform Evidence-Based Aquaculture Guidelines in Canada. Front. Vet. Sci. 2022, 8, 768558. [Google Scholar] [CrossRef]
- Rehman, S.; Gora, A.H.; Ahmad, I.; Rasool, S.I. Stress in Aquaculture Hatcheries: Source, Impact and Mitigation. Int. J. Curr. Microbiol. App. Sci. 2017, 6, 3030–3045. [Google Scholar] [CrossRef]
- Bonga, S.E.W. The Stress Response in Fish. Physiol. Rev. 1997, 77, 591–625. [Google Scholar] [CrossRef]
- Balasch, J.C.; Tort, L. Netting the Stress Responses in Fish. Front. Endocrinol. 2019, 10, 62. [Google Scholar] [CrossRef]
- Romero, L.M.; Gormally, B.M.G. How Truly Conserved Is the “Well-Conserved” Vertebrate Stress Response? Integr. Comp. Biol. 2019, 59, 273–281. [Google Scholar] [CrossRef]
- Schreck, C.B.; Tort, L. The Concept of Stress in Fish. In Fish Physiology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 35, pp. 1–34. ISBN 978-0-12-802728-8. [Google Scholar]
- Aluru, N.; Vijayan, M.M. Stress Transcriptomics in Fish: A Role for Genomic Cortisol Signaling. Gen. Comp. Endocrinol. 2009, 164, 142–150. [Google Scholar] [CrossRef]
- Faught, E.; Aluru, N.; Vijayan, M.M. The Molecular Stress Response. In Fish Physiology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 35, pp. 113–166. ISBN 978-0-12-802728-8. [Google Scholar]
- Sadoul, B.; Vijayan, M.M. Stress and Growth. In Fish Physiology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 35, pp. 167–205. ISBN 978-0-12-802728-8. [Google Scholar]
- Manneken, J.D.; Dauer, M.V.P.; Currie, P.D. Dynamics of Muscle Growth and Regeneration: Lessons from the Teleost. Exp. Cell Res. 2022, 411, 112991. [Google Scholar] [CrossRef]
- Rossi, G.; Messina, G. Comparative Myogenesis in Teleosts and Mammals. Cell. Mol. Life Sci. 2014, 71, 3081–3099. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.A.; Ponce, C.; Zuloaga, R.; González, P.; Avendaño-Herrera, R.; Valdés, J.A.; Molina, A. Effects of Crowding on the Three Main Proteolytic Mechanisms of Skeletal Muscle in Rainbow Trout (Oncorhynchus mykiss). BMC Vet. Res. 2020, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.A.; Zuloaga, R.; Mercado, L.; Einarsdottir, I.E.; Björnsson, B.T.; Valdés, J.A.; Molina, A. Chronic Stress Inhibits Growth and Induces Proteolytic Mechanisms through Two Different Nonoverlapping Pathways in the Skeletal Muscle of a Teleost Fish. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2018, 314, R102–R113. [Google Scholar] [CrossRef]
- Aravena-Canales, D.; Valenzuela-Muñoz, V.; Gallardo-Escarate, C.; Molina, A.; Valdés, J.A. Transcriptomic and Epigenomic Responses to Cortisol-Mediated Stress in Rainbow Trout (Oncorhynchus mykiss) Skeletal Muscle. Int. J. Mol. Sci. 2024, 25, 7586. [Google Scholar] [CrossRef] [PubMed]
- Aravena-Canales, D.; Valenzuela-Muñoz, V.; Gallardo-Escarate, C.; Molina, A.; Valdés, J.A. Molecular and Epigenetic Responses to Crowding Stress in Rainbow Trout (Oncorhynchus mykiss) Skeletal Muscle. Front. Endocrinol. 2025, 16, 1571111. [Google Scholar] [CrossRef]
- Fuentes, E.N.; Zuloaga, R.; Nardocci, G.; Fernandez De La Reguera, C.; Simonet, N.; Fumeron, R.; Valdes, J.A.; Molina, A.; Alvarez, M. Skeletal Muscle Plasticity Induced by Seasonal Acclimatization in Carp Involves Differential Expression of rRNA and Molecules that Epigenetically Regulate Its Synthesis. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2014, 172–173, 57–66. [Google Scholar] [CrossRef]
- Moghadam, H.K.; Johnsen, H.; Robinson, N.; Andersen, Ø.; Jørgensen, E.H.; Johnsen, H.K.; Bæhr, V.J.; Tveiten, H. Impacts of Early Life Stress on the Methylome and Transcriptome of Atlantic Salmon. Sci. Rep. 2017, 7, 5023. [Google Scholar] [CrossRef]
- Uren Webster, T.M.; Rodriguez-Barreto, D.; Martin, S.A.M.; Van Oosterhout, C.; Orozco-terWengel, P.; Cable, J.; Hamilton, A.; Garcia De Leaniz, C.; Consuegra, S. Contrasting Effects of Acute and Chronic Stress on the Transcriptome, Epigenome, and Immune Response of Atlantic Salmon. Epigenetics 2018, 13, 1191–1207. [Google Scholar] [CrossRef]
- Wellband, K.; Roth, D.; Linnansaari, T.; Curry, R.A.; Bernatchez, L. Environment-Driven Reprogramming of Gamete DNA Methylation Occurs during Maturation and Is Transmitted Intergenerationally in Atlantic Salmon. G3 Genes|Genomes|Genet. 2021, 11, jkab353. [Google Scholar] [CrossRef]
- Ibragić, S.; Dahija, S.; Karalija, E. The Good, the Bad, and the Epigenetic: Stress-Induced Metabolite Regulation and Transgenerational Effects. Epigenomes 2025, 9, 10. [Google Scholar] [CrossRef]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin Accessibility Profiling by ATAC-Seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Miao, N.; Sun, T. Detect Accessible Chromatin Using ATAC-Sequencing, from Principle to Applications. Hereditas 2019, 156, 29. [Google Scholar] [CrossRef] [PubMed]
- Mehta, T.K.; Man, A.; Ciezarek, A.; Ranson, K.; Penman, D.; Di-Palma, F.; Haerty, W. Chromatin Accessibility in Gill Tissue Identifies Candidate Genes and Loci Associated with Aquaculture Relevant Traits in Tilapia. Genomics 2023, 115, 110633. [Google Scholar] [CrossRef]
- Guerrero-Peña, L.; Suarez-Bregua, P.; Gil-Gálvez, A.; Naranjo, S.; Méndez-Martínez, L.; Tur, R.; García-Fernández, P.; Tena, J.J.; Rotllant, J. Genome-Wide Chromatin Accessibility and Gene Expression Profiling during Flatfish Metamorphosis. Sci. Data 2023, 10, 196. [Google Scholar] [CrossRef]
- Jiang, H.; Sun, M.; Zhao, Y.; Liu, G.; Zhong, L.; Xue, H.; Chen, X.; Zheng, Y.; Wang, M. The Early Function of Cortisol in Liver during Aeromonas hydrophila Infection: Dynamics of the Transcriptome and Accessible Chromatin Landscapes. Front. Immunol. 2022, 13, 989075. [Google Scholar] [CrossRef]
- Hartig, E.I.; Zhu, S.; King, B.L.; Coffman, J.A. Chronic Cortisol Exposure in Early Development Leads to Neuroendocrine Dysregulation in Adulthood. BMC Res. Notes 2020, 13, 366. [Google Scholar] [CrossRef]
- Sundaray, J.K.; Dixit, S.; Rather, A.; Rasal, K.D.; Sahoo, L. Aquaculture Omics: An Update on the Current Status of Research and Data Analysis. Mar. Genom. 2022, 64, 100967. [Google Scholar] [CrossRef]
- Cavallino, L.; Rincón, L.; Scaia, M.F. Social Behaviors as Welfare Indicators in Teleost Fish. Front. Vet. Sci. 2023, 10, 1050510. [Google Scholar] [CrossRef]
- Aguiar, D.; Marques, C.; Pereira, A.C. The Importance of Monitoring Cortisol in the Agri-Food Sector—A Systematic Review. Metabolites 2023, 13, 692. [Google Scholar] [CrossRef]
- Das, C.; Thraya, M.; Vijayan, M.M. Nongenomic Cortisol Signaling in Fish. Gen. Comp. Endocrinol. 2018, 265, 121–127. [Google Scholar] [CrossRef]
- Faught, E.; Vijayan, M.M. Mechanisms of Cortisol Action in Fish Hepatocytes. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 199, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Aedo, J.E.; Zuloaga, R.; Bastías-Molina, M.; Meneses, C.; Boltaña, S.; Molina, A.; Valdés, J.A. Early Transcriptomic Responses Associated with the Membrane-Initiated Action of Cortisol in the Skeletal Muscle of Rainbow Trout (Oncorhynchus mykiss). Physiol. Genom. 2019, 51, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Aedo, J.E.; Zuloaga, R.; Aravena-Canales, D.; Molina, A.; Valdés, J.A. Role of Glucocorticoid and Mineralocorticoid Receptors in Rainbow Trout (Oncorhynchus mykiss) Skeletal Muscle: A Transcriptomic Perspective of Cortisol Action. Front. Physiol. 2023, 13, 1048008. [Google Scholar] [CrossRef]
- Bothe, M.; Buschow, R.; Meijsing, S.H. Glucocorticoid Signaling Induces Transcriptional Memory and Universally Reversible Chromatin Changes. Life Sci. Alliance 2021, 4, e202101080. [Google Scholar] [CrossRef]
- Oh, K.-S.; Patel, H.; Gottschalk, R.A.; Lee, W.S.; Baek, S.; Fraser, I.D.C.; Hager, G.L.; Sung, M.-H. Anti-Inflammatory Chromatinscape Suggests Alternative Mechanisms of Glucocorticoid Receptor Action. Immunity 2017, 47, 298–309.e5. [Google Scholar] [CrossRef]
- Stavreva, D.A.; Coulon, A.; Baek, S.; Sung, M.-H.; John, S.; Stixova, L.; Tesikova, M.; Hakim, O.; Miranda, T.; Hawkins, M.; et al. Dynamics of Chromatin Accessibility and Long-Range Interactions in Response to Glucocorticoid Pulsing. Genome Res. 2015, 25, 845–857. [Google Scholar] [CrossRef]
- Jiao, J.P.; Qiao, T.F.; Huang, D.D.; Zhu, Z.X.; De Liu, T.; Xia, J.H. Genome-Wide Chromatin Accessibility Profiles in Spleen of GIFT Strain of Nile Tilapia (Oreochromis niloticus) in Response to Streptococcus agalactiae Infection as Revealed by ATAC-Seq and RNA-Seq. Aquaculture 2025, 598, 742079. [Google Scholar] [CrossRef]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-Seq and RNA-Seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Li, J.; Chen, Z.; Bai, Y.; Wei, Y.; Guo, D.; Liu, Z.; Niu, Y.; Shi, B.; Zhang, X.; Cai, Y.; et al. Integration of ATAC-Seq and RNA-Seq Analysis to Identify Key Genes in the Longissimus Dorsi Muscle Development of the Tianzhu White Yak. Int. J. Mol. Sci. 2023, 25, 158. [Google Scholar] [CrossRef]
- Lee, J.-W.; Cho, J.-Y. Comparative Epigenetics of Domestic Animals: Focusing on DNA Accessibility and Its Impact on Gene Regulation and Traits. J. Vet. Sci. 2025, 26, e9. [Google Scholar] [CrossRef]
- Hou, X.; Niu, N.; Liu, X.; Wang, L.; Wang, L.; Zhang, L. Dynamic Changes in Chromatin Accessibility and Gene Expression Involved in Fetal Myogenesis of Min Pig. Anim. Biosci. 2025. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Z.; Zhang, Z.; Pan, H.; Li, H.; Li, X.; An, Q.; Cheng, Y.; Chen, S.; Man, C.; et al. Profiling Chromatin Accessibility Responses in Goat Bronchial Epithelial Cells Infected with Pasteurella multocida. Int. J. Mol. Sci. 2023, 24, 1312. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Bullard, S.A.; Basisty, N.; Marcotte, G.R.; Skopec, Z.P.; Dierdorff, J.M.; Al-Zougbi, A.; Tomcheck, K.C.; DeLau, A.D.; Rathmacher, J.A.; et al. Activating Transcription Factor 4 (ATF4) Promotes Skeletal Muscle Atrophy by Forming a Heterodimer with the Transcriptional Regulator C/EBPβ. J. Biol. Chem. 2020, 295, 2787–2803. [Google Scholar] [CrossRef] [PubMed]
- Gopal Krishnan, P.D.; Lee, W.X.; Goh, K.Y.; Choy, S.M.; Turqueza, L.R.R.; Lim, Z.H.; Tang, H.-W. Transcriptional Regulation of Autophagy in Skeletal Muscle Stem Cells. Dis. Models Mech. 2025, 18, DMM052007. [Google Scholar] [CrossRef]
- Fuentes, E.N.; Ruiz, P.; Valdes, J.A.; Molina, A. Correction: Catabolic Signaling Pathways, Atrogenes, and Ubiquitinated Proteins Are Regulated by the Nutritional Status in the Muscle of the Fine Flounder. PLoS ONE 2020, 15, e0244410. [Google Scholar] [CrossRef]
- Torres-Velarde, J.; Llera-Herrera, R.; García-Gasca, T.; García-Gasca, A. Mechanisms of Stress-Related Muscle Atrophy in Fish: An Ex Vivo Approach. Mech. Dev. 2018, 154, 162–169. [Google Scholar] [CrossRef]
- Liu, L.; Koike, H.; Ono, T.; Hayashi, S.; Kudo, F.; Kaneda, A.; Kagechika, H.; Manabe, I.; Nakashima, T.; Oishi, Y. Identification of a KLF5-Dependent Program and Drug Development for Skeletal Muscle Atrophy. Proc. Natl. Acad. Sci. USA 2021, 118, e2102895118. [Google Scholar] [CrossRef]
- Zhu, X.; Hu, J.; Zhang, J.; Liu, J.; Bao, L.; Pan, Y.; Cheng, J.; Chu, W. Effect of Short-Term Fasting and Glucocorticoids on KLF15 Expression and Branched-Chain Amino Acids Metabolism in Chinese Perch. Aquac. Rep. 2021, 19, 100617. [Google Scholar] [CrossRef]
- Liu, Q.; Tang, X.; Yang, B.; Hao, T.; Han, S.; Xu, X.; Zhao, Z.; Lai, W.; Li, Y.; Du, J.; et al. Autophagy and Endoplasmic Reticulum Stress-Related Protein Homeostasis Links Palmitic Acid to Hepatic Lipotoxicity in Zebrafish (Danio rerio), Counteracted by Linoleic Acid. Free. Radic. Biol. Med. 2025, 233, 148–161. [Google Scholar] [CrossRef]
- He, L.; Cho, S.; Blenis, J. mTORC1, the Maestro of Cell Metabolism and Growth. Genes Dev. 2025, 39, 109–131. [Google Scholar] [CrossRef]
- Tang, H.; Inoki, K.; Brooks, S.V.; Okazawa, H.; Lee, M.; Wang, J.; Kim, M.; Kennedy, C.L.; Macpherson, P.C.D.; Ji, X.; et al. mTORC1 Underlies Age-related Muscle Fiber Damage and Loss by Inducing Oxidative Stress and Catabolism. Aging Cell 2019, 18, e12943. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR Is an S-Adenosylmethionine Sensor for the mTORC1 Pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Mechanism of Activation of Mechanistic Target of Rapamycin Complex 1 by Methionine. Front. Cell Dev. Biol. 2020, 8, 715. [Google Scholar] [CrossRef]
- Fang, H.; Shi, X.; Wan, J.; Zhong, X. Role of Sestrins in Metabolic and Aging-Related Diseases. Biogerontology 2024, 25, 9–22. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, M. Muscular Sestrins: Roles in Exercise Physiology and Stress Resistance. Biomolecules 2023, 13, 722. [Google Scholar] [CrossRef]
- De Souza, S.S.; Da Costa, J.C.; Da Silva, G.S.; De Almeida-Val, V.M.F. Malathion Alters the Transcription of Target Genes of the Tumour Suppressor Tp53 and Cancerous Processes in Colossoma Macropomum: Mechanisms of DNA Damage Response, Oxidative Stress and Apoptosis. Chem.-Biol. Interact. 2023, 374, 110405. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, X.; Huang, Y.; Liu, S.; Niu, Y.; Fu, L. Resistance Exercise Alleviates Dexamethasone-Induced Muscle Atrophy via Sestrin2/MSTN Pathway in C57BL/6J Mice. Exp. Cell Res. 2023, 432, 113779. [Google Scholar] [CrossRef]
- Hwang, J.; Lauinger, L.; Kaiser, P. Distinct Stress Regulators in the CRL Family: Emerging Roles of F-Box Proteins: Cullin-RING Ligases and Stress-Sensing. BioEssays 2025, 47, e202400249. [Google Scholar] [CrossRef]
- Blondelle, J.; Biju, A.; Lange, S. The Role of Cullin-RING Ligases in Striated Muscle Development, Function, and Disease. Int. J. Mol. Sci. 2020, 21, 7936. [Google Scholar] [CrossRef]
- Tamai, S.; Fujita, S.; Komine, R.; Kanki, Y.; Aoki, K.; Watanabe, K.; Takekoshi, K.; Sugasawa, T. Acute Cold Stress Induces Transient MuRF1 Upregulation in the Skeletal Muscle of Zebrafish. Biochem. Biophys. Res. Commun. 2022, 608, 59–65. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Zweig, A.S.; Barber, G.; Hinrichs, A.S.; Karolchik, D. BigWig and BigBed: Enabling Browsing of Large Distributed Datasets. Bioinformatics 2010, 26, 2204–2207. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Number of Reads | Avg. Length | Number of Reads After Trimming | Avg. Length After Trimming | Mapping Rate |
|---|---|---|---|---|---|
| Control 1 | 92,778,260 | 151 | 92,704,676 | 100 | 94.4 |
| Control 2 | 91,565,508 | 151 | 91,452,112 | 100 | 93.2 |
| Control 3 | 90,996,060 | 151 | 90,891,586 | 100 | 95.3 |
| Cortisol 1 | 92,689,336 | 151 | 92,589,652 | 100 | 93.2 |
| Cortisol 2 | 92,413,506 | 151 | 92,300,120 | 100 | 93.5 |
| Cortisol 3 | 90,648,416 | 151 | 90,536,442 | 100 | 94.8 |
| Average/total | 551,091,086 | 151 | 550,474,588 | 100 | 94.1 |
| Name | Number of Reads | Avg. Length | Number of Reads After Trimming | Avg. Length After Trimming | Mapping Rate |
|---|---|---|---|---|---|
| Control 1 | 54,417,314 | 101 | 54,404,316 | 95.7 | 88.9 |
| Control 2 | 61,509,806 | 101 | 61,498,920 | 95.6 | 87.3 |
| Control 3 | 48,703,772 | 101 | 48,690,750 | 94.6 | 86.4 |
| Cortisol 1 | 53,524,650 | 101 | 53,514,862 | 95.3 | 87.1 |
| Cortisol 2 | 58,841,502 | 101 | 58,826,215 | 94.7 | 89.3 |
| Cortisol 3 | 58,696,858 | 101 | 58,688,933 | 95.2 | 88.6 |
| Total/average | 335,693,902 | 101 | 335,623,996 | 95.5 | 87.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuloaga, R.; Garrido, C.; Ahumada-Langer, L.; Galaz, J.L.; Ugarte, G.D.; Molina, A.; Valdés, J.A. Cortisol-Induced Chromatin Remodeling and Gene Expression in Skeletal Muscle of Rainbow Trout: Integrative ATAC-Seq and RNA-Seq Analysis. Int. J. Mol. Sci. 2025, 26, 6079. https://doi.org/10.3390/ijms26136079
Zuloaga R, Garrido C, Ahumada-Langer L, Galaz JL, Ugarte GD, Molina A, Valdés JA. Cortisol-Induced Chromatin Remodeling and Gene Expression in Skeletal Muscle of Rainbow Trout: Integrative ATAC-Seq and RNA-Seq Analysis. International Journal of Molecular Sciences. 2025; 26(13):6079. https://doi.org/10.3390/ijms26136079
Chicago/Turabian StyleZuloaga, Rodrigo, Camila Garrido, Luciano Ahumada-Langer, José Luis Galaz, Giorgia Daniela Ugarte, Alfredo Molina, and Juan Antonio Valdés. 2025. "Cortisol-Induced Chromatin Remodeling and Gene Expression in Skeletal Muscle of Rainbow Trout: Integrative ATAC-Seq and RNA-Seq Analysis" International Journal of Molecular Sciences 26, no. 13: 6079. https://doi.org/10.3390/ijms26136079
APA StyleZuloaga, R., Garrido, C., Ahumada-Langer, L., Galaz, J. L., Ugarte, G. D., Molina, A., & Valdés, J. A. (2025). Cortisol-Induced Chromatin Remodeling and Gene Expression in Skeletal Muscle of Rainbow Trout: Integrative ATAC-Seq and RNA-Seq Analysis. International Journal of Molecular Sciences, 26(13), 6079. https://doi.org/10.3390/ijms26136079