
Dysregulation of Inositol Polyphosphate 5-Phosphatase OCRL in Alzheimer’s Disease: Implications for Autophagy Dysfunction
 ,
, 
 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
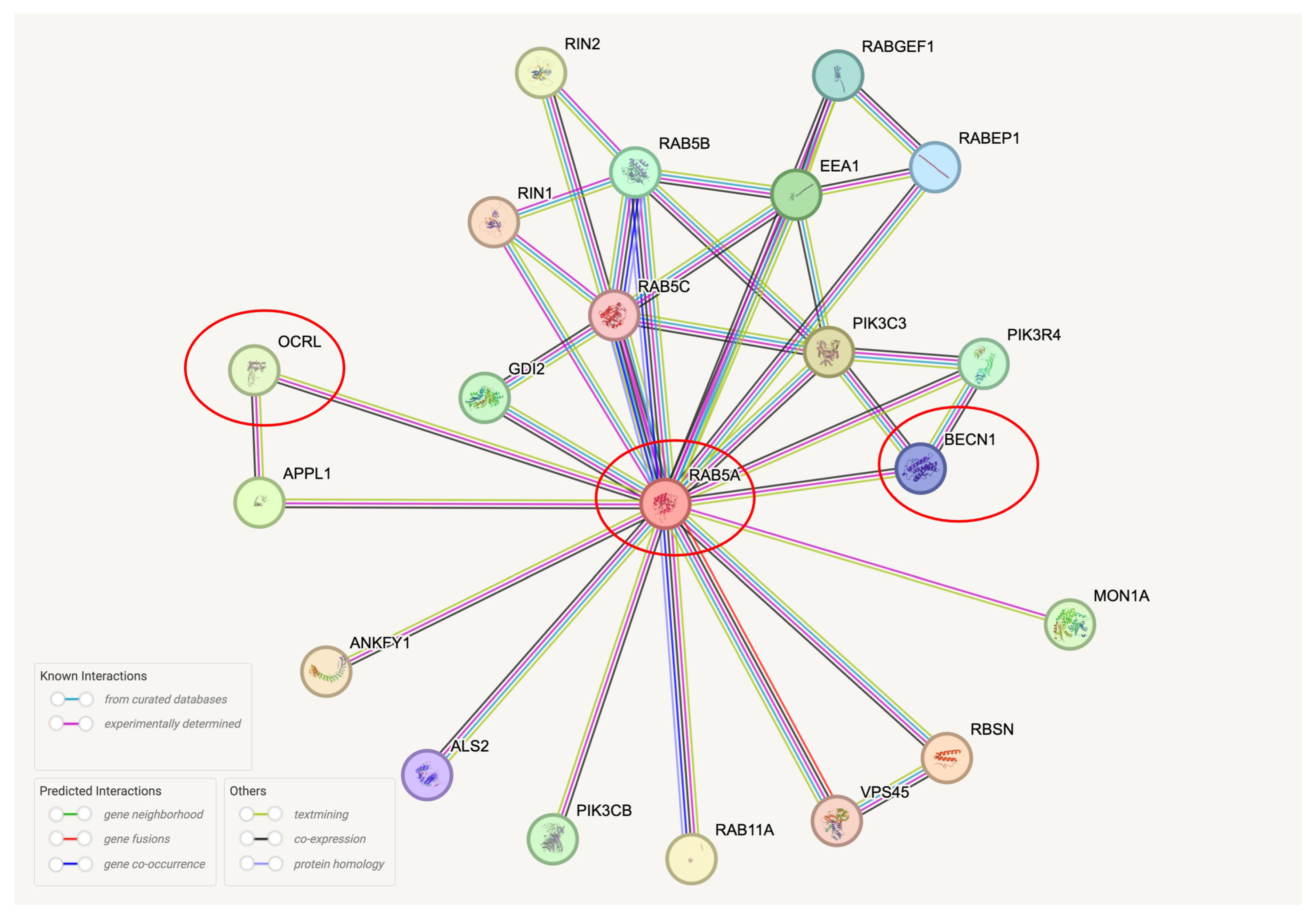
2. Results
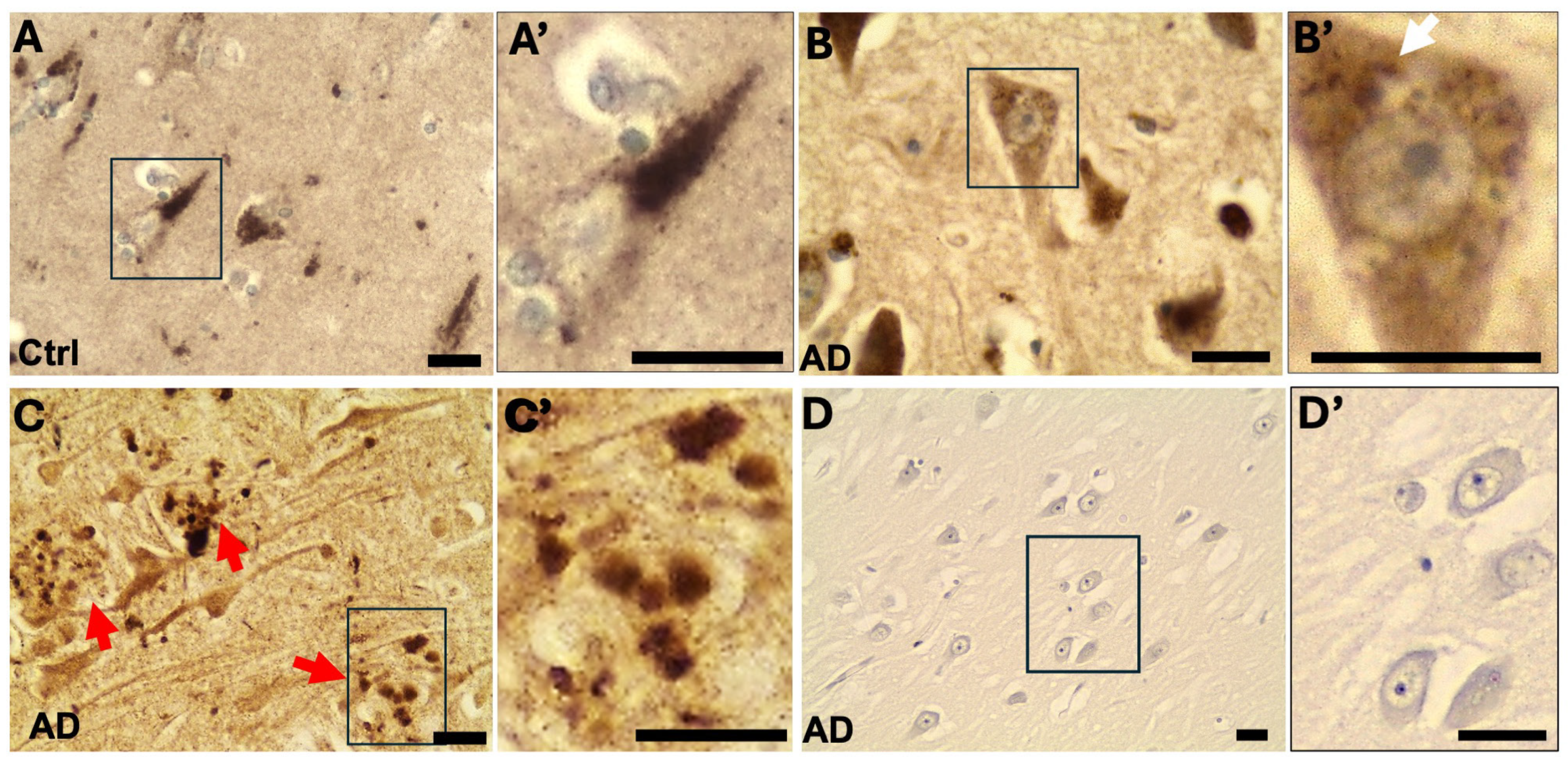
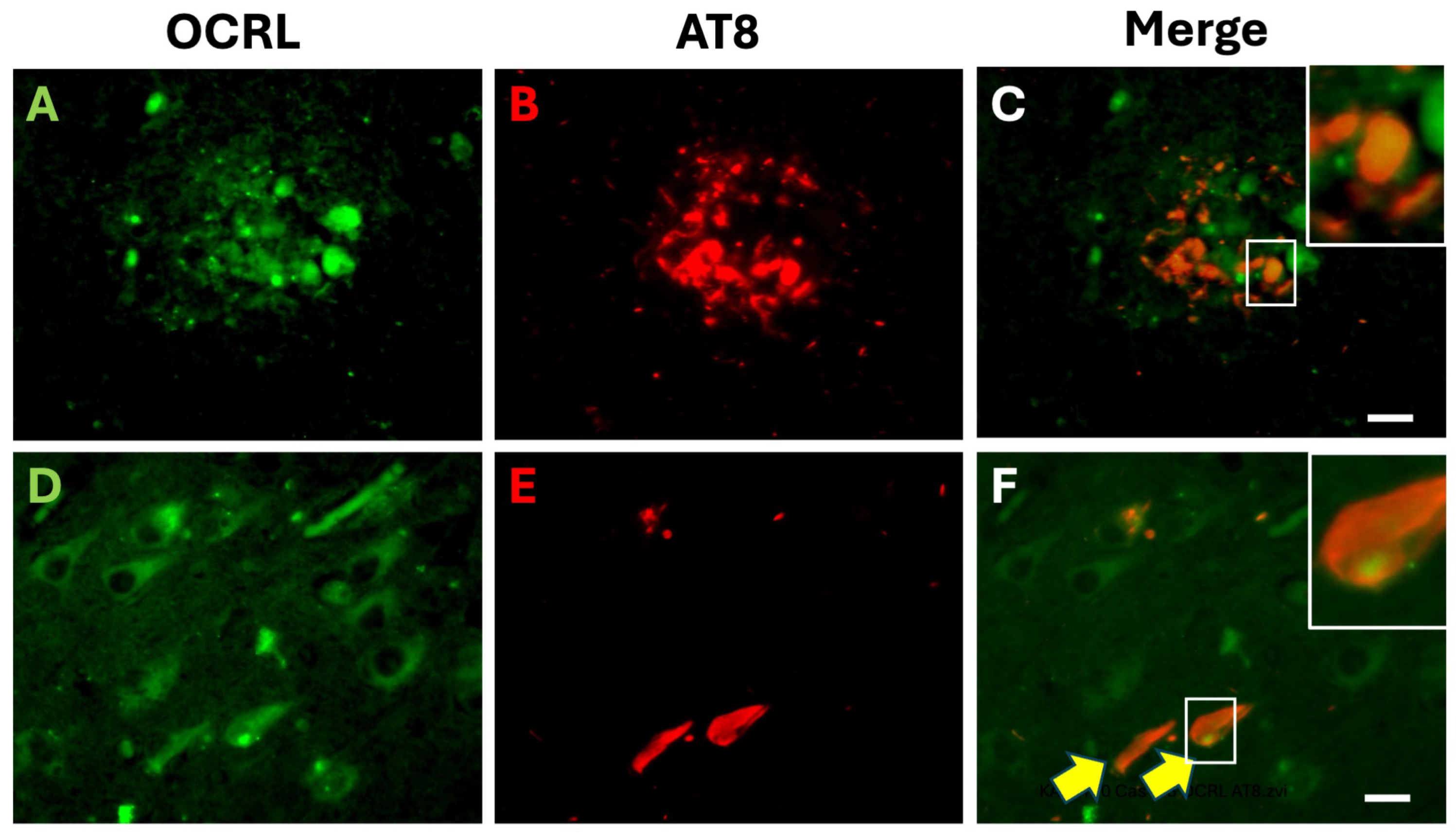
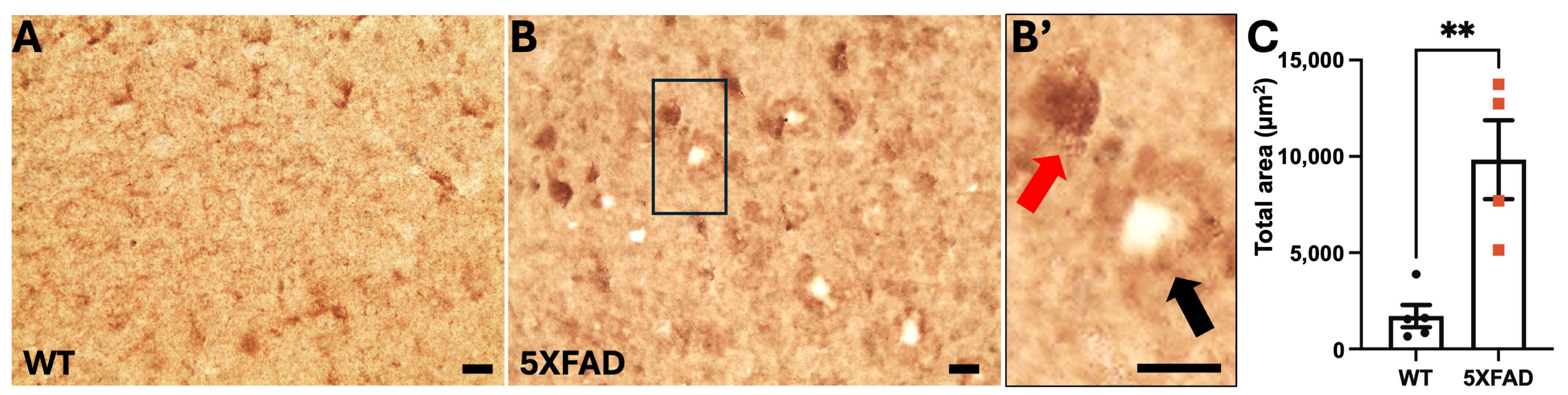
2.1. OCRL Protein Accumulates in Dystrophic Neurites in AD Brains
2.2. RNA Levels, Protein Solubility, and Post-Translational Modifications (PTMs) of OCRL in AD Brains
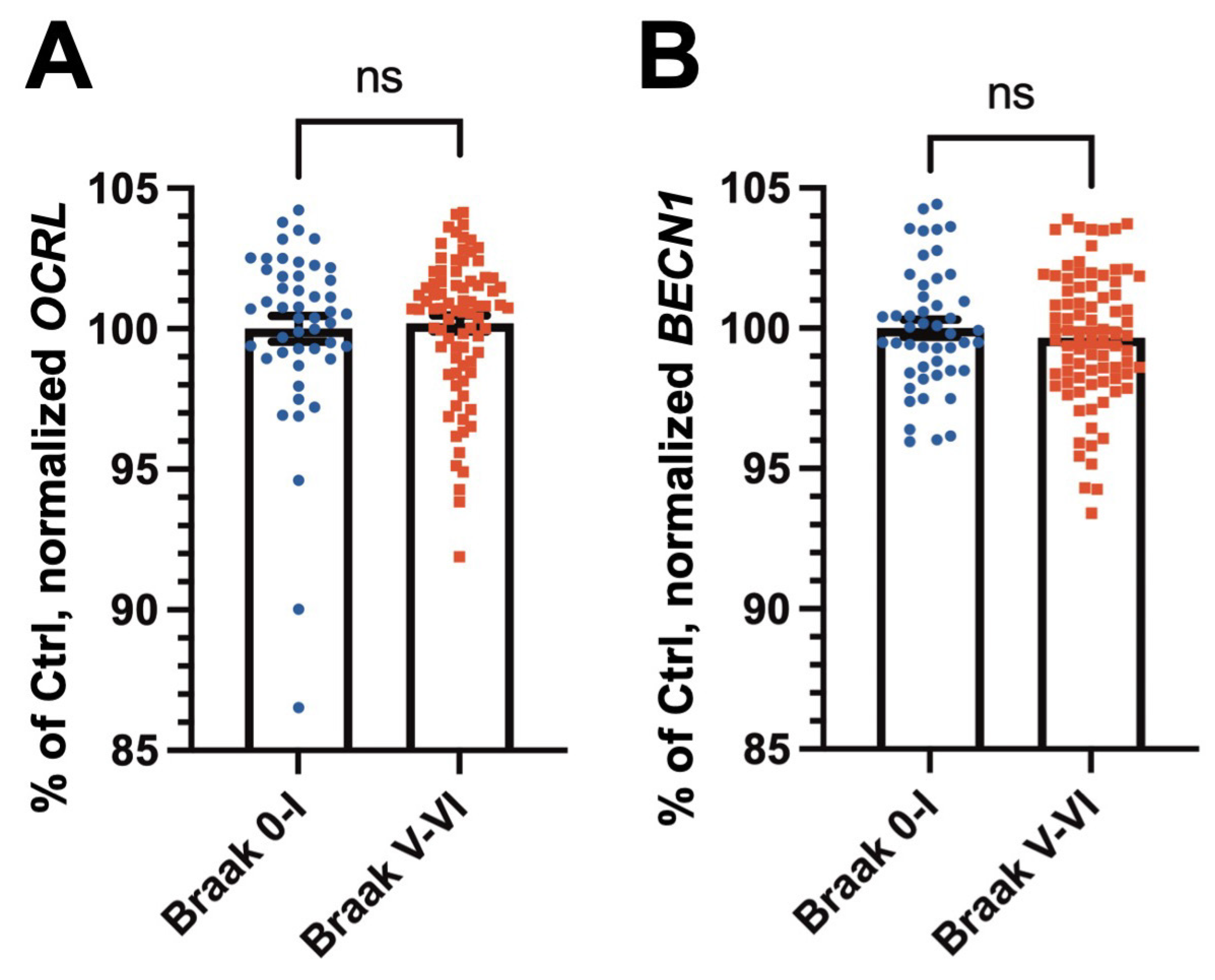
2.2.1. RNA Expression Levels of OCRL and the Autophagy Marker BECN1 Are Not Significantly Altered in AD Brains
2.2.2. OCRL Is Depleted from the RIPA-Soluble Fraction and Correlates with Beclin1 Levels in Control and AD Brains
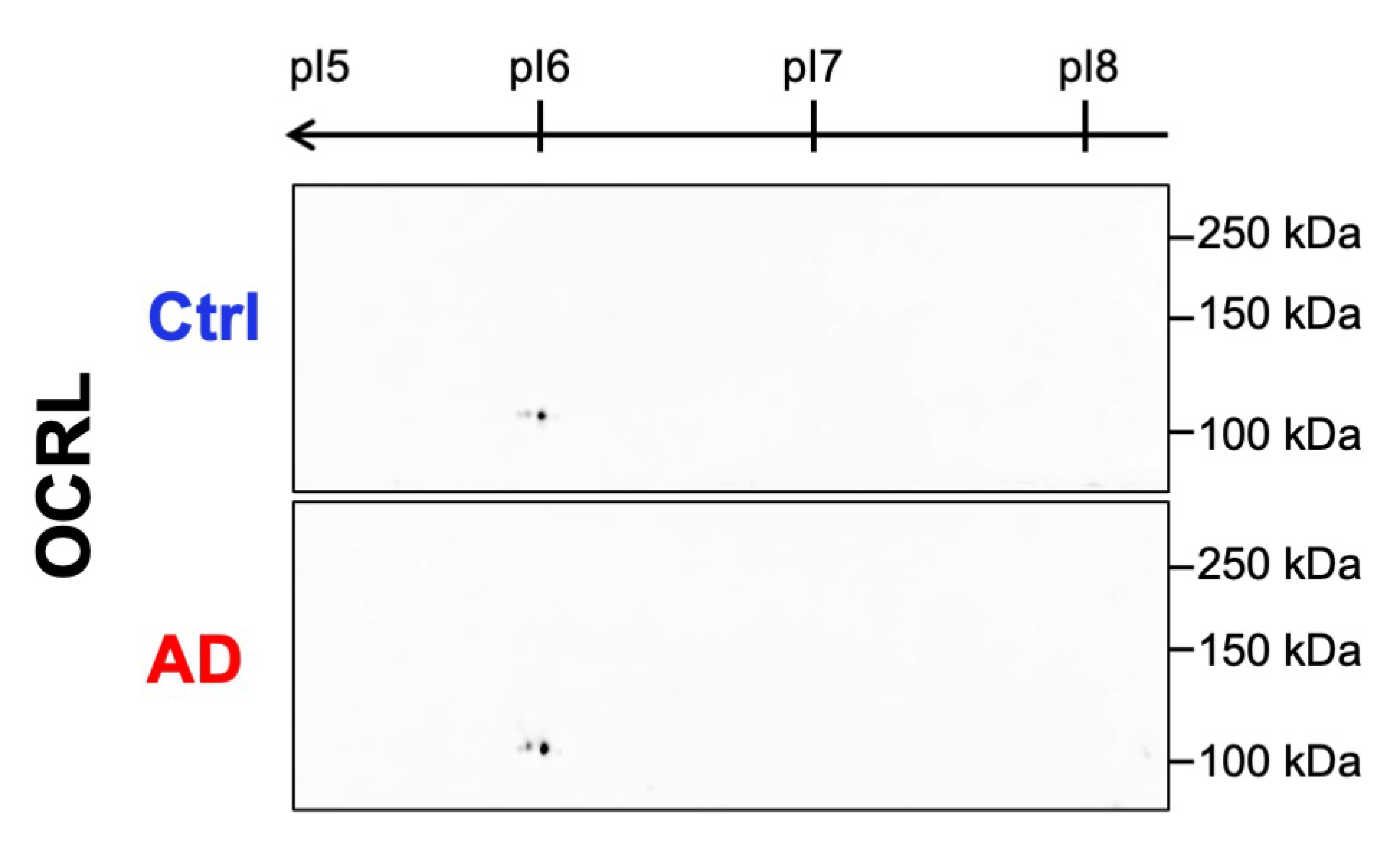
2.2.3. No Aberrant Post-Translational Modifications of OCRL Were Detected in AD Samples by 2D WB
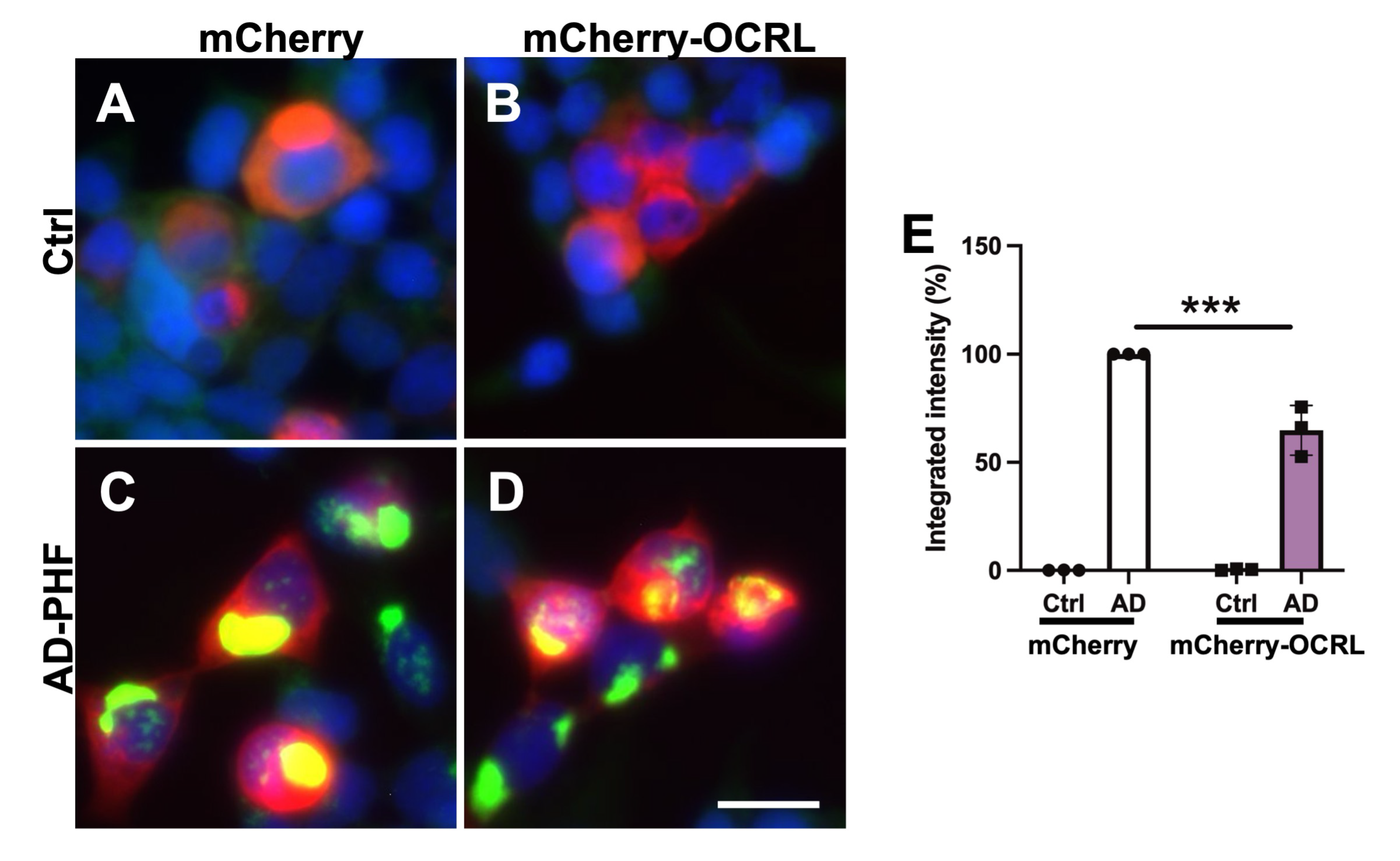
2.3. OCRL Overexpression Attenuates FRET-Positive Tau Inclusions in HEK Tau RD P301L FRET Biosensor Cells
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Human Brain Tissues
4.3. Preparation of Brain Homogenates for Biochemical Analysis
4.4. Preparation of Human Sarkosyl Insoluble PHF-Tau Fraction
4.5. Analyses of RNA Expressions Human and Mouse Data Sets
4.6. WB
4.7. Mouse Lines
4.8. Immunohistochemistry
4.9. Cell Culture
4.10. Liposome-Mediated Transduction of Sarkosyl Insoluble Fraction Containing AD-PHF in Tau RD P301S FRET Biosensor Cells
4.11. Fluorescence-Activated Cell Sorting (FACS)
4.12. Fixation and Counterstaining of Tau RD P301S FRET Biosensor Cells
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Couck, A.M.; Passareiro, E.; Flament-Durand, J. Neurofibrillary tangles of Alzheimer’s disease: An immunohistochemical study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar] [PubMed]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Nishikawa, T.; Takahashi, T.; Nakamori, M.; Yamazaki, Y.; Kurashige, T.; Nagano, Y.; Nishida, Y.; Izumi, Y.; Matsumoto, M. Phosphatidylinositol-4,5-bisphosphate is enriched in granulovacuolar degeneration bodies and neurofibrillary tangles. Neuropathol. Appl. Neurobiol. 2014, 40, 489–501. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Ando, K.; Erneux, C.; Homa, M.; Houben, S.; de Fisenne, M.A.; Brion, J.P.; Leroy, K. Dysregulation of Phosphoinositide 5-phosphatases and phosphoinositides in Alzheimer’s disease. Front. Neurosci. 2021, 15, 614855. [Google Scholar] [CrossRef]
- Kawano, T.; Indo, Y.; Nakazato, H.; Shimadzu, M.; Matsuda, I. Oculocerebrorenal syndrome of Lowe: Three mutations in the OCRL1 gene derived from three patients with different phenotypes. Am. J. Med. Genet. 1998, 77, 348–355. [Google Scholar] [CrossRef]
- Pirruccello, M.; De Camilli, P. Inositol 5-phosphatases: Insights from the Lowe syndrome protein OCRL. Trends Biochem. Sci. 2012, 37, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jefferson, A.B.; Auethavekiat, V.; Majerus, P.W. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1995, 92, 4853–4856. [Google Scholar] [CrossRef]
- De Leo, M.G.; Staiano, L.; Vicinanza, M.; Luciani, A.; Carissimo, A.; Mutarelli, M.; Di Campli, A.; Polishchuk, E.; Di Tullio, G.; Morra, V.; et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 2016, 18, 839–850. [Google Scholar] [CrossRef]
- Festa, B.P.; Berquez, M.; Gassama, A.; Amrein, I.; Ismail, H.M.; Samardzija, M.; Staiano, L.; Luciani, A.; Grimm, C.; Nussbaum, R.L.; et al. OCRL deficiency impairs endolysosomal function in a humanized mouse model for Lowe syndrome and Dent disease. Hum. Mol. Genet. 2019, 28, 1931–1946. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Barnes, J.; Salas, F.; Mokhtari, R.; Dolstra, H.; Pedrosa, E.; Lachman, H.M. Modeling the neuropsychiatric manifestations of Lowe syndrome using induced pluripotent stem cells: Defective F-actin polymerization and WAVE-1 expression in neuronal cells. Mol. Autism. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Ijuin, T.; Horinouchi, T.; Yamamura, T.; Nagano, C.; Okada, E.; Ishiko, S.; Aoto, Y.; Rossanti, R.; Ninchoji, T.; et al. Identification of novel OCRL isoforms associated with phenotypic differences between Dent disease-2 and Lowe syndrome. Nephrol Dial Transpl. 2022, 37, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, V.; Vaz-Silva, J.; Zhu, M.; Gomes, P.; Silva, J.M.; Sousa, N.; Sotiropoulos, I.; Waites, C.L. Rab35 and glucocorticoids regulate APP and BACE1 trafficking to modulate Abeta production. Cell Death Dis. 2021, 12, 1137. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Barnes, J.; Pedrosa, E.; Herman, N.S.; Salas, F.; Wang, P.; Zheng, D.; Lachman, H.M. Transcriptome analysis of neural progenitor cells derived from Lowe syndrome induced pluripotent stem cells: Identification of candidate genes for the neurodevelopmental and eye manifestations. J. Neurodev. Disord. 2020, 12, 14. [Google Scholar] [CrossRef]
- Wiersma, V.I.; Hoozemans, J.J.M.; Scheper, W. Untangling the origin and function of granulovacuolar degeneration bodies in neurodegenerative proteinopathies. Acta Neuropathol. Commun. 2020, 8, 153. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Hatefi, Y.; Hanstein, W.G. Solubilization of particulate proteins and nonelectrolytes by chaotropic agents. Proc. Natl. Acad. Sci. USA 1969, 62, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Ndjim, M.; Turbant, S.; Fontaine, G.; Pregoni, G.; Dauphinot, L.; Yilmaz, Z.; Suain, V.; Mansour, S.; Authelet, M.; et al. The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Tomimura, K.; Sazdovitch, V.; Suain, V.; Yilmaz, Z.; Authelet, M.; Ndjim, M.; Vergara, C.; Belkouch, M.; Potier, M.C.; et al. Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol. Dis. 2016, 94, 32–43. [Google Scholar] [CrossRef]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Franceschini, A.; Lin, J.; von Mering, C.; Jensen, L.J. SVD-phy: Improved prediction of protein functional associations through singular value decomposition of phylogenetic profiles. Bioinformatics 2016, 32, 1085–1087. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- von Mering, C.; Jensen, L.J.; Kuhn, M.; Chaffron, S.; Doerks, T.; Kruger, B.; Snel, B.; Bork, P. STRING 7--recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007, 35, D358–D362. [Google Scholar] [CrossRef]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.G.; Davies, P.; Schein, J.D.; Binder, L.I. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J. Biol. Chem. 1992, 267, 564–569. [Google Scholar] [CrossRef]
- Ando, K.; Dourlen, P.; Sambo, A.V.; Bretteville, A.; Belarbi, K.; Vingtdeux, V.; Eddarkaoui, S.; Drobecq, H.; Ghestem, A.; Begard, S.; et al. Tau pathology modulates Pin1 post-translational modifications and may be relevant as biomarker. Neurobiol. Aging 2013, 34, 757–769. [Google Scholar] [CrossRef]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef]
- PhoshoSitePlus. Available online: https://www.phosphosite.org/ (accessed on 15 March 2025).
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef] [PubMed]
- de Fisenne, M.A.; Yilmaz, Z.; De Decker, R.; Suain, V.; Buee, L.; Ando, K.; Brion, J.P.; Leroy, K. Alzheimer PHF-tau aggregates do not spread tau pathology to the brain via the Retino-tectal projection after intraocular injection in male mouse models. Neurobiol. Dis. 2022, 174, 105875. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef]
- Vicinanza, M.; Di Campli, A.; Polishchuk, E.; Santoro, M.; Di Tullio, G.; Godi, A.; Levtchenko, E.; De Leo, M.G.; Polishchuk, R.; Sandoval, L.; et al. OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J. 2011, 30, 4970–4985. [Google Scholar] [CrossRef]
- Funk, K.E.; Mrak, R.E.; Kuret, J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol. Appl. Neurobiol. 2011, 37, 295–306. [Google Scholar] [CrossRef]
- Wang, B.; He, W.; Prosseda, P.P.; Li, L.; Kowal, T.J.; Alvarado, J.A.; Wang, Q.; Hu, Y.; Sun, Y. OCRL regulates lysosome positioning and mTORC1 activity through SSX2IP-mediated microtubule anchoring. EMBO Rep. 2021, 22, e52173. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Vest, R.; Prado, M.A.; Wilson-Grady, J.; Paulo, J.A.; Shibuya, Y.; Moran-Losada, P.; Lee, T.T.; Luo, J.; Gygi, S.P.; et al. Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer’s disease. Nat. Cell Biol. 2025, 27, 619–632. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: Neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef]
- Ungewickell, A.J.; Majerus, P.W. Increased levels of plasma lysosomal enzymes in patients with Lowe syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 13342–13344. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Ando, K.; Nagaraj, S.; Kucukali, F.; de Fisenne, M.A.; Kosa, A.C.; Doeraene, E.; Lopez Gutierrez, L.; Brion, J.P.; Leroy, K. PICALM and Alzheimer’s Disease: An Update and Perspectives. Cells 2022, 11, 3994. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.; Tysnes, O.B.; Alves, G.W.; Nido, G.S.; Tzoulis, C. Altered transcriptome-proteome coupling indicates aberrant proteostasis in Parkinson’s disease. iScience 2023, 26, 105925. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wu, G.; Zhang, W. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: A quantitative analysis. Genetics 2006, 174, 2229–2243. [Google Scholar] [CrossRef] [PubMed]
- Gry, M.; Rimini, R.; Stromberg, S.; Asplund, A.; Ponten, F.; Uhlen, M.; Nilsson, P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genom. 2009, 10, 365. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, W.; Cui, Q.; Zhou, Y. Computational screening of potential regulators for mRNA-protein expression level discrepancy. Biochem. Biophys. Res. Commun. 2020, 523, 196–201. [Google Scholar] [CrossRef]
- Harnik, Y.; Buchauer, L.; Ben-Moshe, S.; Averbukh, I.; Levin, Y.; Savidor, A.; Eilam, R.; Moor, A.E.; Itzkovitz, S. Spatial discordances between mRNAs and proteins in the intestinal epithelium. Nat. Metab. 2021, 3, 1680–1693. [Google Scholar] [CrossRef]
- Arancio, O. PIP2: A new key player in Alzheimer’s disease. Cellscience 2008, 5, 44–47. [Google Scholar]
- Berman, D.E.; Dall’Armi, C.; Voronov, S.V.; McIntire, L.B.; Zhang, H.; Moore, A.Z.; Staniszewski, A.; Arancio, O.; Kim, T.W.; Di Paolo, G. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 2008, 11, 547–554. [Google Scholar] [CrossRef]
- Landman, N.; Jeong, S.Y.; Shin, S.Y.; Voronov, S.V.; Serban, G.; Kang, M.S.; Park, M.K.; Di Paolo, G.; Chung, S.; Kim, T.W. Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 19524–19529. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, M.; Elder, G.A.; Sano, M.; Holtzman, D.M.; Gandy, S.; Cardozo, C.; Haroutunian, V.; Robakis, N.K.; Cai, D. Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 11965–11970. [Google Scholar] [CrossRef]
- Sun, T.; Li, X.; Zhang, P.; Chen, W.D.; Zhang, H.L.; Li, D.D.; Deng, R.; Qian, X.J.; Jiao, L.; Ji, J.; et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 2015, 6, 7215. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, K.S.; Mao, Y.; McCrea, H.J.; Zoncu, R.; Lee, S.; Paradise, S.; Modregger, J.; Biemesderfer, D.; Toomre, D.; De Camilli, P. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev. Cell 2007, 13, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Hagemann, N.; Schoebel, S.; Blankenfeldt, W.; Goody, R.S.; Erdmann, K.S.; Itzen, A. A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. EMBO J. 2011, 30, 1659–1670. [Google Scholar] [CrossRef]
- Hyvola, N.; Diao, A.; McKenzie, E.; Skippen, A.; Cockcroft, S.; Lowe, M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 2006, 25, 3750–3761. [Google Scholar] [CrossRef]
- Maxson, M.E.; Sarantis, H.; Volchuk, A.; Brumell, J.H.; Grinstein, S. Rab5 regulates macropinocytosis by recruiting the inositol 5-phosphatases OCRL and Inpp5b that hydrolyse PtdIns(4,5)P2. J. Cell Sci. 2021, 134, jcs252411. [Google Scholar] [CrossRef] [PubMed]
- de Hoop, M.J.; Huber, L.A.; Stenmark, H.; Williamson, E.; Zerial, M.; Parton, R.G.; Dotti, C.G. The involvement of the small GTP-binding protein Rab5a in neuronal endocytosis. Neuron 1994, 13, 11–22. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Xu, W.; Fang, F.; Ding, J.; Wu, C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic 2018, 19, 253–262. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- Tremel, S.; Ohashi, Y.; Morado, D.R.; Bertram, J.; Perisic, O.; Brandt, L.T.L.; von Wrisberg, M.K.; Chen, Z.A.; Maslen, S.L.; Kovtun, O.; et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 2021, 12, 1564. [Google Scholar] [CrossRef]
- Zhang, X.; Hartz, P.A.; Philip, E.; Racusen, L.C.; Majerus, P.W. Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 1998, 273, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Takahashi, T.; Nakamori, M.; Hosomi, N.; Maruyama, H.; Miyazaki, Y.; Izumi, Y.; Matsumoto, M. The identification of raft-derived tau-associated vesicles that are incorporated into immature tangles and paired helical filaments. Neuropathol. Appl. Neurobiol. 2016, 42, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, H.; Tang, X.Q. Tau internalization: A complex step in tau propagation. Ageing Res. Rev. 2021, 67, 101272. [Google Scholar] [CrossRef]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef]
- Choi, W.Y.; Kim, S.; Aurass, P.; Huo, W.; Creasey, E.A.; Edwards, M.; Lowe, M.; Isberg, R.R. SdhA blocks disruption of the Legionella-containing vacuole by hijacking the OCRL phosphatase. Cell Rep. 2021, 37, 109894. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Jepson, T.; Shukla, S.; Maya-Romero, A.; Kampmann, M.; Xu, K.; Hurley, J.H. Tau fibrils induce nanoscale membrane damage and nucleate cytosolic tau at lysosomes. bioRxiv 2023. [Google Scholar] [CrossRef]
- Ando, K.; Kucukali, F.; Doeraene, E.; Nagaraj, S.; Antonelli, E.M.; Thazin Htut, M.; Yilmaz, Z.; Kosa, A.C.; Lopez-Guitierrez, L.; Quintanilla-Sanchez, C.; et al. Alteration of gene expression and protein solubility of the PI 5-phosphatase SHIP2 are correlated with Alzheimer’s disease pathology progression. Acta Neuropathol. 2024, 147, 94. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Feiner, L.; Lang, E.; Szendrei, G.I.; Goedert, M.; Lee, V.M. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J. Neurosci. Res. 1994, 39, 669–673. [Google Scholar] [CrossRef]
- Ball, M.; Braak, H.; Coleman, P.; Dickson, D.; Duyckaerts, C.; Gambetti, P.; Hansen, L.; Hyman, B.; Jellinger, K.; Markesberg, W.; et al. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S1–S2. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Teipel, S.J.; Cavedo, E.; Weschke, S.; Grothe, M.J.; Rojkova, K.; Fontaine, G.; Dauphinot, L.; Gonzalez-Escamilla, G.; Potier, M.C.; Bertin, H.; et al. Cortical amyloid accumulation is associated with alterations of structural integrity in older people with subjective memory complaints. Neurobiol. Aging 2017, 57, 143–152. [Google Scholar] [CrossRef]
- Ando, K.; Leroy, K.; Heraud, C.; Yilmaz, Z.; Authelet, M.; Suain, V.; De Decker, R.; Brion, J.P. Accelerated human mutant tau aggregation by knocking out murine tau in a transgenic mouse model. Am. J. Pathol. 2011, 178, 803–816. [Google Scholar] [CrossRef]
- Ando, K.; Brion, J.P.; Stygelbout, V.; Suain, V.; Authelet, M.; Dedecker, R.; Chanut, A.; Lacor, P.; Lavaur, J.; Sazdovitch, V.; et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013, 125, 861–878. [Google Scholar] [CrossRef]
- Coulonval, K.; Vercruysse, V.; Paternot, S.; Pita, J.M.; Corman, R.; Raspe, E.; Roger, P.P. Monoclonal antibodies to activated CDK4: Use to investigate normal and cancerous cell cycle regulation and involvement of phosphorylations of p21 and p27. Cell Cycle 2022, 21, 12–32. [Google Scholar] [CrossRef]
- Greenberg, S.G.; Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef]
- Brion, J.P.; Hanger, D.P.; Bruce, M.T.; Couck, A.M.; Flament-Durand, J.; Anderton, B.H. Tau in Alzheimer neurofibrillary tangles. N- and C-terminal regions are differentially associated with paired helical filaments and the location of a putative abnormal phosphorylation site. Biochem. J. 1991, 273 Pt 1, 127–133. [Google Scholar] [CrossRef]
- Ando, K.; Kabova, A.; Stygelbout, V.; Leroy, K.; Heraud, C.; Frederick, C.; Suain, V.; Yilmaz, Z.; Authelet, M.; Dedecker, R.; et al. Vaccination with Sarkosyl Insoluble PHF-Tau Decrease Neurofibrillary Tangles Formation in Aged Tau Transgenic Mouse Model: A Pilot Study. J. Alzheimers Dis. 2014, 40, S135–S145. [Google Scholar] [CrossRef]
- Lee, A.J.; Ma, Y.; Yu, L.; Dawe, R.J.; McCabe, C.; Arfanakis, K.; Mayeux, R.; Bennett, D.A.; Klein, H.U.; De Jager, P.L. Multi-region brain transcriptomes uncover two subtypes of aging individuals with differences in Alzheimer risk and the impact of APOEepsilon4. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; De Decker, R.; Vergara, C.; Yilmaz, Z.; Mansour, S.; Suain, V.; Sleegers, K.; de Fisenne, M.A.; Houben, S.; Potier, M.C.; et al. Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol. 2020, 139, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- Ando, K.; Kosa, A.C.; Mehadji, Y.; Lasri, H.; Lopez-Gutierrez, L.; Quintanilla-Sanchez, C.; Aydin, E.; Doeraene, E.; Wathelet-Depauw, A.; Nagaraj, S.; et al. Deletion of Murine APP Aggravates Tau and Amyloid Pathologies in the 5xFADXTg30 Alzheimer’s Disease Model. Biomolecules 2025, 15, 159. [Google Scholar] [CrossRef]
- Shooshtari, P.; Fortuno, E.S., 3rd; Blimkie, D.; Yu, M.; Gupta, A.; Kollmann, T.R.; Brinkman, R.R. Correlation analysis of intracellular and secreted cytokines via the generalized integrated mean fluorescence intensity. Cytom. A 2010, 77, 873–880. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, K.; Thazin Htut, M.; Antonelli, E.M.; Kosa, A.-C.; Lopez-Gutierrez, L.; Quintanilla-Sánchez, C.; Aydin, E.; Doeraene, E.; Nagaraj, S.; Ramos, A.R.; et al. Dysregulation of Inositol Polyphosphate 5-Phosphatase OCRL in Alzheimer’s Disease: Implications for Autophagy Dysfunction. Int. J. Mol. Sci. 2025, 26, 5827. https://doi.org/10.3390/ijms26125827
Ando K, Thazin Htut M, Antonelli EM, Kosa A-C, Lopez-Gutierrez L, Quintanilla-Sánchez C, Aydin E, Doeraene E, Nagaraj S, Ramos AR, et al. Dysregulation of Inositol Polyphosphate 5-Phosphatase OCRL in Alzheimer’s Disease: Implications for Autophagy Dysfunction. International Journal of Molecular Sciences. 2025; 26(12):5827. https://doi.org/10.3390/ijms26125827
Chicago/Turabian StyleAndo, Kunie, May Thazin Htut, Eugenia Maria Antonelli, Andreea-Claudia Kosa, Lidia Lopez-Gutierrez, Carolina Quintanilla-Sánchez, Emmanuel Aydin, Emilie Doeraene, Siranjeevi Nagaraj, Ana Raquel Ramos, and et al. 2025. "Dysregulation of Inositol Polyphosphate 5-Phosphatase OCRL in Alzheimer’s Disease: Implications for Autophagy Dysfunction" International Journal of Molecular Sciences 26, no. 12: 5827. https://doi.org/10.3390/ijms26125827
APA StyleAndo, K., Thazin Htut, M., Antonelli, E. M., Kosa, A.-C., Lopez-Gutierrez, L., Quintanilla-Sánchez, C., Aydin, E., Doeraene, E., Nagaraj, S., Ramos, A. R., Coulonval, K., Roger, P. P., Brion, J.-P., & Leroy, K. (2025). Dysregulation of Inositol Polyphosphate 5-Phosphatase OCRL in Alzheimer’s Disease: Implications for Autophagy Dysfunction. International Journal of Molecular Sciences, 26(12), 5827. https://doi.org/10.3390/ijms26125827