
Phenotypic Plasticity and Androgen Receptor Bypass Drive Cross-Resistance to Apalutamide in Castration-Resistant Prostate Cancer Cell Models

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
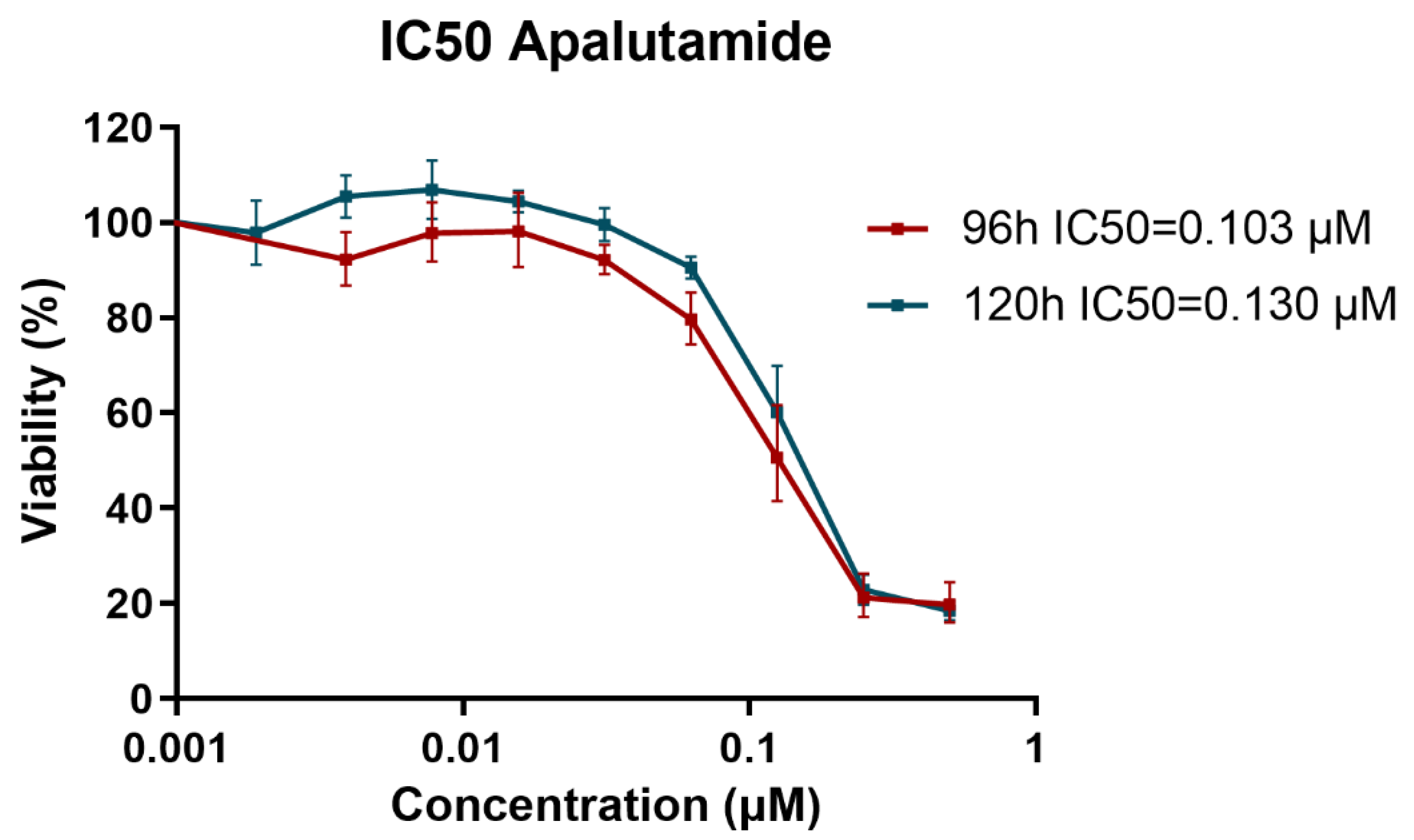
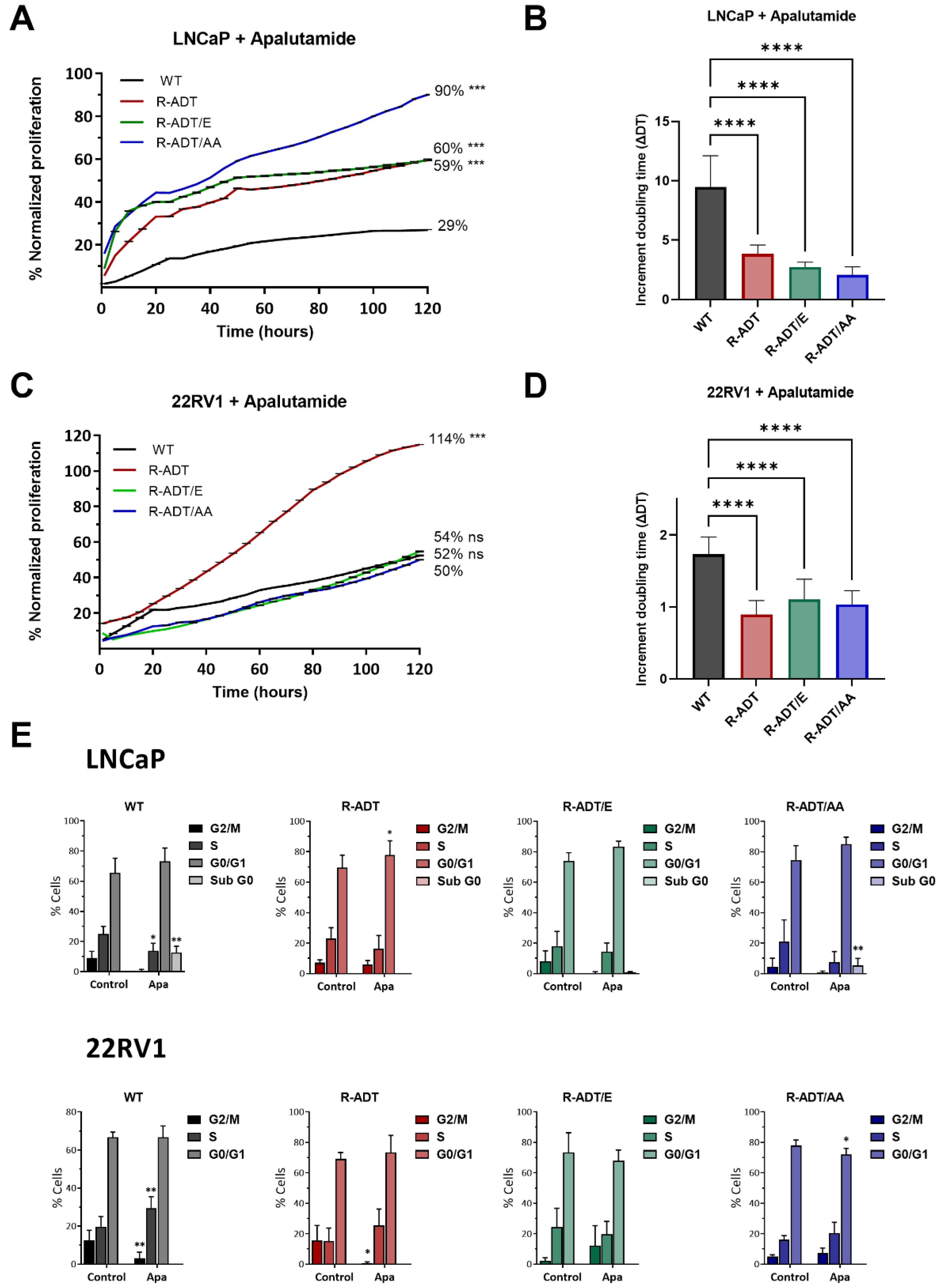
2.1. Proliferation Analysis of CRPC Models in Response to Apalutamide
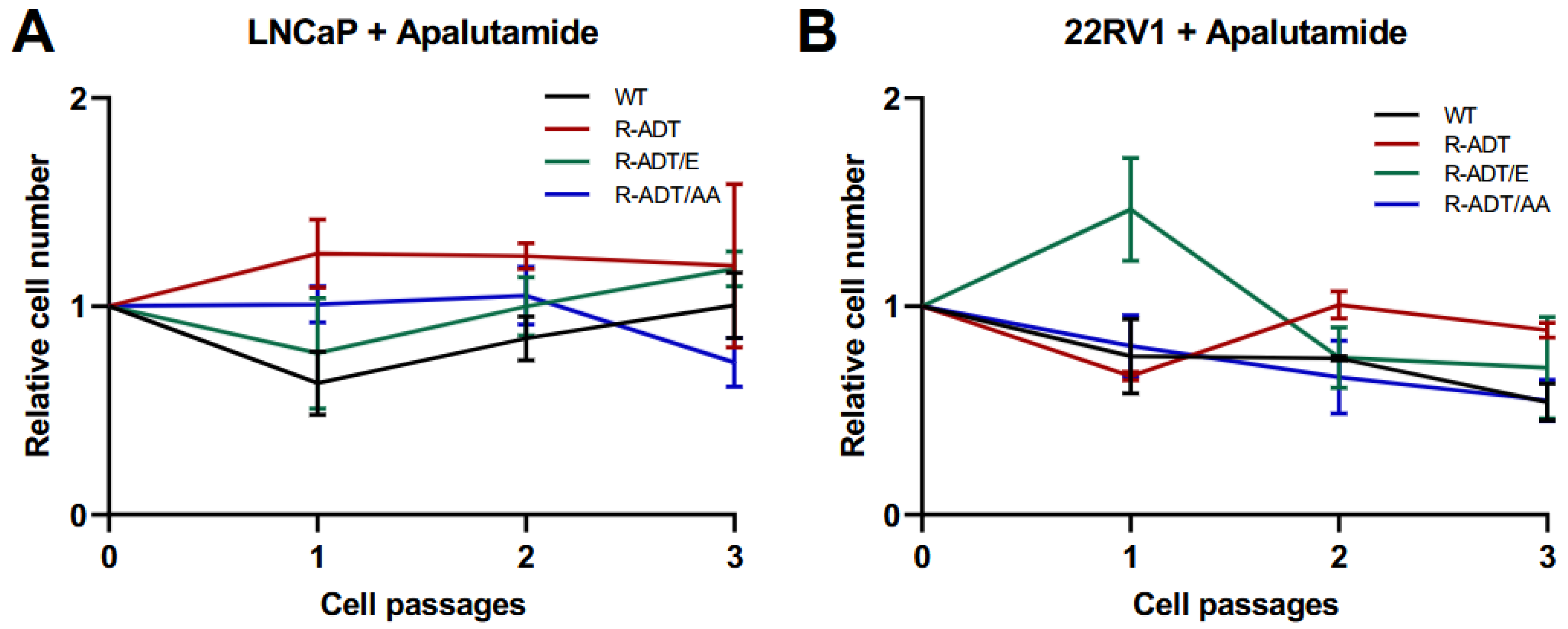
2.2. Colony-Forming Capability Analysis of CRPC Models in Response to Apalutamide
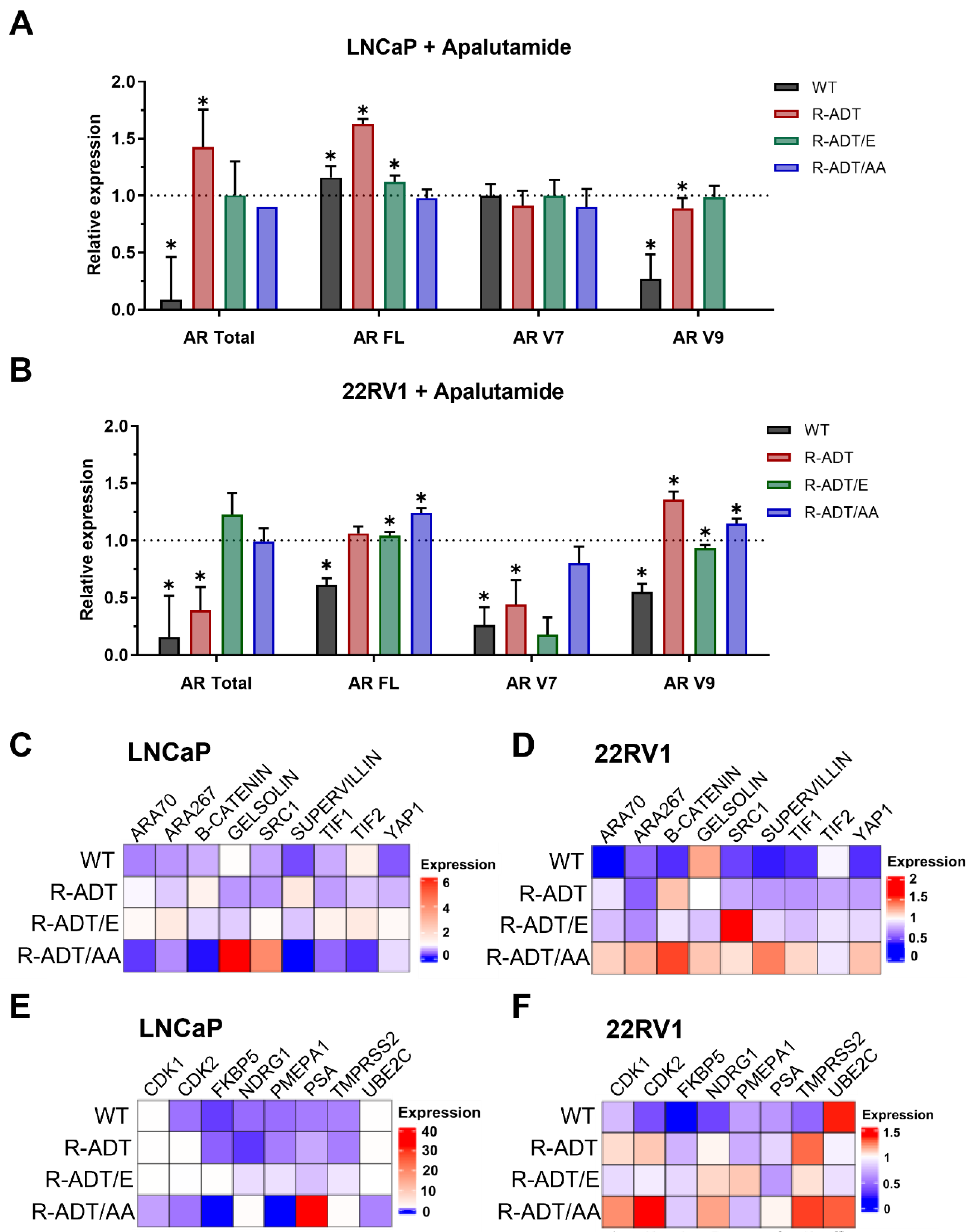
2.3. Gene Expression Analysis of AR Signaling Axis in CRPC Models in Response to Apalutamide
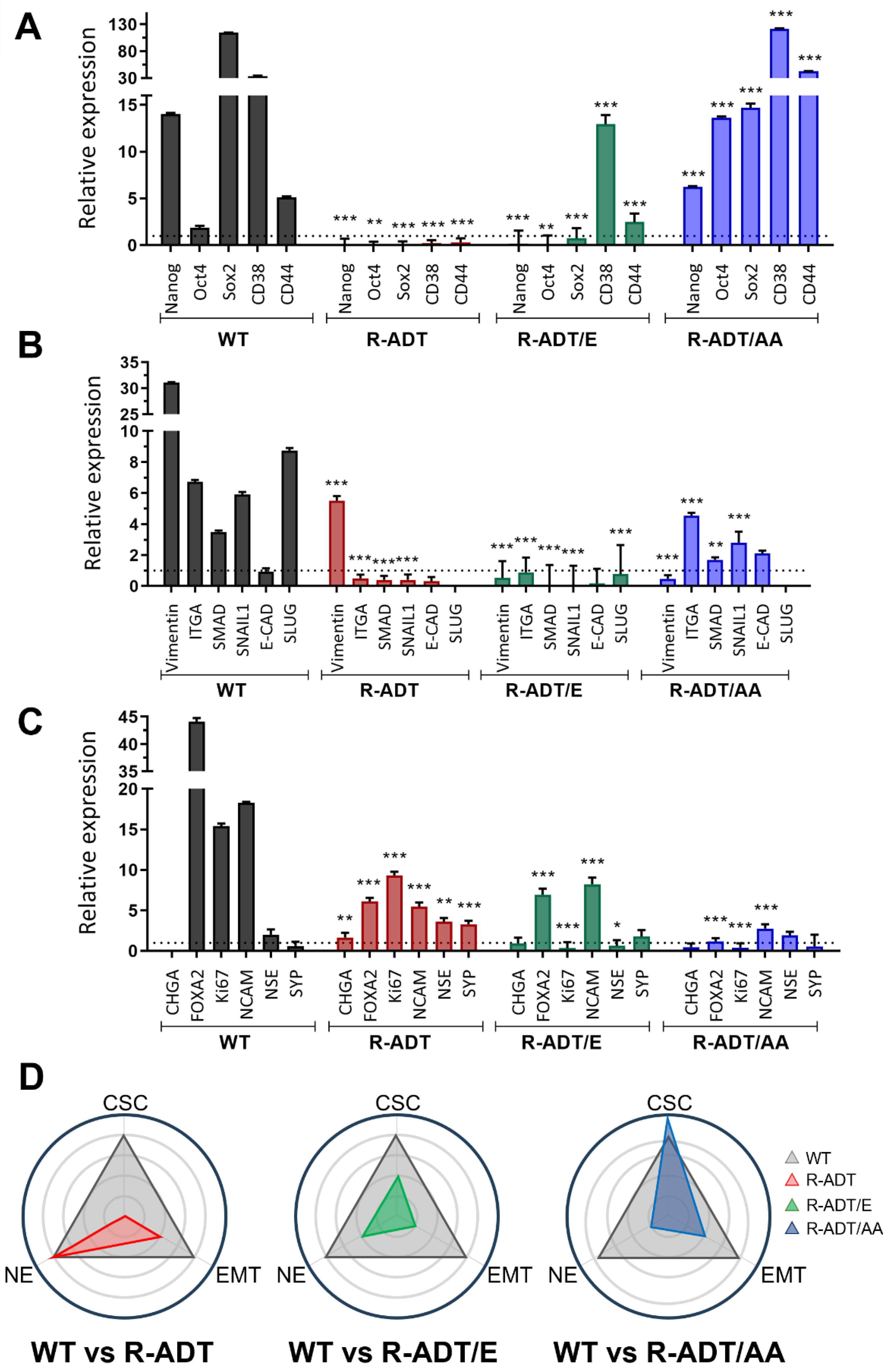
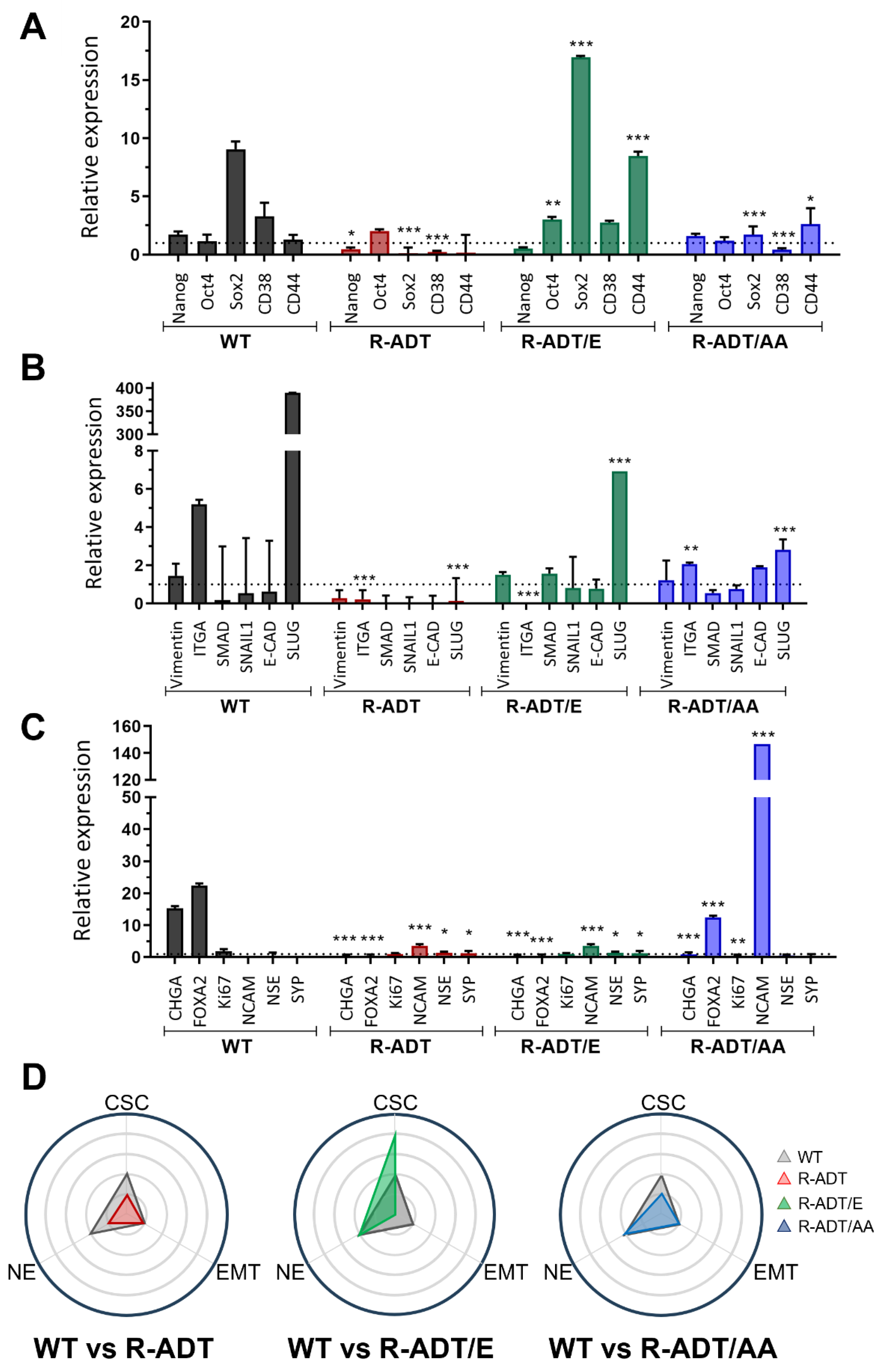
2.4. Gene Expression Analysis of Different Cellular Phenotypes in CRPC Models in Response to Apalutamide
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Treatment with Apalutamide as Second-Line Therapy
4.3. Cell Proliferation Assays
4.4. Sequential Passage Cell Proliferation Monitoring Assay
4.5. Cell Cycle Experiments
4.6. Expression Analysis of the AR Signaling Axis and Associated Transcriptional Coactivators
4.7. Phenotypic Marker Expression
4.8. Clonogenic Assays
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA. Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef]
- Schaeffer, E.M.; Srinivas, S.; Adra, N.; An, Y.; Bitting, R.; Chapin, B.; Cheng, H.H.; D’Amico, A.V.; Desai, N.; Dorff, T.; et al. NCCN Guidelines® Insights: Prostate Cancer, Version 3.2024. J. Natl. Compr. Cancer Netw. JNCCN 2024, 22, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.P.; Banerjee, S.; Brown, T.R.; Zirkin, B.R. Androgen action in prostate function and disease. Am. J. Clin. Exp. Urol. 2018, 6, 62–77. [Google Scholar] [PubMed]
- Xu, J.; Qiu, Y. Role of androgen receptor splice variants in prostate cancer metastasis. Asian J. Urol. 2016, 3, 177–184. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of Advanced Prostate Cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Parker, C.; Sartor, O. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 365, 767. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Yamada, Y.; Matsubara, N.; Tabata, K.-I.; Satoh, T.; Kamiya, N.; Suzuki, H.; Kawahara, T.; Uemura, H.; Yano, A.; Kawakami, S. Abiraterone acetate after progression with enzalutamide in chemotherapy-naïve patients with metastatic castration-resistant prostate cancer: A multi-center retrospective analysis. BMC Res. Notes 2016, 9, 471. [Google Scholar] [CrossRef]
- Stenzl, A.; Dunshee, C.; De Giorgi, U.; Alekseev, B.; Iguchi, T.; Szmulewitz, R.Z.; Flaig, T.W.; Tombal, B.; Morlock, R.; Ivanescu, C.; et al. Effect of Enzalutamide plus Androgen Deprivation Therapy on Health-related Quality of Life in Patients with Metastatic Hormone-sensitive Prostate Cancer: An Analysis of the ARCHES Randomised, Placebo-controlled, Phase 3 Study. Eur. Urol. 2020, 78, 603–614. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.-H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Seitz, A.K.; Thoene, S.; Bietenbeck, A.; Nawroth, R.; Tauber, R.; Thalgott, M.; Schmid, S.; Secci, R.; Retz, M.; Gschwend, J.E.; et al. AR-V7 in Peripheral Whole Blood of Patients with Castration-resistant Prostate Cancer: Association with Treatment-specific Outcome Under Abiraterone and Enzalutamide. Eur. Urol. 2017, 72, 828–834. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen Receptor Variant AR-V9 Is Coexpressed with AR-V7 in Prostate Cancer Metastases and Predicts Abiraterone Resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Naccarato, A.G.; Scatena, C. Recent Advances in Cancer Plasticity: Cellular Mechanisms, Surveillance Strategies, and Therapeutic Optimization. Front. Oncol. 2020, 10, 569. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Simon, I.; Perales, S.; Casado-Medina, L.; Rodríguez-Martínez, A.; Garrido-Navas, M.D.C.; Puche-Sanz, I.; Diaz-Mochon, J.J.; Alaminos, C.; Lupiañez, P.; Lorente, J.A.; et al. Cross-Resistance to Abiraterone and Enzalutamide in Castration Resistance Prostate Cancer Cellular Models Is Mediated by AR Transcriptional Reactivation. Cancers 2021, 13, 1483. [Google Scholar] [CrossRef] [PubMed]
- Badrising, S.; van der Noort, V.; van Oort, I.M.; van den Berg, H.P.; Los, M.; Hamberg, P.; Coenen, J.L.; van den Eertwegh, A.J.M.; de Jong, I.J.; Kerver, E.D.; et al. Clinical activity and tolerability of enzalutamide (MDV3100) in patients with metastatic, castration-resistant prostate cancer who progress after docetaxel and abiraterone treatment. Cancer 2014, 120, 968–975. [Google Scholar] [CrossRef]
- Suzman, D.L.; Luber, B.; Schweizer, M.T.; Nadal, R.; Antonarakis, E.S. Clinical activity of enzalutamide versus docetaxel in men with castration-resistant prostate cancer progressing after abiraterone. Prostate 2014, 74, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Bianchini, D.; Ileana, E.; Sandhu, S.; Patrikidou, A.; Pezaro, C.; Albiges, L.; Attard, G.; Fizazi, K.; De Bono, J.S.; et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann. Oncol. 2013, 24, 1807–1812. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, L.; Cucchiara, V.; Liu, C.; Armstrong, C.M.; Zhao, R.; Yang, J.C.; Lou, W.; Evans, C.P.; Gao, A.C. Intra versus Inter Cross-resistance Determines Treatment Sequence between Taxane and AR-Targeting Therapies in Advanced Prostate Cancer. Mol. Cancer Ther. 2018, 17, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- van Soest, R.J.; van Royen, M.E.; de Morrée, E.S.; Moll, J.M.; Teubel, W.; Wiemer, E.a.C.; Mathijssen, R.H.J.; de Wit, R.; van Weerden, W.M. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2013, 49, 3821–3830. [Google Scholar] [CrossRef]
- Schaaf, Z.A.; Ning, S.; Leslie, A.R.; Sharifi, M.; Han, X.; Armstrong, C.; Lou, W.; Lombard, A.P.; Liu, C.; Gao, A.C. Therapeutic Resistance Models and Treatment Sequencing in Advanced Prostate Cancer. Cancers 2023, 15, 5273. [Google Scholar] [CrossRef]
- Fizazi, K.; Gillessen, S.; ESMO Guidelines Committee. Updated treatment recommendations for prostate cancer from the ESMO Clinical Practice Guideline considering treatment intensification and use of novel systemic agents. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2023, 34, 557–563. [Google Scholar] [CrossRef]
- González del Alba, A.; Méndez-Vidal, M.J.; Vazquez, S.; Castro, E.; Climent, M.A.; Gallardo, E.; Gonzalez-Billalabeitia, E.; Lorente, D.; Maroto, J.P.; Arranz, J.A. SEOM clinical guidelines for the treatment of advanced prostate cancer (2020). Clin. Transl. Oncol. 2021, 23, 969–979. [Google Scholar] [CrossRef]
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to second-generation androgen receptor antagonists in prostate cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ning, S.; Lou, W.; Yang, J.C.; Armstrong, C.M.; Lombard, A.P.; D’Abronzo, L.S.; Evans, C.P.; Gao, A.C.; Liu, C. Cross-Resistance Among Next-Generation Antiandrogen Drugs Through the AKR1C3/AR-V7 Axis in Advanced Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Chi, K.N.; de Wit, R.; de Jonge, M.J.A.; Attard, G.; Friedlander, T.W.; Yu, M.K.; Hellemans, P.; Chien, C.; Abrams, C.; et al. Pharmacokinetics, Safety, and Antitumor Effect of Apalutamide with Abiraterone Acetate plus Prednisone in Metastatic Castration-Resistant Prostate Cancer: Phase Ib Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.A.J.; Koolen, S.L.W.; Mathijssen, R.H.J.; de Wit, R.; van Soest, R.J. Cross-resistance and drug sequence in prostate cancer. Drug Resist. Updat. 2021, 56, 100761. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef]
- Rahman, M.; Miyamoto, H.; Chang, C. Androgen Receptor Coregulators in Prostate Cancer: Mechanisms and Clinical Implications. Clin Cancer Res 2004, 10, 2208–2219. [Google Scholar] [CrossRef]
- Brooke, G.N.; Gamble, S.C.; Hough, M.A.; Begum, S.; Dart, D.A.; Odontiadis, M.; Powell, S.M.; Fioretti, F.M.; Bryan, R.A.; Waxman, J.; et al. Antiandrogens Act as Selective Androgen Receptor Modulators at the Proteome Level in Prostate Cancer Cells*. Mol. Cell. Proteomics 2015, 14, 1201–1216. [Google Scholar] [CrossRef]
- Nouri, M.; Ratther, E.; Stylianou, N.; Nelson, C.C.; Hollier, B.G.; Williams, E.D. Androgen-targeted therapy-induced epithelial mesenchymal plasticity and neuroendocrine transdifferentiation in prostate cancer: An opportunity for intervention. Front. Oncol. 2014, 4, 370. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef]
- Olatunde, D.; De Benedetti, A. TLK1>Nek1 Axis Promotes Nuclear Retention and Activation of YAP with Implications for Castration-Resistant Prostate Cancer. Cancers 2024, 16, 2918. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Tso, C.L.; McBride, W.H.; Sun, J.; Patel, B.; Tsui, K.H.; Paik, S.H.; Gitlitz, B.; Caliliw, R.; van Ophoven, A.; Wu, L.; et al. Androgen deprivation induces selective outgrowth of aggressive hormone-refractory prostate cancer clones expressing distinct cellular and molecular properties not present in parental androgen-dependent cancer cells. Cancer J. Sudbury Mass 2000, 6, 220–233. [Google Scholar]
- Merkens, L.; Sailer, V.; Lessel, D.; Janzen, E.; Greimeier, S.; Kirfel, J.; Perner, S.; Pantel, K.; Werner, S.; von Amsberg, G. Aggressive variants of prostate cancer: Underlying mechanisms of neuroendocrine transdifferentiation. J. Exp. Clin. Cancer Res. CR 2022, 41, 46. [Google Scholar] [CrossRef]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef]
- Paranjape, A.N.; Soundararajan, R.; Werden, S.J.; Joseph, R.; Taube, J.H.; Liu, H.; Rodriguez-Canales, J.; Sphyris, N.; Wistuba, I.; Miura, N.; et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene 2016, 35, 5963–5976. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide Inhibits Androgen Receptor Variants Expression and Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Smith, R.; Liu, M.; Liby, T.; Bayani, N.; Bucher, E.; Chiotti, K.; Derrick, D.; Chauchereau, A.; Heiser, L.; Alumkal, J.; et al. Enzalutamide response in a panel of prostate cancer cell lines reveals a role for glucocorticoid receptor in enzalutamide resistant disease. Sci. Rep. 2020, 10, 21750. [Google Scholar] [CrossRef]
- Grossebrummel, H.; Peter, T.; Mandelkow, R.; Weiss, M.; Muzzio, D.; Zimmermann, U.; Walther, R.; Jensen, F.; Knabbe, C.; Zygmunt, M.; et al. Cytochrome P450 17A1 inhibitor abiraterone attenuates cellular growth of prostate cancer cells independently from androgen receptor signaling by modulation of oncogenic and apoptotic pathways. Int. J. Oncol. 2016, 48, 793–800. [Google Scholar] [CrossRef]
- Barbier, R.H.; McCrea, E.M.; Lee, K.Y.; Strope, J.D.; Risdon, E.N.; Price, D.K.; Chau, C.H.; Figg, W.D. Abiraterone induces SLCO1B3 expression in prostate cancer via microRNA-579-3p. Sci. Rep. 2021, 11, 10765. [Google Scholar] [CrossRef] [PubMed]
- Soylu, H.; Kırca, M.; Avcı, S.; Ozpolat, B.; Ustunel, I. Antiandrogen abiraterone and docetaxel treatments affect Notch1, Jagged1 and Hes1 expressions in metastatic prostate cancer cells. Exp. Mol. Pathol. 2021, 119, 104607. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, I.; Sanchez-Manas, J.M.; Perales, S.; Martinez-Navajas, G.; Ceron-Hernandez, J.; Real, P.J. Phenotypic Plasticity and Androgen Receptor Bypass Drive Cross-Resistance to Apalutamide in Castration-Resistant Prostate Cancer Cell Models. Int. J. Mol. Sci. 2025, 26, 5939. https://doi.org/10.3390/ijms26135939
Simon I, Sanchez-Manas JM, Perales S, Martinez-Navajas G, Ceron-Hernandez J, Real PJ. Phenotypic Plasticity and Androgen Receptor Bypass Drive Cross-Resistance to Apalutamide in Castration-Resistant Prostate Cancer Cell Models. International Journal of Molecular Sciences. 2025; 26(13):5939. https://doi.org/10.3390/ijms26135939
Chicago/Turabian StyleSimon, Iris, Jose Manuel Sanchez-Manas, Sonia Perales, Gonzalo Martinez-Navajas, Jorge Ceron-Hernandez, and Pedro J. Real. 2025. "Phenotypic Plasticity and Androgen Receptor Bypass Drive Cross-Resistance to Apalutamide in Castration-Resistant Prostate Cancer Cell Models" International Journal of Molecular Sciences 26, no. 13: 5939. https://doi.org/10.3390/ijms26135939
APA StyleSimon, I., Sanchez-Manas, J. M., Perales, S., Martinez-Navajas, G., Ceron-Hernandez, J., & Real, P. J. (2025). Phenotypic Plasticity and Androgen Receptor Bypass Drive Cross-Resistance to Apalutamide in Castration-Resistant Prostate Cancer Cell Models. International Journal of Molecular Sciences, 26(13), 5939. https://doi.org/10.3390/ijms26135939