
Comprehensive Evaluation of a 1021-Gene Panel in FFPE and Liquid Biopsy for Analytical and Clinical Use
 , , , , , ,
, , , , , ,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Assay Performance Quality Metrics
2.2. Validation Materials
2.2.1. Reference Materials Results
2.2.2. Tissue Cohort Results
2.2.3. FFPE and Liquid Biopsy Comparative Analysis Results
2.3. MSI Results’ Validation
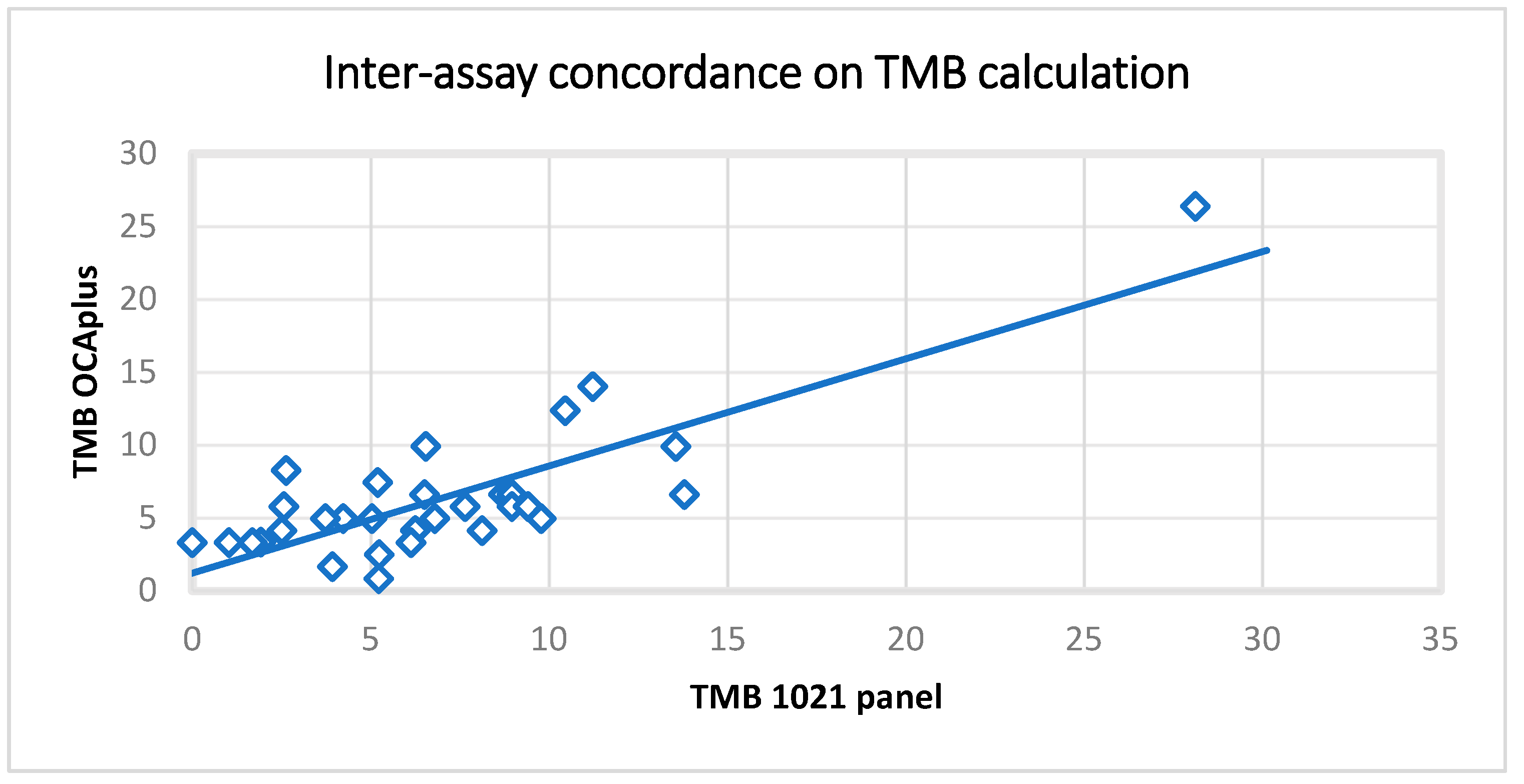
2.4. TMB Results’ Validation
2.5. LOH
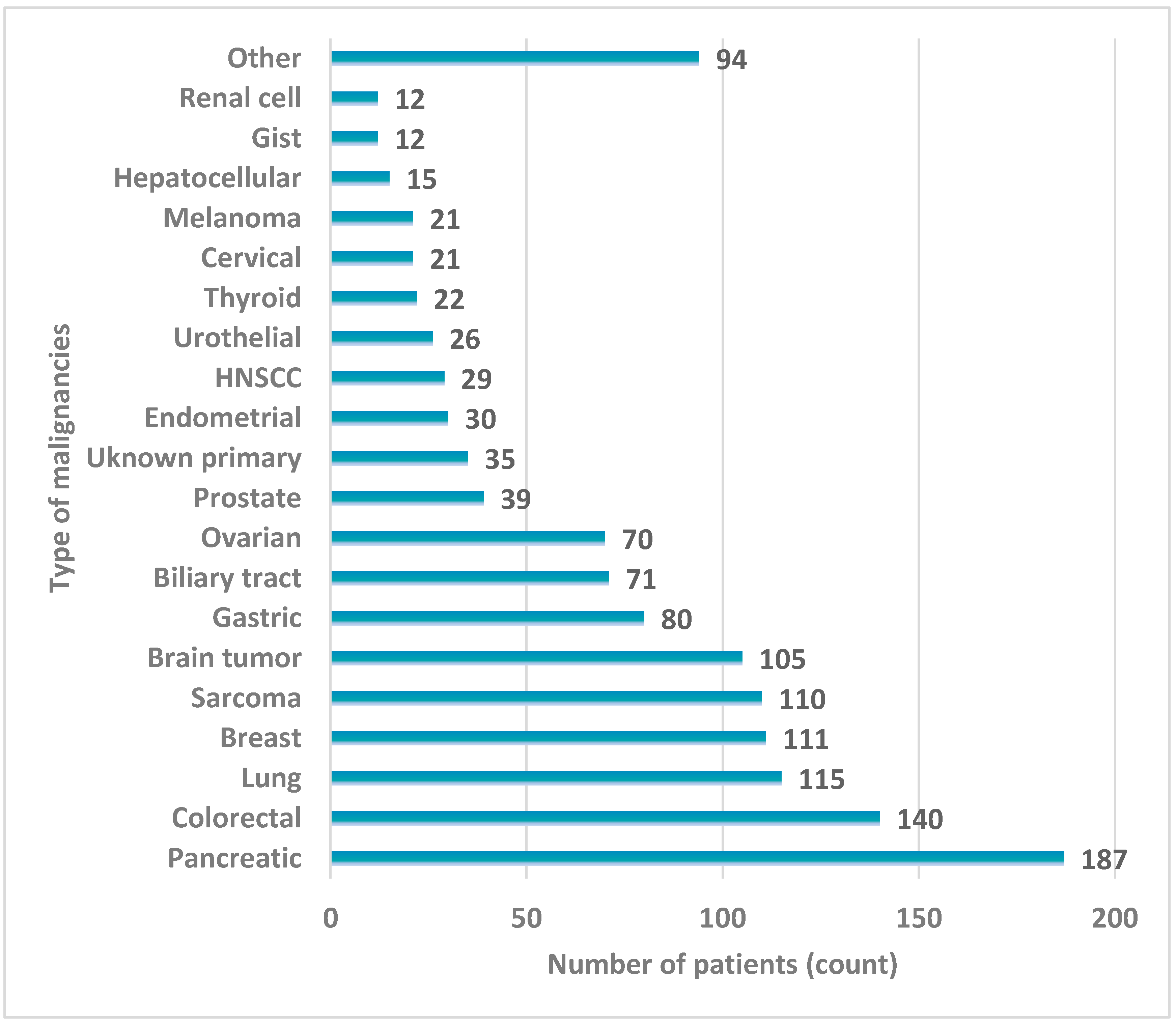
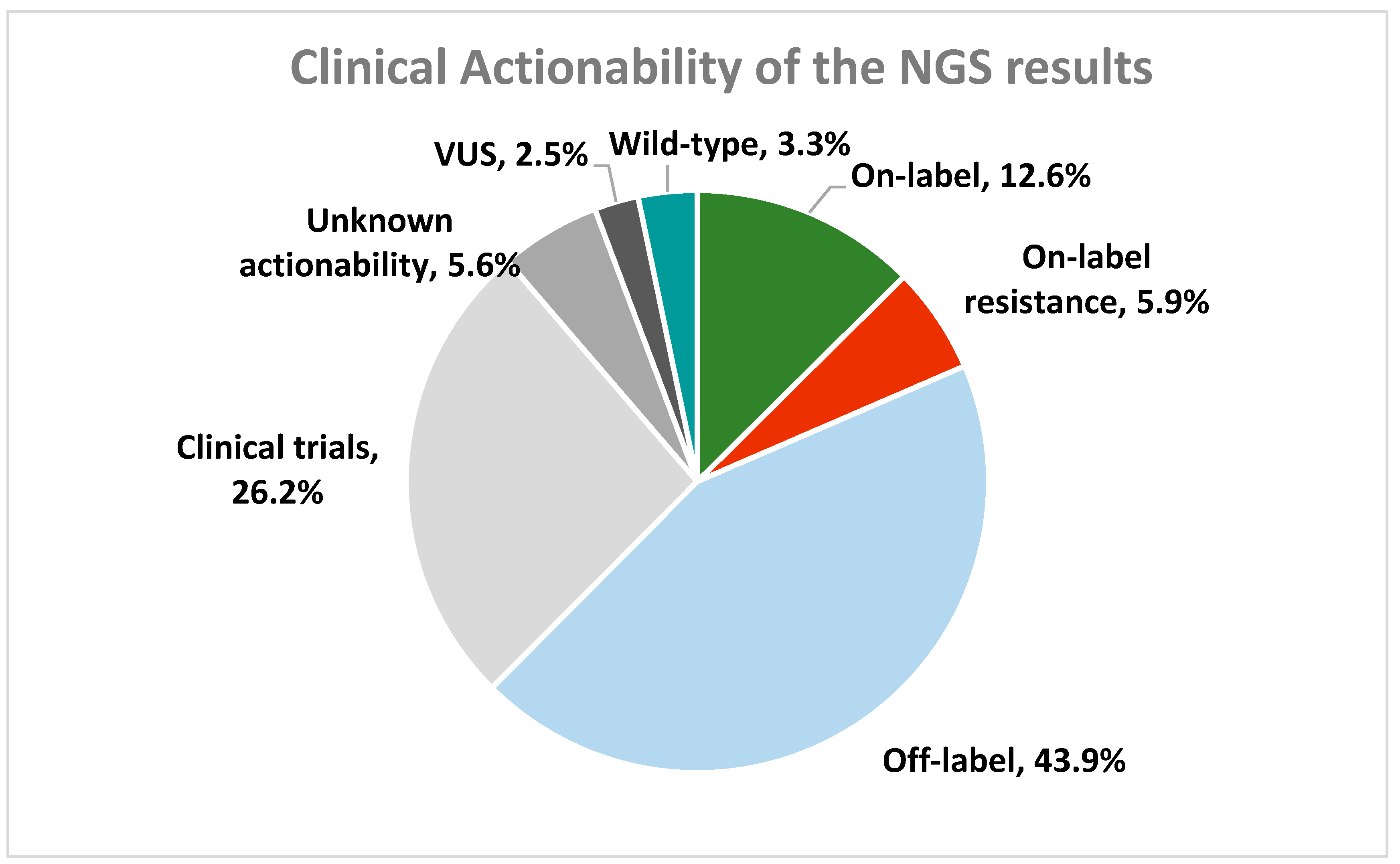
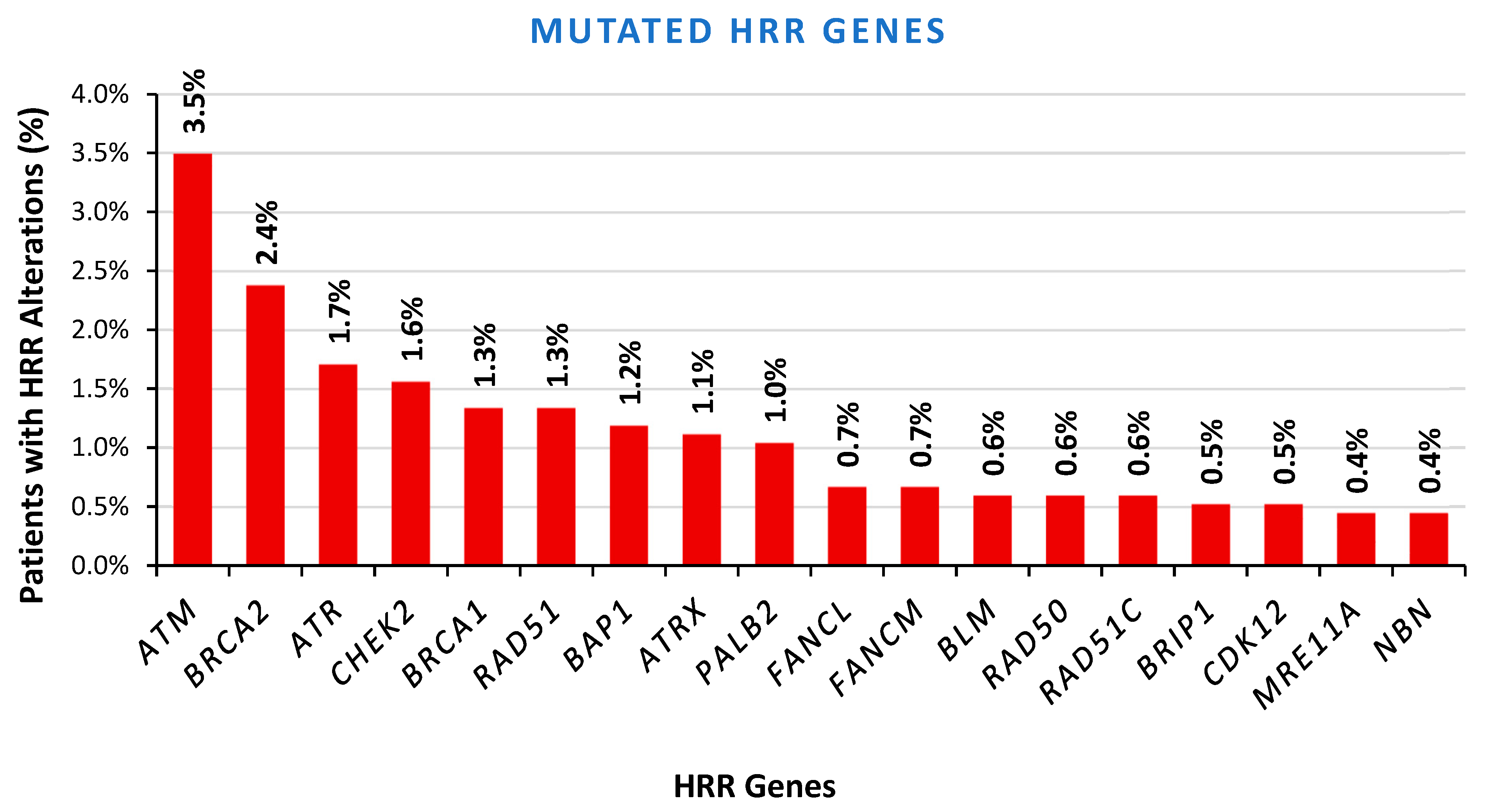
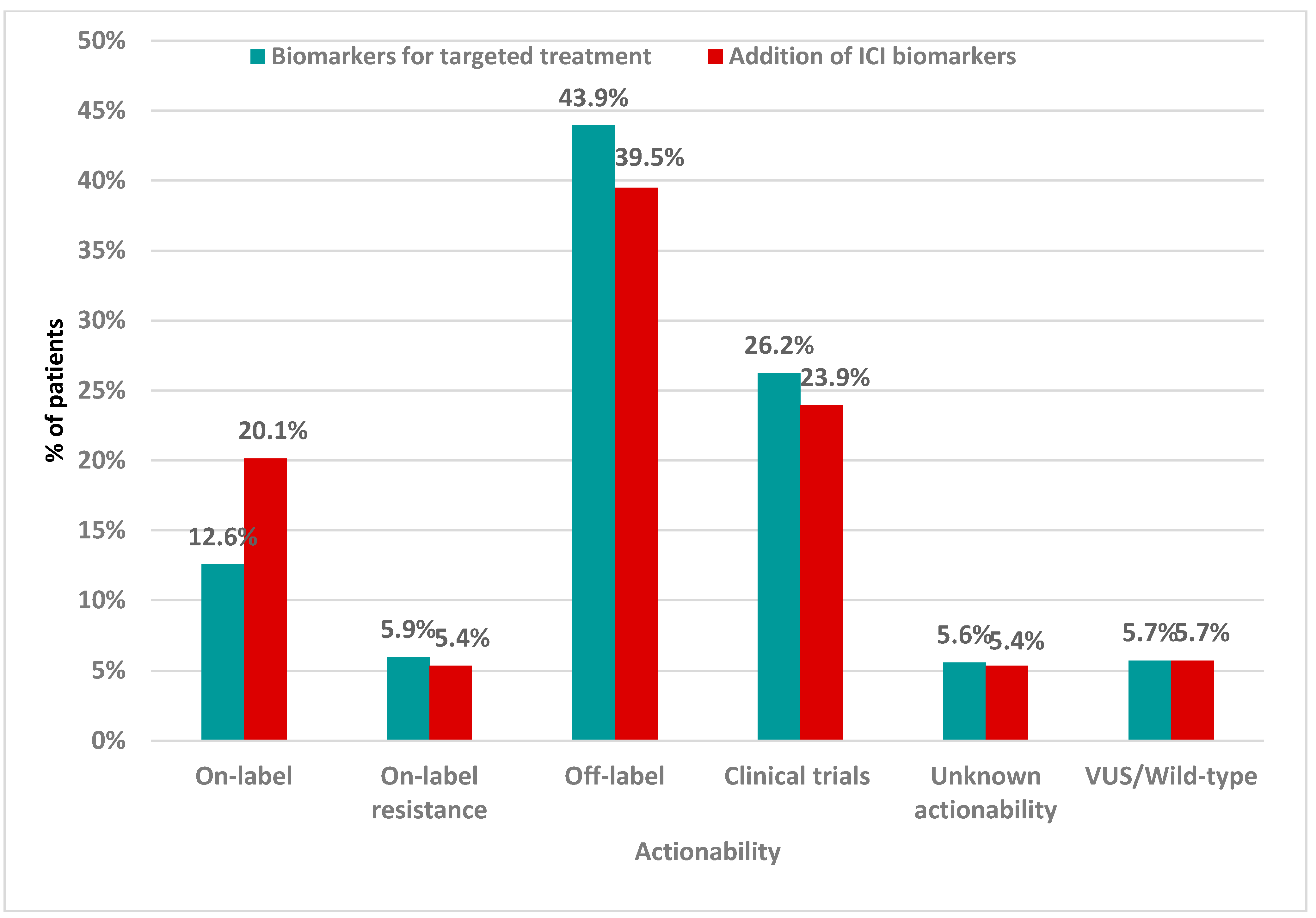
2.6. Clinical Validation Results
3. Discussion
4. Material and Methods
4.1. Analytical Validation
4.2. Patients
4.3. Tissue Selection and Nucleic Acid Isolation
4.4. NGS Procedure
- Library Preparation Prior to Capture: The genomic DNA (gDNA) extracted from FFPE tissue or leukocytes (regarded as the control) was sheared into small fragments and then gDNA fragments were purified with magnetic beads. For the cfDNA extracted from plasma (regarded as the case), the steps involving fragmentation and purification were not necessary. End repair and A-tailing were performed for the selected gDNA fragments. Universal adaptors with known sequences were ligated onto the gDNA fragments. The P5 and P7 index sequences were incorporated into each library at the ends of the repaired gDNA fragments. The indexes include a unique sequence for identifying each individual sample. The pre-capture libraries were obtained after further purification.
- Target Enrichment: Pre-capture libraries were enriched for specific genes of interest using a hybridization capture-based method. Specially designed, biotinylated probes spanning the gene regions of interest were hybridized with the libraries. After hybridization and incubation, the probes and hybridized targeted libraries were isolated from non-targeted libraries using streptavidin magnetic beads. Enriched, targeted libraries were obtained after the application of the washing, amplification, and purification steps.
- Library Circularization: Enriched libraries were denatured into single-stranded DNA at high temperature and circularized into single-stranded circular DNA.
- Sequencing: The single-stranded DNA nanoball (DNB) was prepared for sequencing by using the original single-stranded circular DNA as a template for isothermal amplification (called rolling circle amplification (RCA)). DNA nanoballs were pumped by the fluidics system, loaded onto a patterned array chip (sequencing flow cell) and sequenced using cPAS (combinatorial probe anchor synthesis)-based sequencing on an MGI sequencing platform (DNBSEQ-G400).
4.5. Data Analysis and Result Interpretation
4.6. LOH
4.7. OncoScan CNV Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NGS | Next-Generation Sequencing |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| TCC | Tumor Cell Content |
| MSI | Microsatellite Instability |
| TMB | Tumor Mutational Burden |
| RCA | Rolling Circle Amplification |
| UMIs | Unique Molecular Identifiers |
| SNP | Single-Nucleotide Polymorphism |
| CNV | Copy Number Variation |
| LOH | Loss of Heterozygosity |
| VAF | Variant Allele Frequency |
| HR-HRR | Homologous Recombination (Repair) |
| PPA | Positive Percent Agreement |
| NPA | Negative Percent Agreement |
References
- Mosele, M.F.; Westphalen, C.B.; Stenzinger, A.; Barlesi, F.; Bayle, A.; Bièche, I.; Bonastre, J.; Castro, E.; Dienstmann, R.; Krämer, A.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Advanced Cancer in 2024: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2024, 35, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Lee, H.Y. Molecular Targeted Therapy for Anticancer Treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Catalano, M.; Iannone, L.F.; Nesi, G.; Nobili, S.; Mini, E.; Roviello, G. Immunotherapy-Related Biomarkers: Confirmations and Uncertainties. Crit. Rev. Oncol. Hematol. 2023, 192, 104135. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.; Jochum, W.; Padberg, B.; Demmer, I.; Mertz, K.D.; Joerger, M.; Britschgi, C.; Matter, M.S.; Rothschild, S.I.; Omlin, A. How to Read a Next-Generation Sequencing Report—What Oncologists Need to Know. ESMO Open 2022, 7, 100570. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-Cancer Circulating Tumor DNA Detection in over 10,000 Chinese Patients. Nat. Commun. 2021, 12, 11. [Google Scholar] [CrossRef]
- Vanni, I.; Pastorino, L.; Andreotti, V.; Comandini, D.; Fornarini, G.; Grassi, M.; Puccini, A.; Tanda, E.T.; Pastorino, A.; Martelli, V.; et al. Combining Germline, Tissue and Liquid Biopsy Analysis by Comprehensive Genomic Profiling to Improve the Yield of Actionable Variants in a Real-World Cancer Cohort. J. Transl. Med. 2024, 22, 462. [Google Scholar] [CrossRef]
- Dumur, C.I.; Krishnan, R.; Almenara, J.A.; Brown, K.E.; Dugan, K.R.; Farni, C.; Ibrahim, F.Z.; Sanchez, N.A.; Rathore, S.; Pradhan, D.; et al. Analytical Validation and Clinical Utilization of the Oncomine Comprehensive Assay Plus Panel for Comprehensive Genomic Profiling in Solid Tumors. J. Mol. Pathol. 2023, 4, 109–127. [Google Scholar] [CrossRef]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the Classification of Pathogenicity of Somatic Variants in Cancer (Oncogenicity): Joint Recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022, 24, 986–998. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The Functions and Regulation of the PTEN Tumour Suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Wang, Y.; Rozen, V.; Zhao, Y.; Wang, Z. Oncogenic Activation of PIK3CA in Cancers: Emerging Targeted Therapies in Precision Oncology. Genes. Dis. 2025, 12, 101430. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Guan, X.; Zhang, X.; Luan, X.; Song, Z.; Cheng, X.; Zhang, W.; Qin, J.-J. Targeting KRAS Mutant Cancers: From Druggable Therapy to Drug Resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Thongyoo, P.; Chindaprasirt, J.; Aphivatanasiri, C.; Intarawichian, P.; Kunprom, W.; Kongpetch, S.; Techasen, A.; Loilome, W.; Namwat, N.; Titapun, A.; et al. KRAS Mutations in Cholangiocarcinoma: Prevalence, Prognostic Value, and KRAS G12/G13 Detection in Cell-Free DNA. Cancer Genom. Proteom. 2025, 22, 112–126. [Google Scholar] [CrossRef]
- Lucero, M.; Riley, I.D.; Hazard, R.H.; Sanvictores, D.; Tallo, V.; Dumaluan, D.G.M.; Ugpo, J.M.; Lopez, A.D. Assessing the Quality of Medical Death Certification: A Case Study of Concordance between National Statistics and Results from a Medical Record Review in a Regional Hospital in the Philippines. Popul. Health Metr. 2018, 16, 23. [Google Scholar] [CrossRef]
- Foreman, R.; Wollman, R. Mammalian Gene Expression Variability Is Explained by Underlying Cell State. Mol. Syst. Biol. 2020, 16, e9146. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ozaki, H.; Ueno, M.; Komatsu, Y.; Yuki, S.; Esaki, T.; Taniguchi, H.; Sunakawa, Y.; Yamaguchi, K.; Kato, K.; et al. Targeted Therapy Guided by Circulating Tumor DNA Analysis in Advanced Gastrointestinal Tumors. Nat. Med. 2024, 31, 165–175. [Google Scholar] [CrossRef]
- Singal, G.; Miller, P.G.; Agarwala, V.; Li, G.; Kaushik, G.; Backenroth, D.; Gossai, A.; Frampton, G.M.; Torres, A.Z.; Lehnert, E.M.; et al. Association of Patient Characteristics and Tumor Genomics with Clinical Outcomes Among Patients with Non-Small Cell Lung Cancer Using a Clinicogenomic Database. JAMA 2019, 321, 1391–1399. [Google Scholar] [CrossRef]
- Kästner, A.; Kron, A.; van den Berg, N.; Moon, K.; Scheffler, M.; Schillinger, G.; Pelusi, N.; Hartmann, N.; Rieke, D.T.; Stephan-Falkenau, S.; et al. Evaluation of the Effectiveness of a Nationwide Precision Medicine Program for Patients with Advanced Non-Small Cell Lung Cancer in Germany: A Historical Cohort Analysis. Lancet Reg. Health Eur. 2024, 36, 100788. [Google Scholar] [CrossRef]
- Subbiah, V.; Gouda, M.A.; Ryll, B.; Burris, H.A.; Kurzrock, R. The Evolving Landscape of Tissue-agnostic Therapies in Precision Oncology. CA Cancer J. Clin. 2024, 74, 433–452. [Google Scholar] [CrossRef]
- Passaro, A.; Al Bakir, M.; Hamilton, E.G.; Diehn, M.; André, F.; Roy-Chowdhuri, S.; Mountzios, G.; Wistuba, I.I.; Swanton, C.; Peters, S. Cancer Biomarkers: Emerging Trends and Clinical Implications for Personalized Treatment. Cell 2024, 187, 1617–1635. [Google Scholar] [CrossRef]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Jantus-Lewintre, E.; Rappa, A.; Ruano, D.; van Egmond, D.; Gallach, S.; Gozuyasli, D.; Durães, C.; Costa, J.L.; Camps, C.; Lacroix, L.; et al. Multicenter In-House Evaluation of an Amplicon-Based Next-Generation Sequencing Panel for Comprehensive Molecular Profiling. Mol. Diagn. Ther. 2025, 29, 249–261. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.-C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO Recommendations on the Use of Circulating Tumour DNA Assays for Patients with Cancer: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Niitsu, H.; Nakahara, H.; Ishida, K.; Kaneko, Y.; Hayes, C.N.; Arihiro, K.; Hinoi, T. Real-World Data Analysis for Factors Influencing the Quality Check Status in FoundationOne CDx Cancer Genomic Profiling Tests. Sci. Rep. 2025, 15, 5167. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef]
- Bozzi, F.; Conca, E.; Silvestri, M.; Dagrada, G.; Ardore, A.; Penso, D.; Lorenzini, D.; Volpi, C.C.; Trupia, D.V.; Busico, A.; et al. Detecting Gene Copy Number Alterations by Oncomine Comprehensive Genomic Profiling in a Comparative Study on FFPE Tumor Samples. Sci. Rep. 2025, 15, 4314. [Google Scholar] [CrossRef] [PubMed]
- Milbury, C.A.; Creeden, J.; Yip, W.-K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and Analytical Validation of FoundationOne®CDx, a Comprehensive Genomic Profiling Assay for Solid Tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef]
- Beaubier, N.; Tell, R.; Lau, D.; Parsons, J.R.; Bush, S.; Perera, J.; Sorrells, S.; Baker, T.; Chang, A.; Michuda, J.; et al. Clinical Validation of the Tempus XT Next-Generation Targeted Oncology Sequencing Assay. Oncotarget 2019, 10, 2384–2396. [Google Scholar] [CrossRef]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and Analytical Validation of FoundationOne Liquid CDx, a Novel 324-Gene CfDNA-Based Comprehensive Genomic Profiling Assay for Cancers of Solid Tumor Origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Froyen, G.; Volders, P.; Geerdens, E.; Berden, S.; Van der Meulen, J.; De Cock, A.; Vermeire, S.; Van Huysse, J.; de Barsy, M.; Beniuga, G.; et al. Analysis of Comprehensive Genomic Profiling of Solid Tumors with a Novel Assay for Broad Analysis in Clinical Diagnostics. Mol. Oncol. 2025, 19, 1797–1810. [Google Scholar] [CrossRef]
- Froyen, G.; Geerdens, E.; Berden, S.; Cruys, B.; Maes, B. Diagnostic Validation of a Comprehensive Targeted Panel for Broad Mutational and Biomarker Analysis in Solid Tumors. Cancers 2022, 14, 2457. [Google Scholar] [CrossRef]
- Werner, T.V.; Kock, S.; Weber, I.; Kayser, G.; Werner, M.; Lassmann, S. Validation of a DNA-Based Next-Generation Sequencing Test for Molecular Diagnostic Variant and Fusion Detection in Formalin-Fixed, Paraffin-Embedded Tissue Specimens and Liquid Biopsy Plasma/Cell-Free DNA Samples. J. Mol. Diagn. 2022, 24, 784–802. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.; Ben-Shachar, R.; Feliciano, J.; Gai, L.; Beauchamp, K.A.; Rivers, Z.; Hockenberry, A.J.; Harrison, G.; Guittar, J.; Catela, C.; et al. Actionable Structural Variant Detection via RNA-NGS and DNA-NGS in Patients With Advanced Non–Small Cell Lung Cancer. JAMA Netw. Open 2024, 7, e2442970. [Google Scholar] [CrossRef] [PubMed]
- Mack, P.C.; Keller-Evans, R.B.; Li, G.; Lofgren, K.T.; Schrock, A.B.; Trabucco, S.E.; Allen, J.M.; Tolba, K.; Oxnard, G.R.; Huang, R.S.P. Real-World Clinical Performance of a DNA-Based Comprehensive Genomic Profiling Assay for Detecting Targetable Fusions in Nonsquamous NSCLC. Oncologist 2024, 29, e984–e996. [Google Scholar] [CrossRef]
- Parisi, C.; Tagliamento, M.; Belcaid, L.; Aldea, M.; Bayle, A.; Remon-Masip, J.; Italiano, A.; Planchard, D.; Besse, B.; Barlesi, F. Circulating Tumor DNA in Clinical Trials for Solid Tumors: Challenges and Current Applications. J. Liq. Biopsy 2023, 1, 100007. [Google Scholar] [CrossRef]
- MimixTM OncoSpan FFPE Reference Standard. Available online: https://horizondiscovery.com/en/reference-standards/products/oncospan-ffpe (accessed on 20 May 2025).
- MimixTM Structural Multiplex (FFPE) Reference Standard. Available online: https://horizondiscovery.com/en/reference-standards/products/structural-multiplex-reference-standard-ffpe (accessed on 20 May 2025).
- Dupain, C.; Gutman, T.; Girard, E.; Kamoun, C.; Marret, G.; Castel-Ajgal, Z.; Sablin, M.P.; Neuzillet, C.; Borcoman, E.; Hescot, S.; et al. Tumor Mutational Burden Assessment and Standardized Bioinformatics Approach Using Custom NGS Panels in Clinical Routine. BMC Biol. 2024, 22, 43. [Google Scholar] [CrossRef]
- Tsantikidi, A.; Papazisis, K.; Floros, T.; Gazouli, M.; Papadopoulou, E.; Tsaousis, G.; Nasioulas, G.; Mester, A.; Milan, K.P.; Gozman, B.; et al. RediScore: Prospective Validation of a Pipeline for Homologous Recombination Deficiency Analysis. Oncol. Lett. 2023, 26, 480. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.M.; Haase, K.; Van Loo, P.; Markowetz, F. Allele-Specific Multi-Sample Copy Number Segmentation in ASCAT. Bioinformatics 2021, 37, 1909–1911. [Google Scholar] [CrossRef]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-Specific Copy Number Analysis of Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Imanishi, S.; Naoi, Y.; Shimazu, K.; Shimoda, M.; Kagara, N.; Tanei, T.; Miyake, T.; Kim, S.J.; Noguchi, S. Clinicopathological Analysis of Homologous Recombination-Deficient Breast Cancers with Special Reference to Response to Neoadjuvant Paclitaxel Followed by FEC. Breast Cancer Res. Treat. 2019, 174, 627–637. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Control Index | Criterion | |
|---|---|---|
| DNA Quality Assessment | DNA amount (ng) | ≥50 |
| DNA Library Quality Assessment | DNA library amount (ng) | ≥600 |
| Sequencing Quality Assessment | Average effective sequencing depth | ≥500 (for 2% VAF)/1000 (for 0.5% VAF) |
| Fraction of target covered with ≥50× | ≥99% | |
| Fraction of base quality ≥Q30 | ≥80% | |
| Reference Sample | Type of Variation | Number of True-Positive Variants | Number of False-Positive Variants | Number of False-Negative Variants | Sensitivity | Specificity | Mean Depth | VAF |
|---|---|---|---|---|---|---|---|---|
| S800-1 | SNV/INDEL | 27 | 0 | 0 | 100% | 100% | 1000× | 2.00% |
| CNV | 3 | 0 | 0 | 100% | 100% | 2.00% | ||
| Fusion | 6 | 0 | 0 | 100% | 100% | 2.00% | ||
| S800-2 | SNV/INDEL | 27 | 0 | 0 | 100% | 100% | 2000× | 0.50% |
| CNV | 3 | 0 | 0 | 100% | 100% | 0.50% | ||
| Fusion | 6 | 0 | 0 | 100% | 100% | 0.50% | ||
| Tru-Q 7 | SNV/INDEL | 39 | 0 | 1 | 97.50% | 100% | 993× | 1–1.3% |
| Tru-Q 7 | SNV/INDEL | 37 | 0 | 4 | 93% | 100% | 560× | 1–1.3% |
| mix of Tru-Q 7+ Tru-Q0 (wild type) | SNV/INDEL | 34 | 0 | 0 | 85% | 100% | 794× | 0.5–0.65% |
| Patient | Tissue Findings Using the OCA Plus Assay | VAF | Liquid-Biopsy Findings with the 1021 Assay | AF |
|---|---|---|---|---|
| Sample 1 | ERRFI1 c.1007_1016del | 46% | ERRFI1 c.1007_1016del | 0.50% |
| TERT -146 T>C | 35% | TERT -146 T>C | 0.90% | |
| PALB2 c.1A>G | 21% | PALB2 c.1A>G | 0.80% | |
| TP53 c.743G>T | 49.% | TP53 c.743G>T | 1.60% | |
| CCND1 amplification | 10 copies | CCND1 amplification | 3.2 copies | |
| FGF19 amplification | 10 copies | FGF19 amplification | 3.2 copies | |
| FGF4 amplification | 10 copies | FGF4 amplification | 3.2 copies | |
| FGF3 amplification | 10 copies | FGF3 amplification | 3.2 copies | |
| Sample 2 | PIK3CA c.328_330del | 7% | PIK3CA c.328_330del | 4.90% |
| CDH1 c.67C>T | 10% | CDH1 c.67C>T | 8.50% | |
| Sample 3 | EGFR c.2573T>G | 34.% | EGFR c.2573T>G | 32.40% |
| TP53 c.413C>T | 46% | TP53 c.413C>T | 6.50% | |
| Sample 4 | KRAS c.38G>A | 42% | Not Detected | |
| BAP1 c.783+2T>C | 22% | Not Detected | ||
| CDKN2A c.151-1G>T | 29% | Not Detected | ||
| Sample 5 | NRAS c.183A>C | 15% | Not Detected | |
| APC c.4280delC | 60% | Not Detected | ||
| ERBB3 c.695C>T | 16% | Not Detected | ||
| KDMA c.641T>A | 13.20% | Not Detected | ||
| Sample 6 | KRAS c.35G>A | 28.60% | KRAS c.35G>A | 3.00% |
| RET c.2410G>A | 32.44% | RET c.2410G>A | 1.42% | |
| IDH1 c.395G>T | 20.52% | IDH1 c.395G>T | 0.98% | |
| TP53 c.614A>G | 40.66% | TP53 c.614A>G | 2.24% | |
| Sample 7 | EML4(6)–ALK(20) | 2416 copies | EML4(6)–ALK(20) | 1.20% |
| Sample 8 | CTNNB1 c.121A>G | 42.20% | Not detected | |
| CD74-ROS1 fusion | 3895 copies | Not detected | ||
| Sample 9 | TP53 c.814G>T | 44.30% | TP53 c.814G>T | 0.55% |
| Sample 10 | KRAS c.35G>T | 32.25% | KRAS c.35G>T | 0.68% |
| PIK3CA c.3139C>T | 12.69% | PIK3CA c.3139C>T | 1.72% | |
| MAP2K7 c.289C>T | 13.47% | MAP2K7 c.289C>T | 0.30% | |
| TP53 c.734G>A | 11.70% | TP53 c.734G>A | 0.60% |
| Validated Variants | Validation Samples | Source of Validation |
|---|---|---|
| SNVs, Indels, CNVs, Fusions | 50 clinical samples, 6 reference standards | FFPE/Tumor DNA |
| gLOH | 37 clinical samples | FFPE/Tumor DNA |
| MSI | 66 patient samples orthogonally tested for MSI status | FFPE |
| TMB | 59 patient samples orthogonally tested for TMB status, 2 reference standards | FFPE/Tumor DNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meintani, A.; Ozdogan, M.; Touroutoglou, N.; Papazisis, K.; Boukovinas, I.; Bilir, C.; Giassas, S.; Sualp, T.; Lacin, S.; Dan Corneliu, J.; et al. Comprehensive Evaluation of a 1021-Gene Panel in FFPE and Liquid Biopsy for Analytical and Clinical Use. Int. J. Mol. Sci. 2025, 26, 5930. https://doi.org/10.3390/ijms26135930
Meintani A, Ozdogan M, Touroutoglou N, Papazisis K, Boukovinas I, Bilir C, Giassas S, Sualp T, Lacin S, Dan Corneliu J, et al. Comprehensive Evaluation of a 1021-Gene Panel in FFPE and Liquid Biopsy for Analytical and Clinical Use. International Journal of Molecular Sciences. 2025; 26(13):5930. https://doi.org/10.3390/ijms26135930
Chicago/Turabian StyleMeintani, Angeliki, Mustafa Ozdogan, Nikolaos Touroutoglou, Konstantinos Papazisis, Ioannis Boukovinas, Cemil Bilir, Stylianos Giassas, Tansan Sualp, Sahin Lacin, Jinga Dan Corneliu, and et al. 2025. "Comprehensive Evaluation of a 1021-Gene Panel in FFPE and Liquid Biopsy for Analytical and Clinical Use" International Journal of Molecular Sciences 26, no. 13: 5930. https://doi.org/10.3390/ijms26135930
APA StyleMeintani, A., Ozdogan, M., Touroutoglou, N., Papazisis, K., Boukovinas, I., Bilir, C., Giassas, S., Sualp, T., Lacin, S., Dan Corneliu, J., Kosmidis, P., Ozatli, T., Ziogas, D., Theochari, M., Botsolis, K., Kapetsis, G., Tsantikidi, A., Florou-Chatzigiannidou, C., Maxouri, S., ... Nasioulas, G. (2025). Comprehensive Evaluation of a 1021-Gene Panel in FFPE and Liquid Biopsy for Analytical and Clinical Use. International Journal of Molecular Sciences, 26(13), 5930. https://doi.org/10.3390/ijms26135930