
Bacillibactin, a Potential Bacillus-Based Antibacterial Non-Ribosomal Peptide: In Silico Studies for Targeting Common Fish Pathogens
 ,
,  and
and 
Abstract
1. Introduction
2. Results
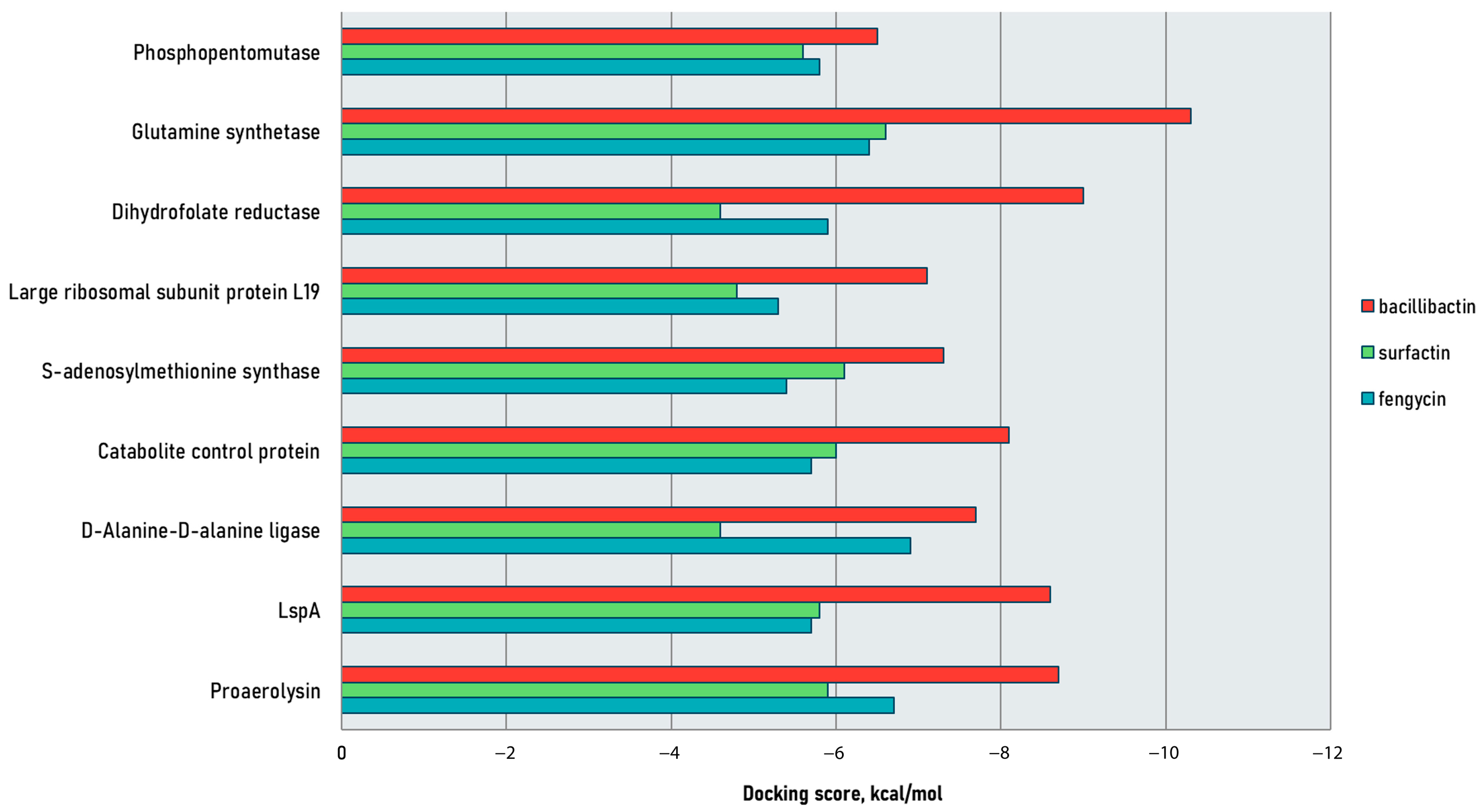
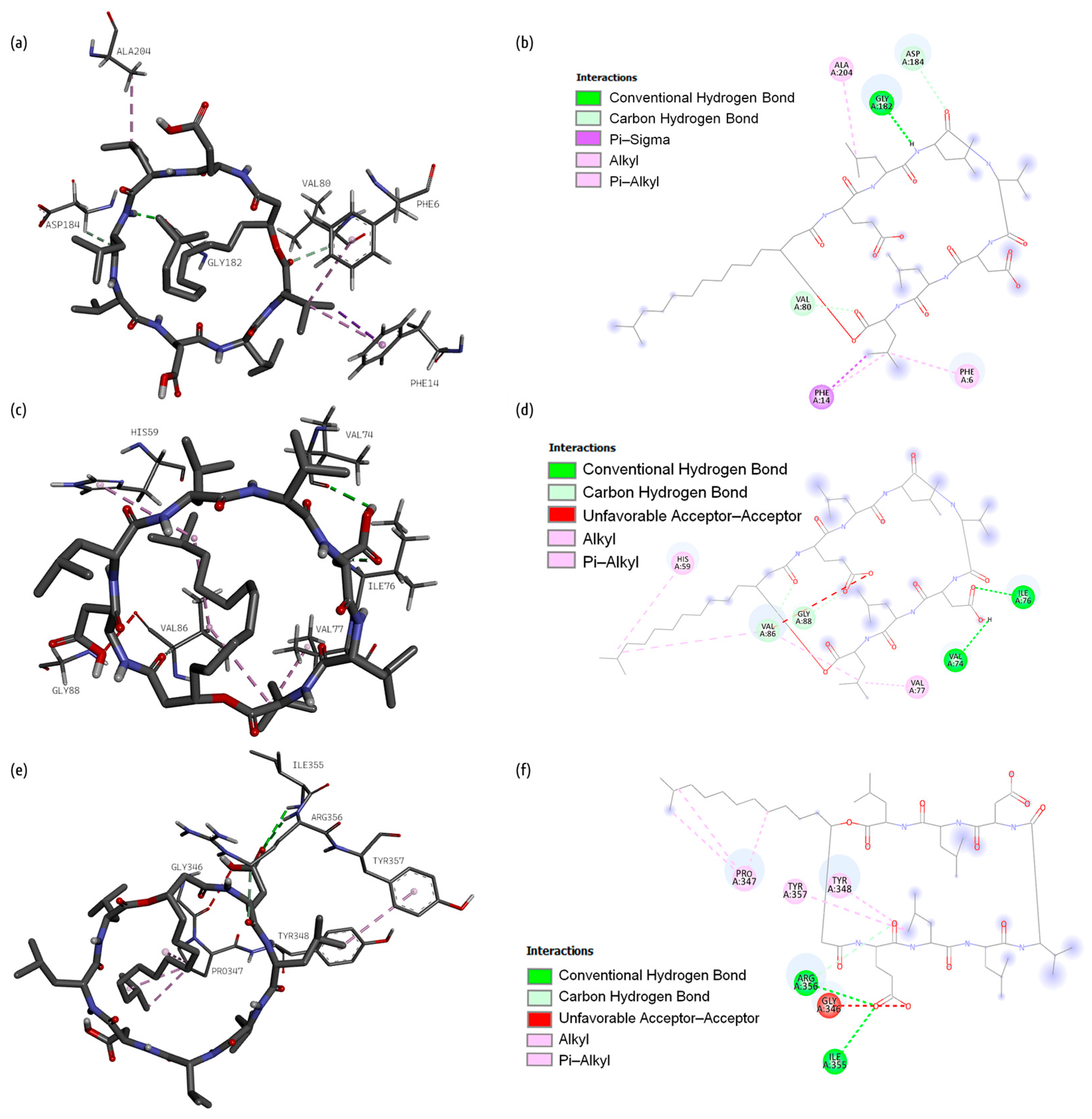
2.1. Docking Studies
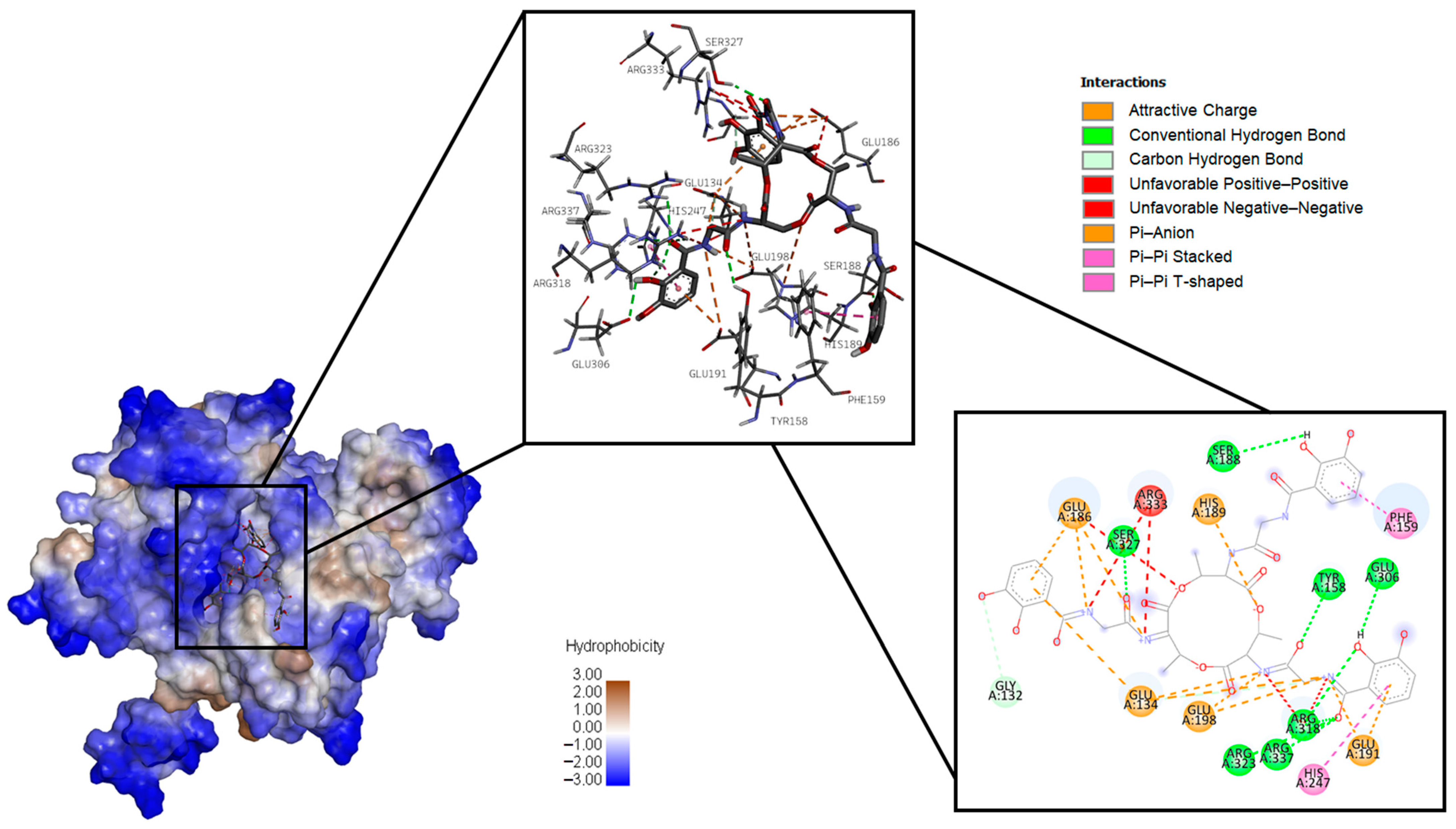
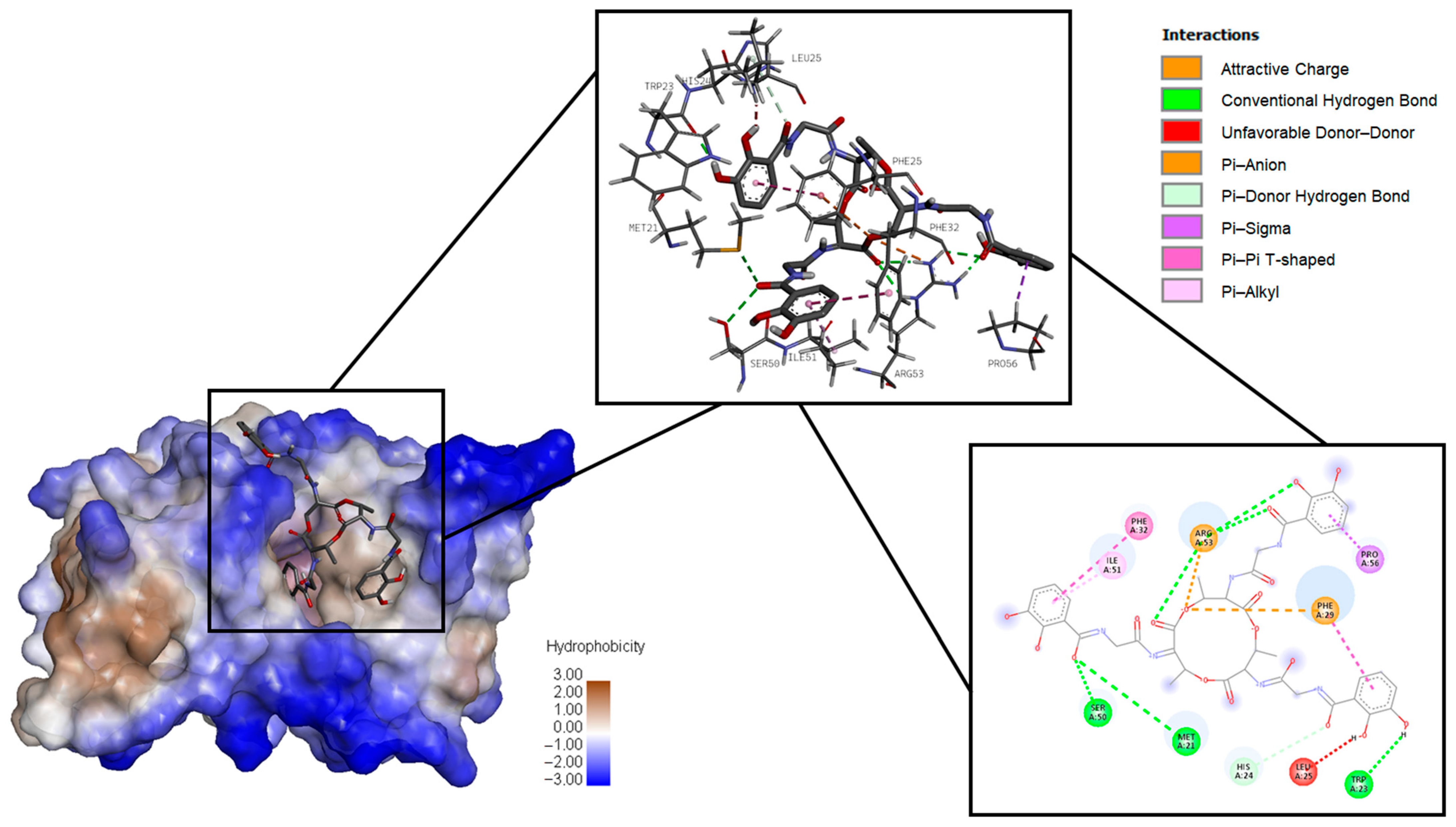
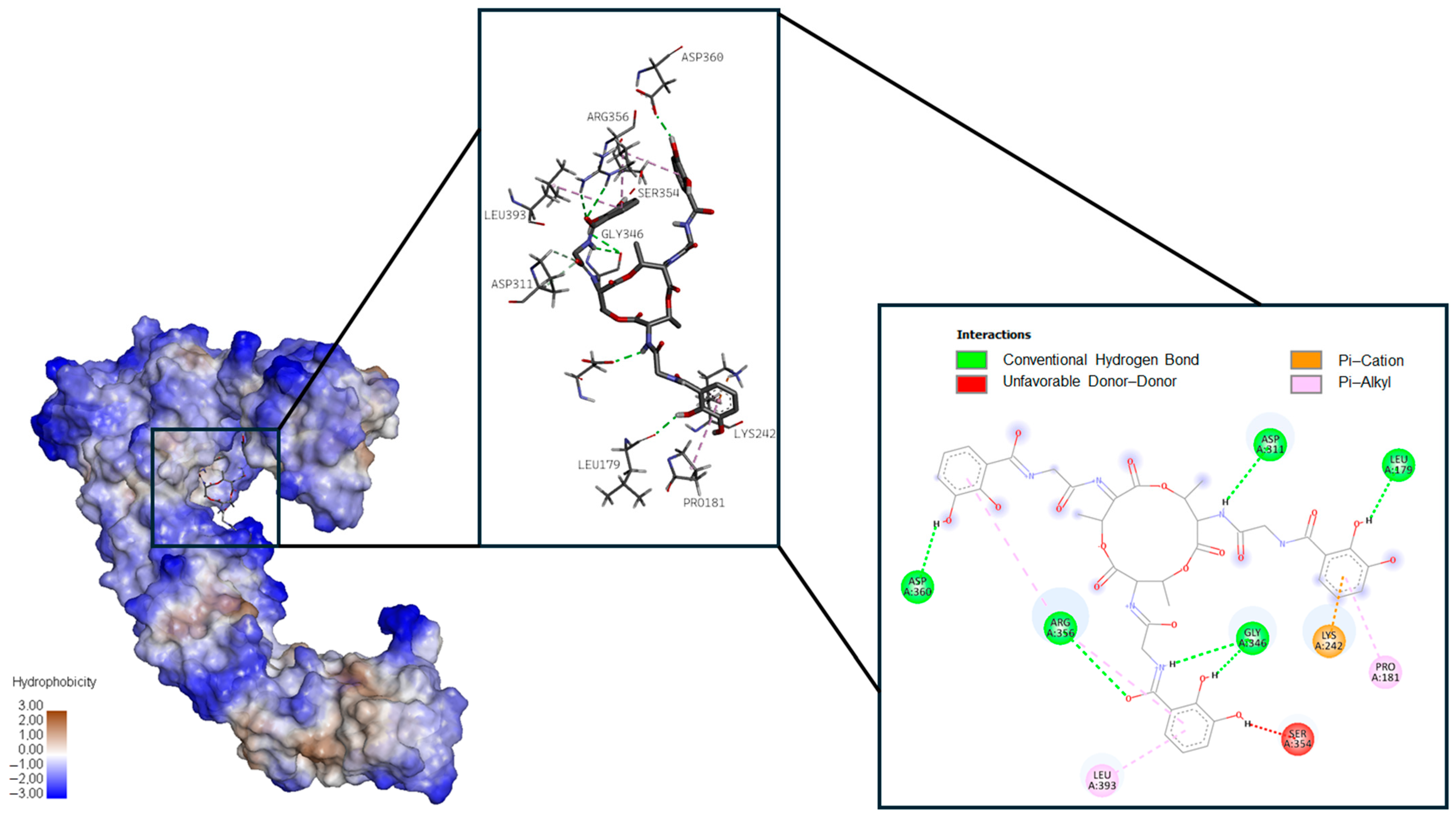
2.1.1. Bacillibactin
2.1.2. Fengycin
2.1.3. Surfactin
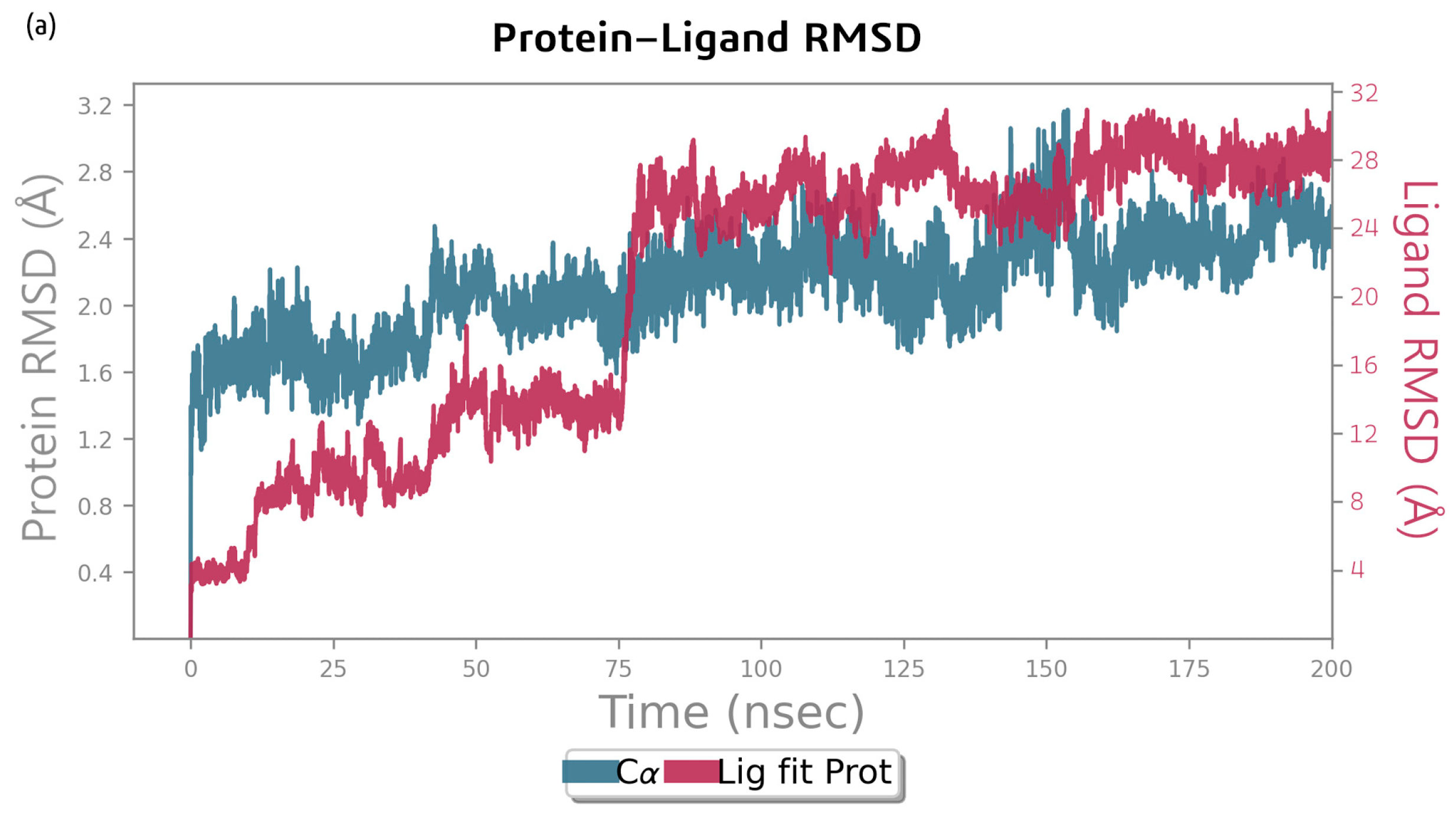
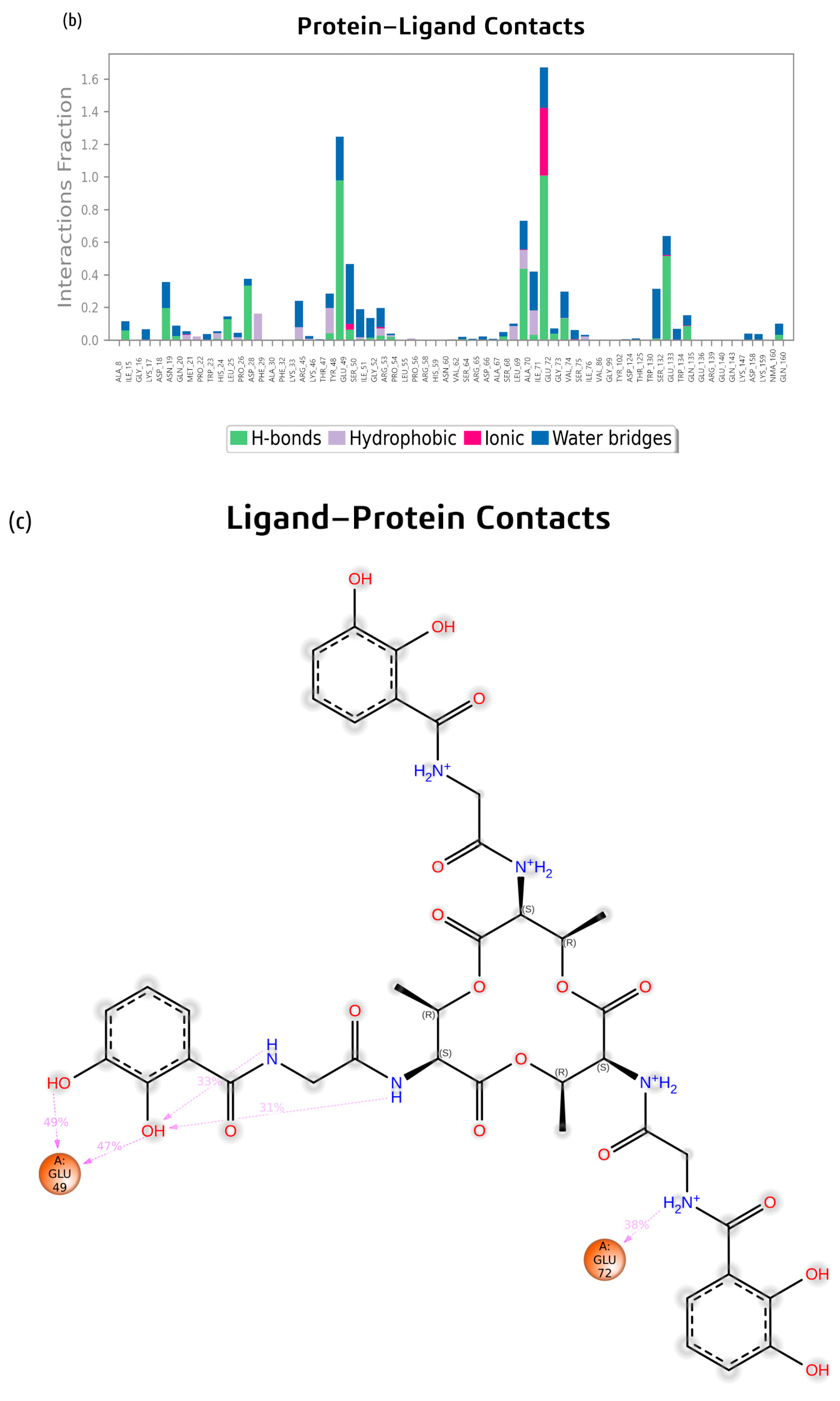
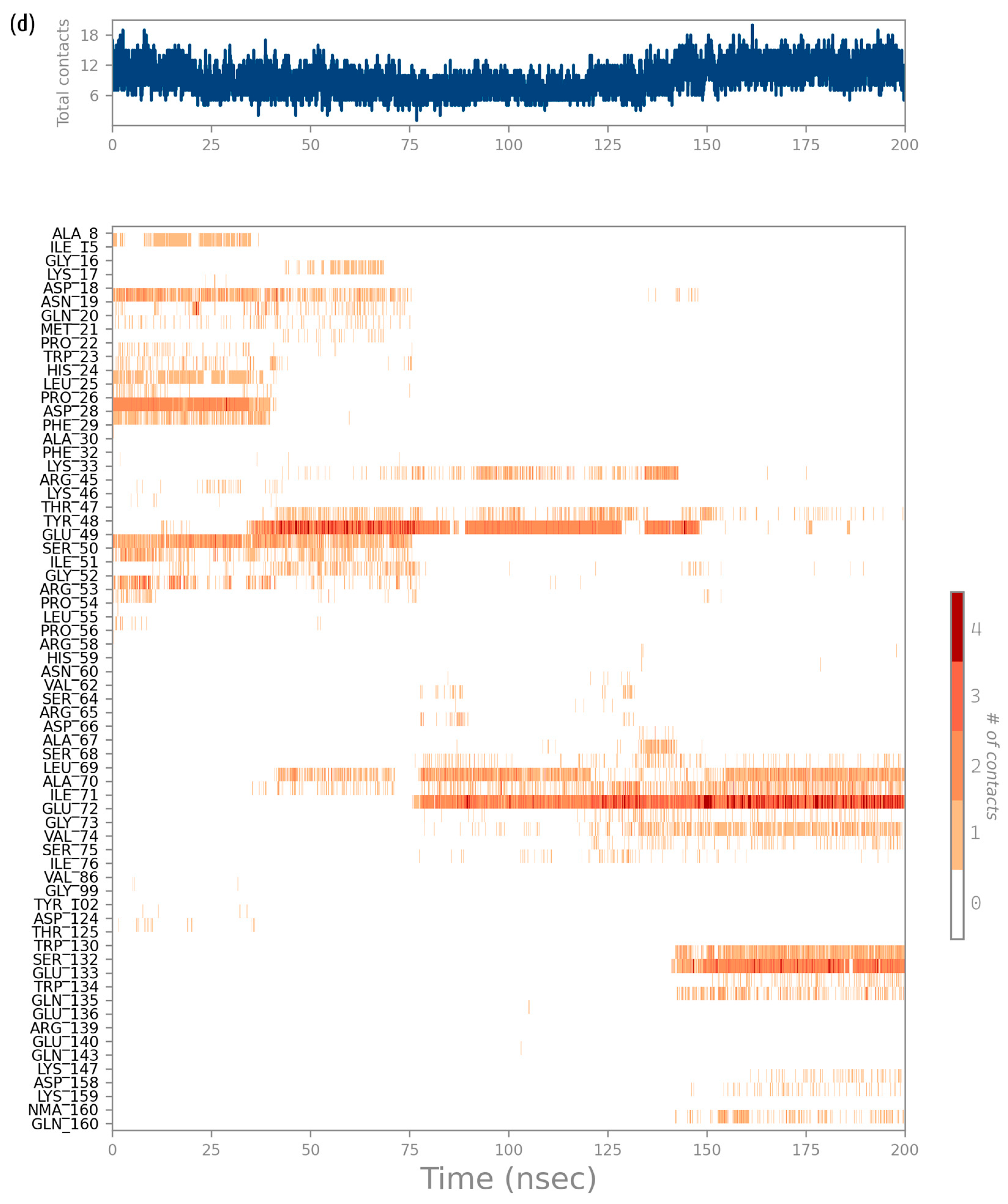
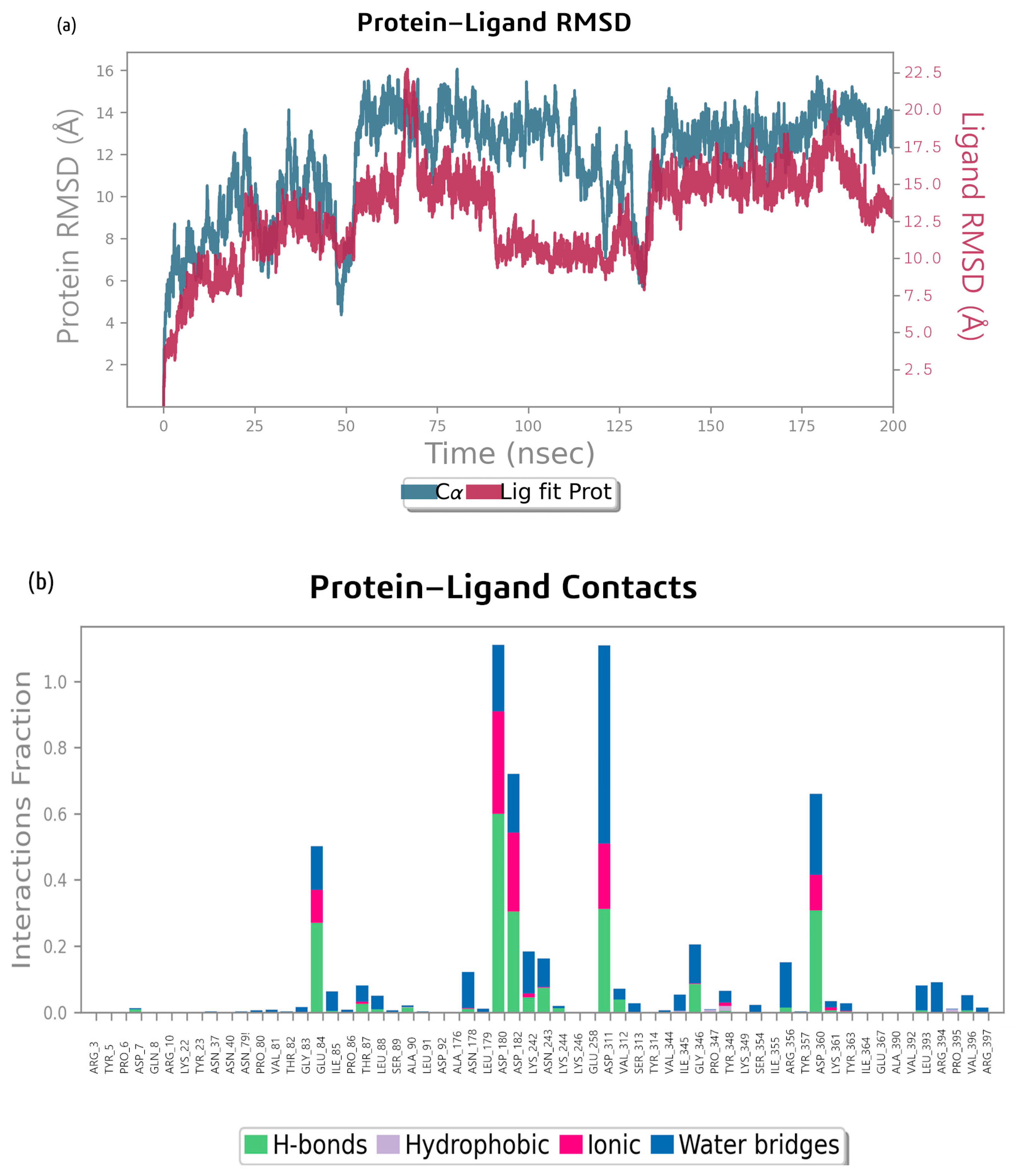
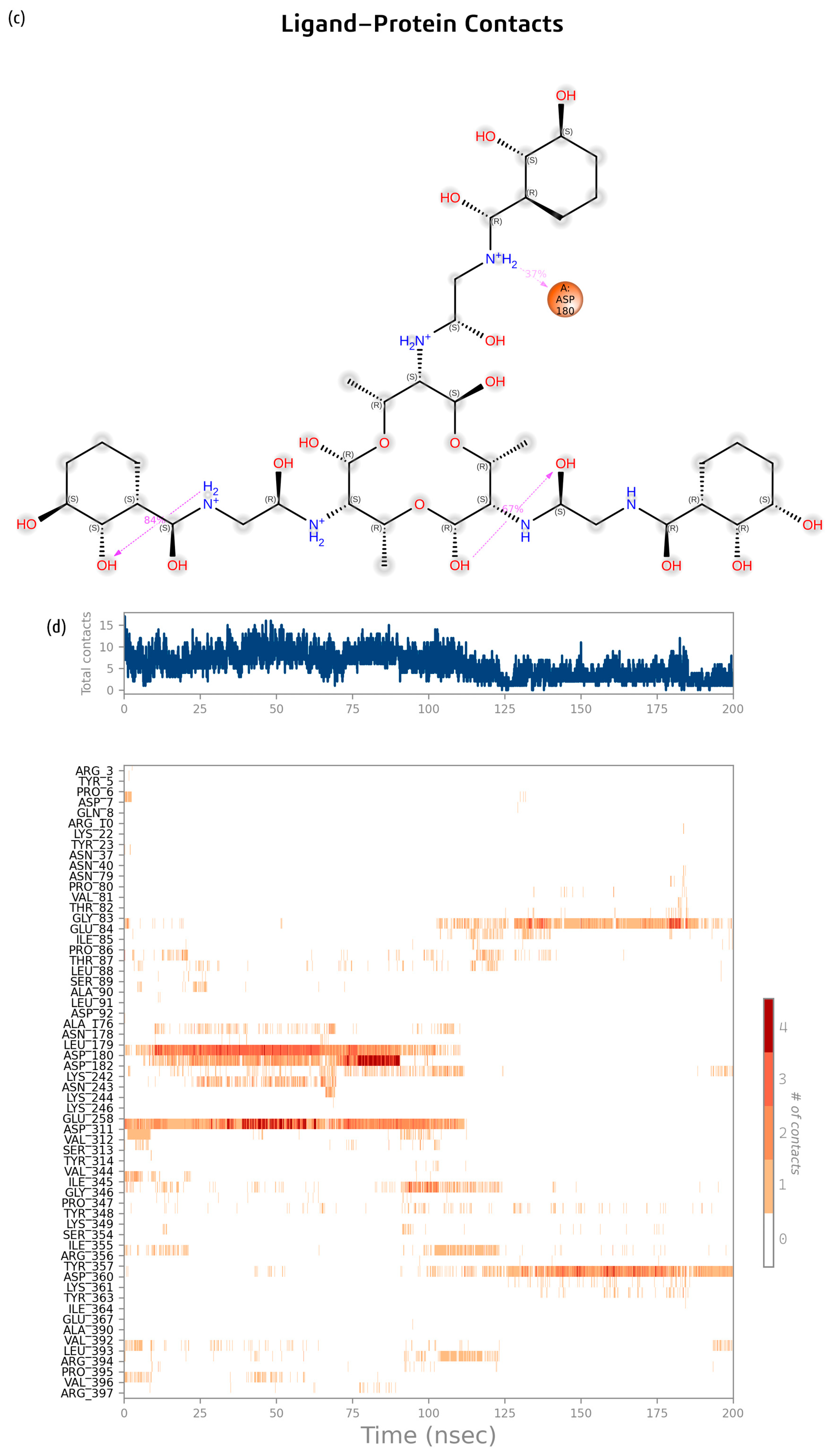
2.2. Molecular Dynamics Simulation
2.3. Bacillibactin Genes in Bacterial Genomes
2.4. In Vitro Exneriments
3. Discussion
4. Materials and Methods
4.1. Preparation of Receptors and Ligands
4.2. Docking Studies
4.3. Molecular Dynamics Simulation
4.4. Probiotics Strains for In Vivo Testing
4.5. Evaluation of Antimicrobial Activity of Strains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caulier, S.; Nannan, C.; Gillis, A.; Licciardi, F.; Bragard, C.; Mahillon, J. Overview of the Antimicrobial Compounds Produced by Members of the Bacillus subtilis Group. Front. Microbiol. 2019, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulou, A.; Theologidis, I.; Benaki, D.; Koukounia, M.; Zervakou, A.; Tzima, A.; Diallinas, G.; Hatzinikolaou, D.G.; Skandalis, N. Direct Antibiotic Activity of Bacillibactin Broadens the Biocontrol Range of Bacillus amyloliquefaciens MBI600. mSphere 2021, 6, e00376-21. [Google Scholar] [CrossRef]
- Hai, N.V. The use of probiotics in aquaculture. J. Appl. Microbiol. 2015, 119, 917–935. [Google Scholar] [CrossRef]
- Ringø, E.; Hoseinifar, S.H.; Ghosh, K.; Doan, H.V.; Beck, B.R.; Song, S.K. Lactic Acid Bacteria in Finfish—An Update. Front. Microbiol. 2018, 9, 1818. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, M.; Karimi, A.; Fallah, F.; Akhavan, M.M. Overview of ribosomal and non-ribosomal antimicrobial peptides produced by Gram positive bacteria. Cell. Mol. Biol. 2017, 63, 20–32. [Google Scholar] [CrossRef]
- Martínez Cruz, P.; Ibáñez, A.L.; Monroy Hermosillo, O.A.; Ramírez Saad, H.C. Use of probiotics in aquaculture. ISRN Microbiol. 2012, 2012, 916845. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Lin, J.; Wang, C. Probiotics in aquaculture: Challenges and outlook. Aquac. Res. 2022, 53, 496–507. [Google Scholar] [CrossRef]
- Ranjan, A.; Rajput, V.D.; Prazdnova, E.V.; Gurnani, M.; Bhardwaj, P.; Sharma, S.; Jindal, T. Nature’s antimicrobial arsenal: Non-ribosomal peptides from PGPB for plant pathogen biocontrol. Fermentation 2023, 9, 597. [Google Scholar] [CrossRef]
- Xu, B.H.; Lu, Y.Q.; Ye, Z.W.; Zheng, Q.W.; Wei, T.; Lin, J.F.; Guo, L.Q. Genomics-guided discovery and structure identification of cyclic lipopeptides from the Bacillus siamensis JFL15. PLoS ONE 2018, 13, e0202893. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.; Zhu, G.; Zhao, K.; Jiang, W.; Li, H.; Tian, X. Mechanism of the Potential therapeutic candidate Bacillus subtilis BSXE-1601 against shrimp pathogenic vibrios and multifunctional metabolites biosynthetic capability of the strain as predicted by genome analysis. Front. Microbiol. 2020, 11, 581802. [Google Scholar] [CrossRef]
- Yan, L.; Li, G.; Liang, Y.; Tan, M.; Fang, J.; Peng, J.; Li, K. Co-production of surfactin and fengycin by Bacillus subtilis BBW1542 isolated from marine sediment: A promising biocontrol agent against foodborne pathogens. J. Food Sci. Technol. 2024, 61, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Bonmatin, J.M.; Laprévote, O.; Peypoux, F. Diversity among microbial cyclic lipopeptides: Iturins and surfactins. Activity-structure relationships to design new bioactive agents. Comb. Chem. High Throughput Screen. 2003, 6, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Guo, C.; Wang, Y.; Liu, D.; Lv, Y.; Lv, F.; Lu, Z. Fengycin inhibits the growth of the human lung cancer cell line 95D through reactive oxygen species production and mitochondria-dependent apoptosis. Anti-Cancer Drugs 2013, 24, 587–598. [Google Scholar] [CrossRef]
- Bing, Y.; Chunmei, D.U. Review on secondary antibiotic metabolites of Bacillus velezensis. Chin. J. Biol. Control 2022, 38, 502. [Google Scholar] [CrossRef]
- Algammal, A.M.; Mabrok, M.; Sivaramasamy, E.; Youssef, F.M.; Atwa, M.H.; El-Kholy, A.W.; Hozzein, W.N. Emerging MDR-Pseudomonas aeruginosa in fish commonly harbor opr L and tox A virulence genes and bla TEM, bla CTX-M, and tet A antibiotic-resistance genes. Sci. Rep. 2020, 10, 15961. [Google Scholar] [CrossRef]
- Li, T.; Raza, S.H.A.; Yang, B.; Sun, Y.; Wang, G.; Sun, W.; Shan, X. Aeromonas veronii infection in commercial freshwater fish: A potential threat to public health. Animals 2020, 10, 608. [Google Scholar] [CrossRef]
- Podobnik, M.; Kisovec, M.; Anderluh, G. Molecular mechanism of pore formation by aerolysin-like proteins. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160209. [Google Scholar] [CrossRef]
- Pederick, J.L.; Thompson, A.P.; Bell, S.G.; Bruning, J.B. d-Alanine–d-alanine ligase as a model for the activation of ATP-grasp enzymes by monovalent cations. J. Biol. Chem. 2020, 295, 7894–7904. [Google Scholar] [CrossRef]
- Fontecave, M.; Atta, M.; Mulliez, E. S-adenosylmethionine: Nothing goes to waste. Trends Biochem. Sci. 2004, 29, 243–249. [Google Scholar] [CrossRef]
- Cui, W.Q.; Qu, Q.W.; Wang, J.P.; Bai, J.W.; Bello-Onaghise, G.S.; Li, Y.A.; Li, Y.H. Discovery of potential anti-infective therapy targeting glutamine synthetase in Staphylococcus xylosus. Front. Chem. 2019, 7, 381. [Google Scholar] [CrossRef]
- Feng, Y.; Chang, S.K.; Portnoy, D.A. The major role of Listeria monocytogenes folic acid metabolism during infection is the generation of N-formylmethionine. mBio 2023, 14, e01074-23. [Google Scholar] [CrossRef] [PubMed]
- Panosian, T.D.; Nannemann, D.P.; Watkins, G.R.; Phelan, V.V.; McDonald, W.H.; Wadzinski, B.E.; Bachmann, B.O.; Iverson, T.M. Bacillus cereus phosphopentomutase is an alkaline phosphatase family member that exhibits an altered entry point into the catalytic cycle. J. Biol. Chem. 2011, 286, 8043–8054. [Google Scholar] [CrossRef]
- Vogeley, L.; El Arnaout, T.; Bailey, J.; Stansfeld, P.J.; Boland, C.; Caffrey, M. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 2016, 351, 876–880. [Google Scholar] [CrossRef]
- Deutscher, J. The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 2008, 11, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M., Jr.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Ramakrishnan, V. Structure of the 30S ribosomal subunit. Nature 2000, 407, 327–339. [Google Scholar] [CrossRef]
- Todea, A.; Fortuna, S.; Ebert, C.; Asaro, F.; Tomada, S.; Cespugli, M.; Gardossi, L. Rational Guidelines for the Two-Step Scalability of Enzymatic Polycondensation: Experimental and Computational Optimization of the Enzymatic Synthesis of Poly (glycerolazelate). ChemSusChem 2022, 15, e202102657. [Google Scholar] [CrossRef]
- Zappaterra, F.; Meola, D.; Presini, F.; Aprile, S.; Venturi, V.; Nosengo, C.; De Luca, C.; Catani, M.; Lerin, L.A.; Giovannini, P.P. Differential effect of nine cinnamic acid derivatives on the biocatalytic activity of Candida antarctica lipase type B. Curr. Res. Biotechnol. 2024, 8, 100231. [Google Scholar] [CrossRef]
- Li, L.; Hu, K.; Hong, B.; Lu, X.; Liu, Y.; Xie, J.; Jin, S.; Zhou, S.; Zhao, Q.; Lu, H.; et al. The inhibitory effect of Bacillus amyloliquefaciens L1 on Aeromonas hydrophila and its mecanism. Aquaculture 2021, 539, 736590. [Google Scholar] [CrossRef]
- Yousuf, S.; Jamal, M.T.; Al-Farawati, R.K.; Al-Mur, B.A.; Singh, R. Evaluation of Bacillus paramycoides Strains Isolated from Channa Fish sp. on Growth Performance of Labeo rohita Fingerlings Challenged by Fish Pathogen Aeromonas hydrophila MTCC 12301. Microorganisms 2023, 11, 842. [Google Scholar] [CrossRef]
- Aleksandrovna, M.M.; Valerevna, P.E.; Mikhailovna, B.V.; Anatolyevich, C.V.; Astghik, P.; Shahlo, M. Bacteria of genus Bacillus as antagonists of pathogens in aquaculture. Vestn. Astrakhan State Tech. Univ. Ser. Fish. 2023, 1, 89–97. [Google Scholar]
- Hoseinifar, S.H.; Sun, Y.-Z.; Wang, A.; Zhou, Z. Probiotics as Means of Diseases Control in Aquaculture, a Review of Current Knowledge and Future Perspectives. Front. Microbiol. 2018, 9, 2429. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Q.; Xu, Z.; Zhang, N.; Shen, Q.; Zhang, R. Responses of beneficial Bacillus amyloliquefaciens SQR9 to different soilborne fungal pathogens through the alteration of antifungal compounds production. Front. Microbiol. 2014, 5, 636. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Meena, K.; Tiwari, M. Differential anti-microbial secondary metabolites in different ESKAPE pathogens explain their adaptation in the hospital setup. Infect. Genet. Evol. 2018, 66, 57–65. [Google Scholar] [CrossRef]
- Bagewadi, Z.K.; Yunus Khan, T.M.; Gangadharappa, B.; Kamalapurkar, A.; Shamsudeen, S.M.; Yaraguppi, D.A. Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach. Saudi J. Biol. Sci. 2023, 30, 103753. [Google Scholar] [CrossRef]
- Carrillo, C.; Teruel, J.A.; Aranda, F.J.; Ortiz, A. Molecular mechanism of membrane permeabilization by the peptide antibiotic surfactin. Biochim. Biophys. Acta-Biomembr. 2003, 1611, 91–97. [Google Scholar] [CrossRef]
- Deleu, M.; Paquot, M.; Nylander, T. Fengycin interaction with lipid monolayers at the air–aqueous interface—Implications for the effect of fengycin on biological membranes. J. Colloid Interface Sci. 2005, 283, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Maget-Dana, R.; Ptak, M. Interactions of surfactin with membrane models. Biophys. J. 1995, 68, 1937–1943. [Google Scholar] [CrossRef]
- Piewngam, P.; Zheng, Y.; Nguyen, T.H.; Dickey, S.W.; Joo, H.S.; Villaruz, A.E.; Glose, K.A.; Fisher, E.L.; Hunt, R.L.; Li, B.; et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature 2018, 562, 532–537. [Google Scholar] [CrossRef]
- Wróbel, A.; Maliszewski, D.; Baradyn, M.; Drozdowska, D. Trimethoprim: An old antibacterial drug as a template to search for new targets. Synthesis, biological activity and molecular modeling study of novel trimethoprim analogs. Molecules 2019, 25, 116. [Google Scholar] [CrossRef]
- Park, J.H.; Reviello, R.E.; Loll, P.L. Crystal structure of vancomycin bound to the resistance determinant D-alanine-D-serine. IUCrJ 2024, 11, 133–139. [Google Scholar] [CrossRef]
- Zhou, T.; Cai, P.; Li, J.; Li, Z.; Dan, X.; Huang, X.; Zhang, X. Whole genome analysis of intestinal source Bacillus and its effect on the prevention and control of hybrid snakehead (Channa maculata ♀ × Channa argus ♂) nocardiosis. Front. Mar. Sci. 2024, 11, 1254806. [Google Scholar] [CrossRef]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.M.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenkoet, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qu, Q.; Liu, X.; Cui, W.; Yu, F.; Chen, X.; Li, Y. 1-Hydroxyanthraquinone exhibited antibacterial activity by regulating glutamine synthetase of Staphylococcus xylosus as a virulence factor. Biomed. Pharmacother. 2020, 123, 109779. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liu, Y.; Xu, N.; Yang, Q.; Ai, X. Morin protects channel catfish from Aeromonas hydrophila infection by blocking aerolysin activity. Front. Microbiol. 2018, 9, 2828. [Google Scholar] [CrossRef] [PubMed]
- Ravi, L.; Girish, S.; Harshini, M.; Sreenivas, B.A. β-Sitosterol: An antibacterial agent in aquaculture management of Vibrio infections. J. Pure Appl. Microbiol. 2020, 14, 2699–2714. [Google Scholar] [CrossRef]
- Prazdnova, E.V.; Mazanko, M.S.; Shevchenko, V.N.; Skripnichenko, R.V.; Kulikov, M.P.; Golovko, L.S.; Grigoriev, V.A.; Maltseva, T.A.; Kulikova, D.B.; Rudoy, D.V. Genomic Characterization of Four Novel Probiotic Strains with Enzymatic Activity and Their Effects on Carp (Cyprinus carpio). Animals 2025. submitted. [Google Scholar]
- Mazanko, M.S.; Kulikov, M.P.; Prazdnova, E.V.; Shevchenko, V.N.; Rudoy, D.V. Functional genomic analysis of new Bacillus strains for aquaculture. Microorganisms 2005. submitted. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogen | Protein Target | Ligand Dock Score, kcal/mol | ||
|---|---|---|---|---|
| Bacillibactin | Surfactin | Fengycin | ||
| Aeromonas hydrophila | Proaerolysin | −8.7 * | −5.9 | −6.7 * |
| D-alanine–D-alanine ligase | −7.7 * | −4.6 | −6.9 * | |
| S-adenosylmethionine synthase | −7.3 * | −6.1 | −5.4 | |
| Streptococcus agalactiae | Lipoprotein signaling peptidase | −8.6 * | −5.8 | −5.7 |
| Catabolite control protein | −8.1 * | −6.0 | −5.7 | |
| Large ribosomal subunit protein L19 | −7.1 * | −4.8 | −5.3 | |
| Phosphopentomutase | −6.5 * | −5.6 | −5.8 | |
| Vibrio anguillarum | Dihydrofolate reductase | −9.0 * | −4.6 | −5.9 |
| Staphylococcus xylosus | Glutamine synthetase | −10.3 * | −6.6 * | −6.4 |
| Strain | Dilution | Pseudomonas aeruginosa | Aeromonas veronii | |
|---|---|---|---|---|
| Control | - | OD600 | 0.356 ± 0.036 | 0.733 ± 0.072 |
| % | 100% | 100% | ||
| % | 97% | 102% | ||
| MT55 | 1/2 | OD600 | 0 | 0 |
| % | 0% * | 0% * | ||
| 1/4 | OD600 | 0 | 0.413 ± 0.029 | |
| % | 0% * | 56% * | ||
| MT155 | 1/2 | OD600 | 0 | 0 |
| % | 0% * | 0% * | ||
| 1/4 | OD600 | 0 | 0.352 ± 0.042 | |
| % | 0% * | 48% * | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prazdnova, E.; Zaikina, A.; Neurov, A.; Mazanko, M.; Ranjan, A.; Rudoy, D. Bacillibactin, a Potential Bacillus-Based Antibacterial Non-Ribosomal Peptide: In Silico Studies for Targeting Common Fish Pathogens. Int. J. Mol. Sci. 2025, 26, 5811. https://doi.org/10.3390/ijms26125811
Prazdnova E, Zaikina A, Neurov A, Mazanko M, Ranjan A, Rudoy D. Bacillibactin, a Potential Bacillus-Based Antibacterial Non-Ribosomal Peptide: In Silico Studies for Targeting Common Fish Pathogens. International Journal of Molecular Sciences. 2025; 26(12):5811. https://doi.org/10.3390/ijms26125811
Chicago/Turabian StylePrazdnova, Evgeniya, Anna Zaikina, Alexey Neurov, Maria Mazanko, Anuj Ranjan, and Dmitry Rudoy. 2025. "Bacillibactin, a Potential Bacillus-Based Antibacterial Non-Ribosomal Peptide: In Silico Studies for Targeting Common Fish Pathogens" International Journal of Molecular Sciences 26, no. 12: 5811. https://doi.org/10.3390/ijms26125811
APA StylePrazdnova, E., Zaikina, A., Neurov, A., Mazanko, M., Ranjan, A., & Rudoy, D. (2025). Bacillibactin, a Potential Bacillus-Based Antibacterial Non-Ribosomal Peptide: In Silico Studies for Targeting Common Fish Pathogens. International Journal of Molecular Sciences, 26(12), 5811. https://doi.org/10.3390/ijms26125811