
The 8-Hydroxyquinoline Derivatives of 1,4-Naphthoquinone: Synthesis, Computational Analysis, and Anticancer Activity

Abstract
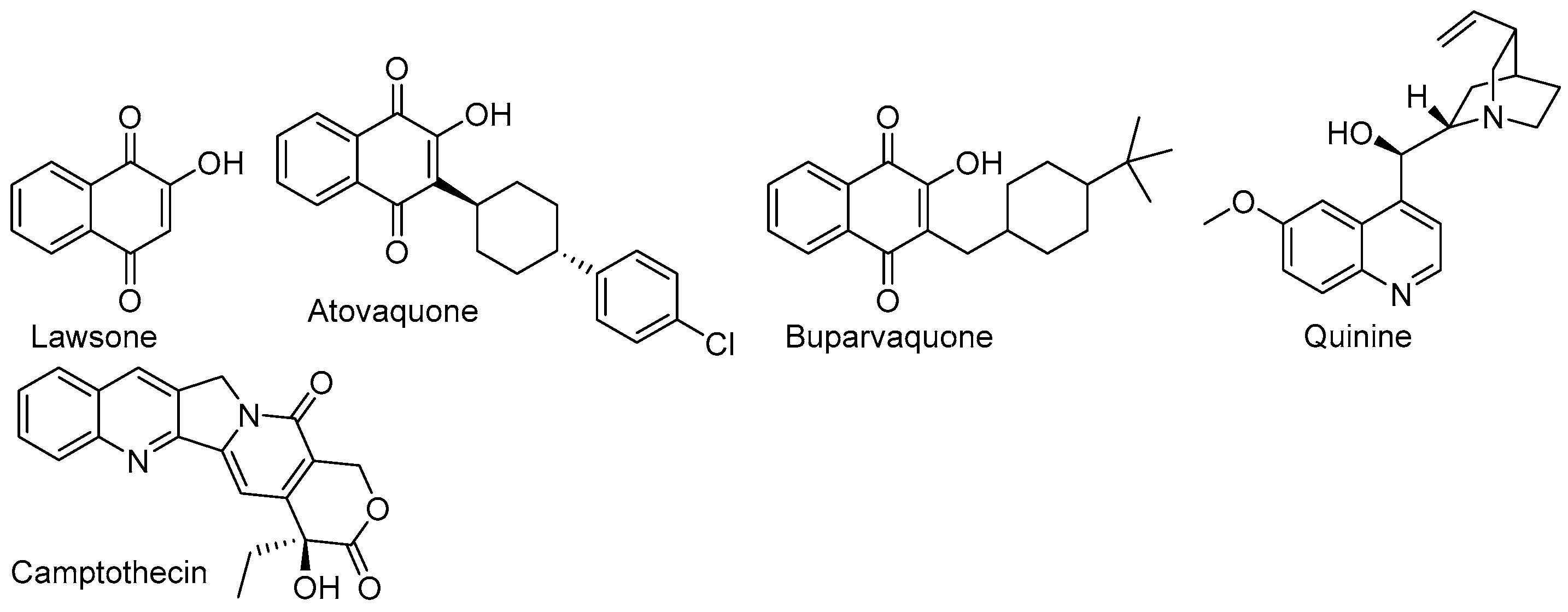
1. Introduction
2. Results and Discussion
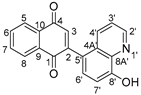
2.1. Synthesis and Structure Analysis
2.1.1. HR-MS Spectroscopy
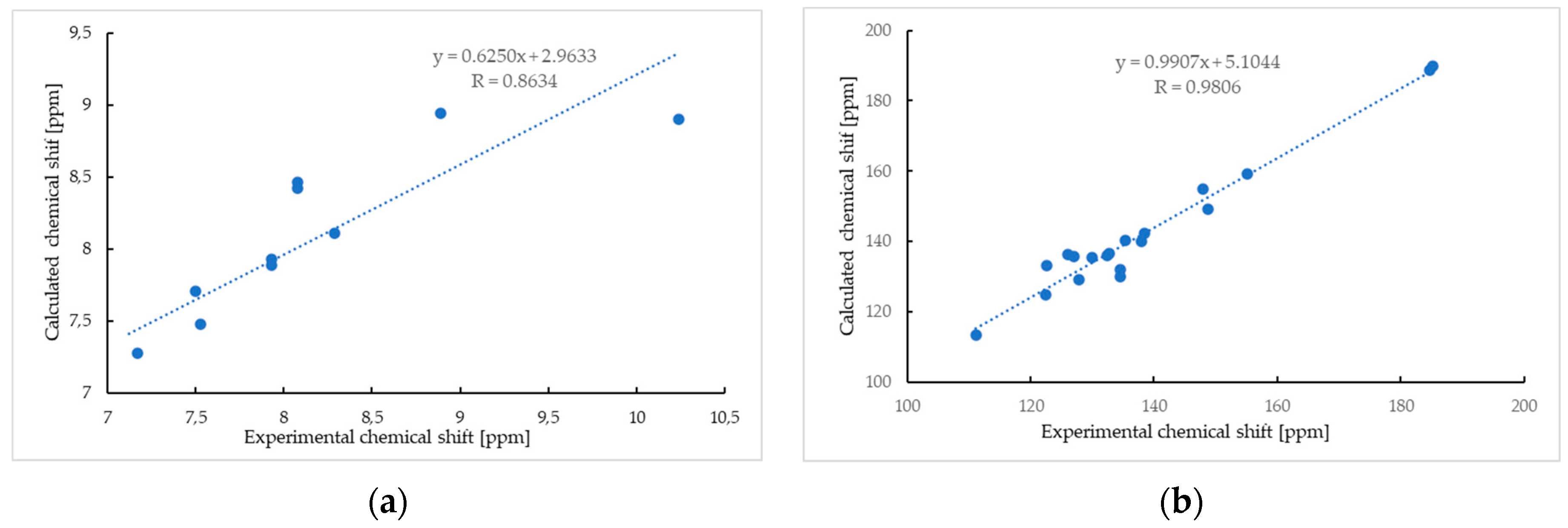
2.1.2. Nuclear Magnetic Resonance Spectroscopy
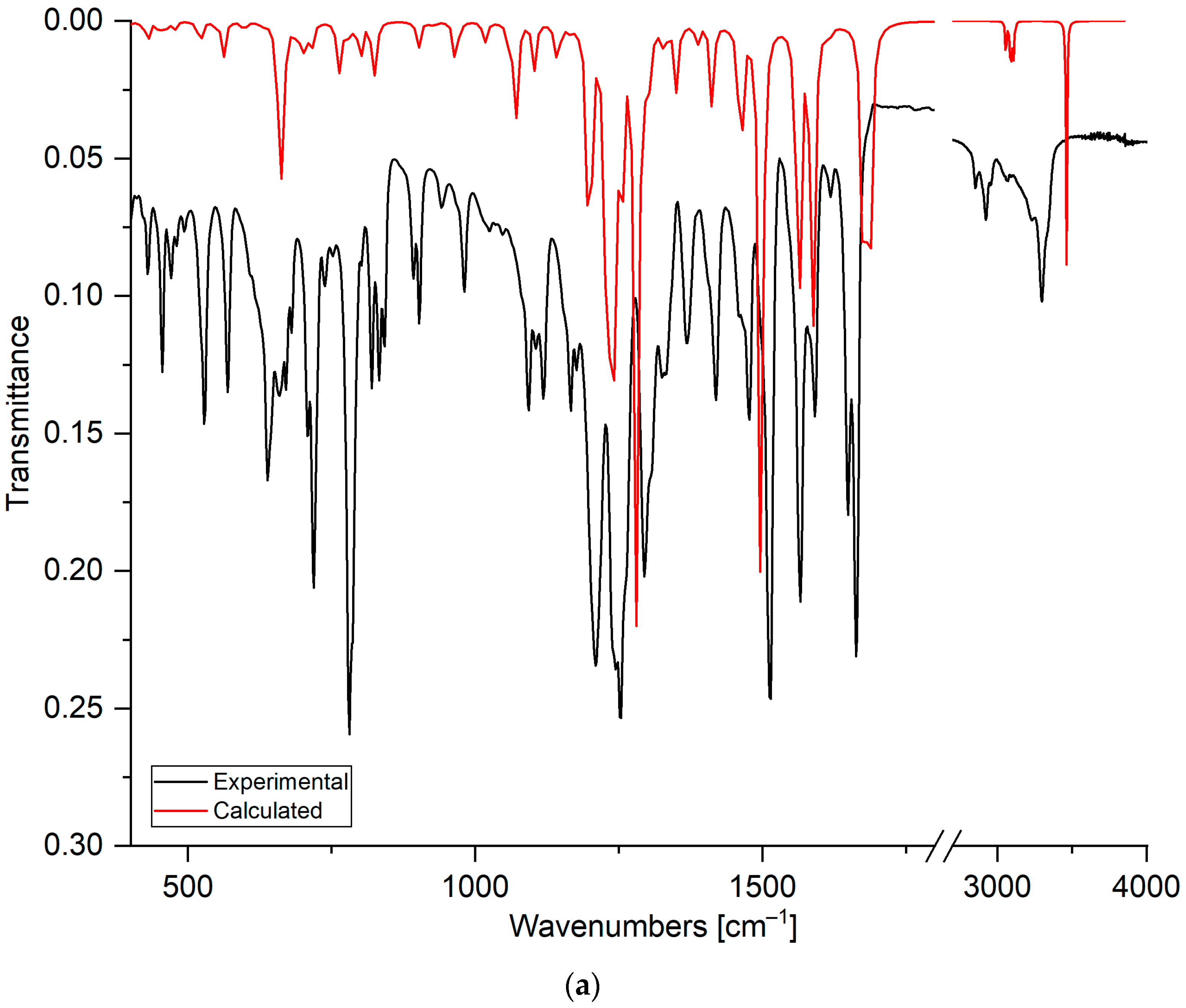
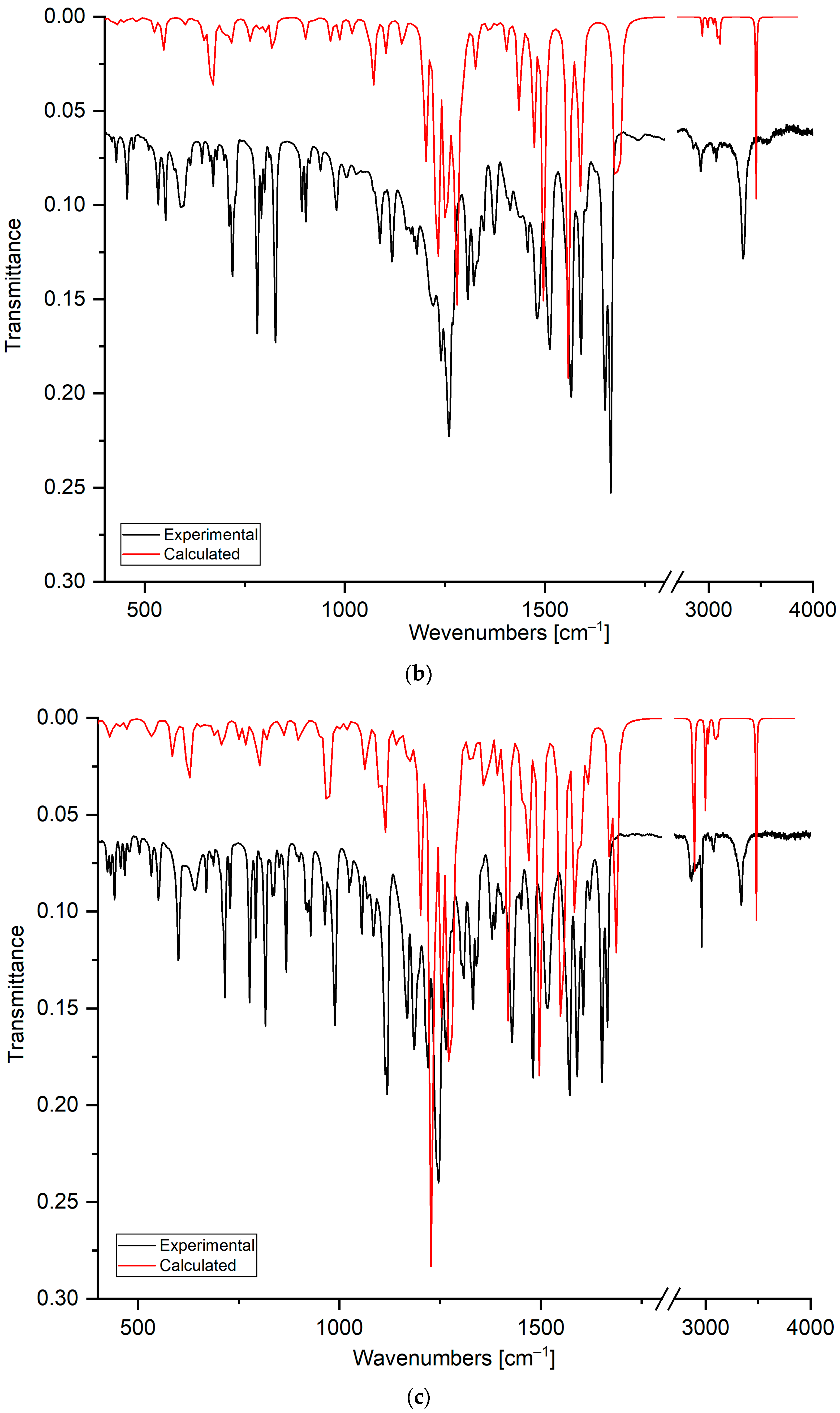
2.1.3. Fourier-Transform Infrared Spectroscopy
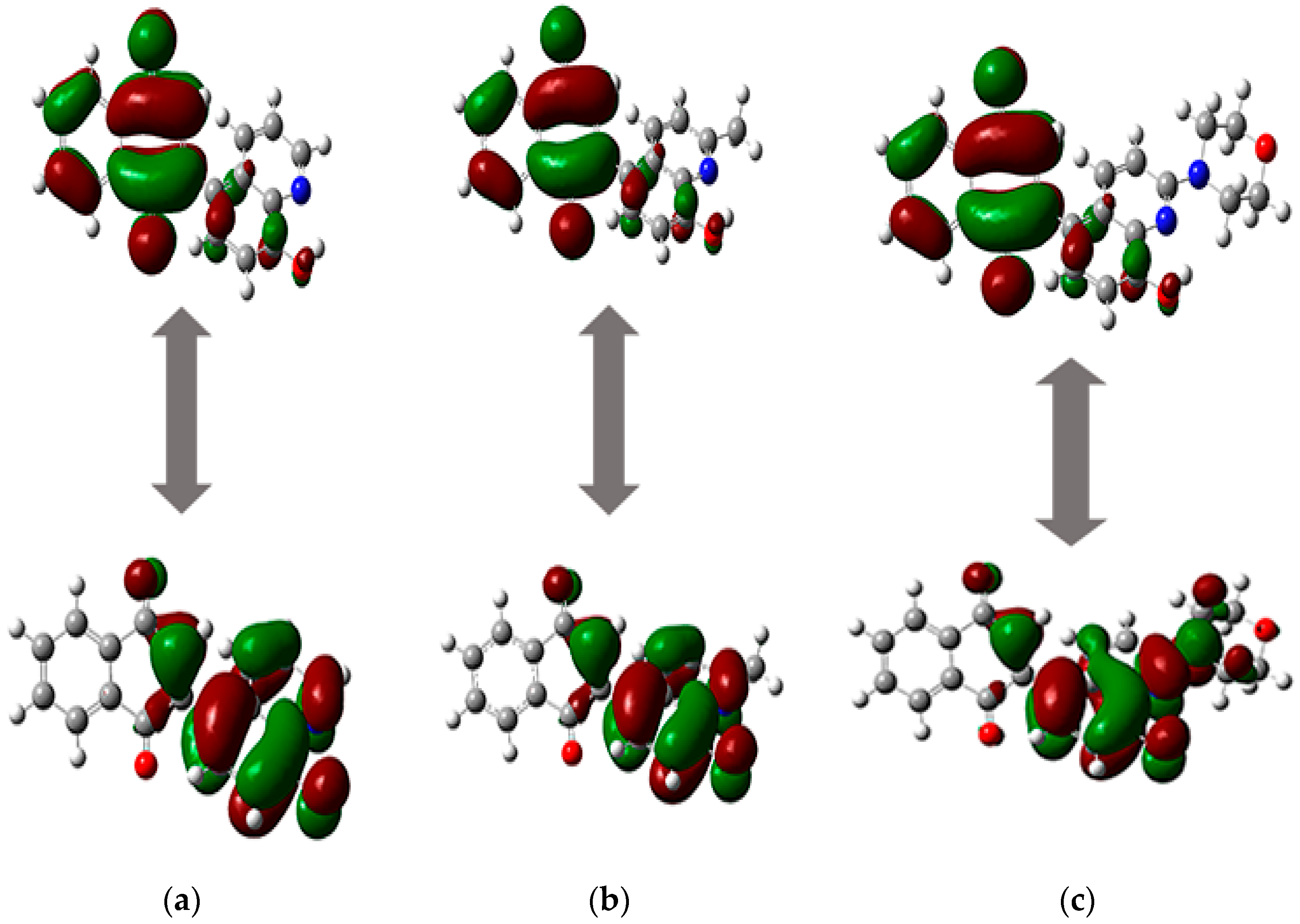
2.2. Analysis of Physicochemical Properties
2.3. Biological Activity
2.3.1. NQO-1 Activity
2.3.2. Anticancer Activity In Vitro
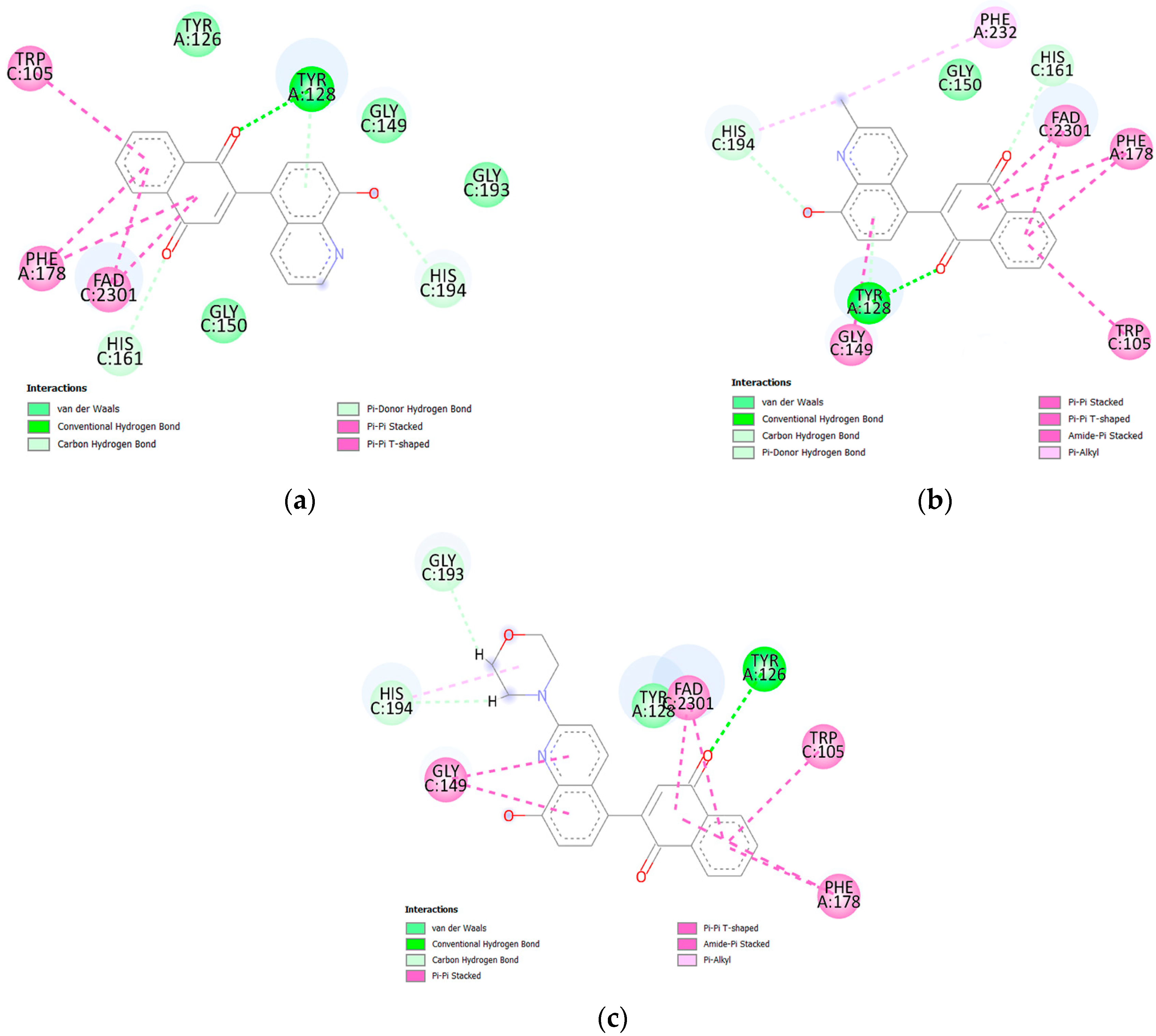
2.4. Molecular Docking Analysis
3. Materials and Methods
3.1. Physical Characterization
3.2. Synthesis of Compounds 5–7
3.3. Computational Details
3.4. The ADMET Study
3.5. Enzymatic Study
3.6. Anticancer Study
3.7. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aminin, D.; Polonik, S. 1,4-Naphthoquinones: Some Biological Properties and Application. Chem. Pharm. Bull. 2020, 68, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.S.; Sekar, M.; Gan, S.H.; Kumarasamy, V.; Subramaniyan, V.; Wu, Y.S.; Mat Rani, N.N.I.; Ravi, S.; Wong, L.S. Lawsone Unleashed: A Comprehensive Review on Chemistry, Biosynthesis, and Therapeutic Potentials. Drug Des. Devel Ther. 2024, 18, 295–3313. [Google Scholar] [CrossRef]
- Lopez, L.; Nary Flores, S.D.; Silva Belmares, S.Y.; Saenz Galindo, A. Naphthoquinones: Biological properties and synthesis of lawsone and derivates—A structured review. Vitae 2014, 21, 248–258. [Google Scholar] [CrossRef]
- Mone, N.; Bhagwat, S.; Sharma, D.; Chaskar, M.; Patil, R.; Zamboni, P.; Nawani, N.S. Satpute, aphthoquinones and Their Derivatives: Emerging Trends in Combating Microbial Pathogens. Coatings 2021, 11, 434. [Google Scholar] [CrossRef]
- Majdi, C.; Duvauchelle, V.; Meffre, P.; Benfodda, Z. An overview on the antibacterial properties of juglone, naphthazarin, plumbagin and lawsone derivatives and their metal complexes. Biomed. Pharmacother. 2023, 162, 114690. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.; Lima, A.; Sousa, J.; Pereira, G.; da Silva, G.; Brandão, G. 8-Methoxy-α-lapachone and lawsone: Antiproliferative effects on bladder tumour cells. Nat. Prod. Res. 2023, 21, 1058–1064. [Google Scholar] [CrossRef]
- Osman, A.M.; van Noort, P.C. Evidence for redox cycling of lawsone (2-hydroxy-1,4-naphthoquinone) in the presence of the hypoxanthine/xanthine oxidase system. J. Appl. Toxicol. 2023, 23, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Sauriasari, R.; Wang, D.H.; Takemura, Y.; Tsutsui, K.; Masuoka, N.; Sano, K.; Horita, M.; Wang, B.L.; Ogino, K. Cytotoxicity of lawsone and cytoprotective activity of antioxidants in catalase mutant Escherichia coli. Toxicology 2007, 235, 103–111. [Google Scholar] [CrossRef]
- Kirkland, D.; Marzin, D. An assessment of the genotoxicity of 2-hydroxy-1,4-naphthoquinone, the natural dye ingredient of Henna. Mutat. Res. 2003, 537, 183–199. [Google Scholar] [CrossRef]
- Kumari, P.; Singh, V.; Kant, V.; Ahuja, M. Current status of 1,4-naphthoquinones and their derivatives for wound healing. EJMCER 2024, 12, 100194. [Google Scholar] [CrossRef]
- Durán, A.; Chinchilla, N.; Simonet, A.; Gutiérrez, M.; Bolívar, J.; Valdivia, M.; Molinillo, J.; Macías, F. Biological Activity of Naphthoquinones Derivatives in the Search of Anticancer Lead Compounds. Toxins 2023, 15, 348. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Carballo, K.; Lamoureux, G.V.; Perez, A.L.; Cruz, A.B.; Filho, V.C. Novel one-pot synthesis of a library of 2-aryloxy-1,4-naphthoquinone derivatives. Determination of antifungal and antibacterial activity. RSC Adv. 2022, 12, 18507. [Google Scholar] [CrossRef] [PubMed]
- Lomlim, L.; Manuschai, J.; Ratti, P.; Kara, J.; Sakunphueak, A.; Panichayupakaranant, P.; Naorungroj, S. Effect of alkynyloxy derivatives of lawsone as an antifungal spray for acrylic denture base: An in vitro study. Heliyon 2023, 9, 13919. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Liang, F.M.; Zhang, W.J.; Zhang, Y.; Cui, J.H.; Dai, Y.Y.; Qiu, X.M.; Wang, W.H.; Zhou, Y.; Chen, D.P.; et al. Novel 2-amino-1,4-naphthoquinone derivatives induce A549 cell death through autophagy. Molecules 2023, 28, 3289. [Google Scholar] [CrossRef]
- Costa Souza, R.M.; Montenegro Pimentel, L.M.L.; Ferreira, L.K.M.; Pereira, V.R.A.; Santos, A.C.D.S.; Dantas, W.M.; Silva, C.J.O.; De Medeiros Brito, R.M.; Andrade, J.L.; De Andrade-Neto, V.F.; et al. Biological activity of 1,2,3-triazole-2-amino-1,4-naphthoquinone derivatives and their evaluation as therapeutic strategy for malaria control. Eur. J. Med. Chem. 2023, 55, 115400. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Krzykawski, K.; Halama, A.; Kubina, R. Hybrids of 1,4-Naphthoquinone with Thymidine Derivatives: Synthesis, Anticancer Activity, and Molecular Docking Study. Molecules 2023, 28, 6644. [Google Scholar] [CrossRef]
- Angulo-Elizari, E.; Henriquez-Figuereo, A.; Morán-Serradilla, C.; Plano, D.; Sanmartín, C. Unlocking the potential of 1,4-naphthoquinones: A comprehensive review of their anticancer properties. Eur. J. Med. Chem. 2024, 268, 116249. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E.; Latocha, M. Hybrids of 1,4-Quinone with Quinoline Derivatives: Synthesis, Biological Activity, and Molecular Docking with DT-Diaphorase (NQO1). Molecules 2022, 27, 6206. [Google Scholar] [CrossRef]
- Choudhary, D.; Rani, P.; Rangra, N.K.; Gupta, G.K.; Khokra, S.L.; Bhandare, R.R.; Shaik, A.B. Designing novel anti-plasmodial quinoline-furanone hybrids: Computational insights, synthesis, and biological evaluation targeting Plasmodium falciparum lactate dehydrogenase. RSC Adv. 2024, 14, 18764–18776. [Google Scholar] [CrossRef]
- Pinheiro, L.C.; Boechat, N.; Ferreira Mde, L.; Júnior, C.C.; Jesus, A.M.; Leite, M.M.; Souza, N.B.; Krettli, A.U. Anti-Plasmodium falciparum activity of quinoline-sulfonamide hybrids. Bioorg. Med. Chem. 2015, 23, 5979–5984. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Zhao, C.; Zuo, L.; Ke, X.; Wang, X. Michael addition reaction in C-5 of Camptothecin. J. Saudi Chem. Soc. 2024, 28, 101867. [Google Scholar] [CrossRef]
- Silva, V.L.; Saxena, J.; Nicolini, F.; Hoare, J.I.; Metcalf, S.; Martin, S.A.; Lockley, M. Chloroxine overrides DNA damage tolerance to restore platinum sensitivity in high-grade serous ovarian cancer. Cell Death Dis. 2021, 12, 395. [Google Scholar] [CrossRef]
- Zhang, W.; Shao, W.; Dong, Z.; Zhang, S.; Liu, C.; Chen, S. Cloxiquine, a traditional antituberculosis agent, suppresses the growth and metastasis of melanoma cells through activation of PPARγ. Cell Death Dis. 2019, 10, 404. [Google Scholar] [CrossRef]
- Mohan, I.; Ayyakannu Arumugam, N. Review on recent development of quinoline for anticancer activities. Arab. J. Chem. 2022, 15, 104168. [Google Scholar]
- Parmar, M.C.; Patel, B.Y. Green and traditional one-pot synthesis techniques for bioactive quinoline derivatives: A review. Tetrahedron Green. Chem. 2025, 5, 100062. [Google Scholar] [CrossRef]
- Liang, X.; Lin, W.; Chen, Y.; Yang, W.; Zhou, X.; Ai, S.; Qiu, L.; Cao, R.; Wang, J. Synthesis and in vitro and in vivo evaluation of novel bivalent quinolines as antitumor agents via targeting autophagy in cervical cancer. Eur. J. Med. Chem. 2025, 288, 117421. [Google Scholar] [CrossRef]
- Pakhariya, R.P.; Bhatnagar, A.; Pemawat, G. Quinoline analogs: Multifaceted heterocyclic compounds with varied synthetic strategies and potent antitubercular properties. RSC Adv. 2025, 15, 3646. [Google Scholar] [CrossRef]
- Ribeiro, N.; Bulut, I.; Sergi, B.; Pósa, V.; Spengler, G.; Sciortino, G.; André, V.; Ferreira, L.P.; Biver, T.; Ugone, V.; et al. Promising anticancer agents based on 8-hydroxyquinoline hydrazone copper(II) complexes. Front. Chem. 2023, 11, 1106349. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- Bala, I.A.; Al Sharif, O.F.; Asiri, A.M.; El-Shishtawy, R.M. Quinoline: A versatile bioactive scaffold and its molecular hybridization. Results Chem. 2024, 7, 101529. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. Engl. 2003, 42, 3996. [Google Scholar] [CrossRef]
- Shaveta-Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, Q.; Sun, H.; Li, W. Novel diosgenin-1,4-quinone hybrids: Synthesis, antitumor evaluation, and mechanism studies. J. Steroid Biochem. Mol. Biol. 2021, 214, 105993. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorg. Chem. 2021, 106, 104478. [Google Scholar] [CrossRef]
- Mancini, I.; Vigna, J.; Sighel, D.; Defant, A. Hybrid Molecules Containing Naphthoquinone and Quinolinedione Scaffolds as Antineoplastic Agents. Molecules 2022, 27, 4948. [Google Scholar] [CrossRef]
- Wang, S.B.; Deng, X.Q.; Zheng, Y.; Zhang, H.J.; Quan, Z.S. Synthesis and anticonvulsant activity evaluation of 8-alkoxy-5-(4H-1,2,4-triazol-4-yl)quinoline derivatives. Arch. Pharm. Res. 2013, 36, 32–40. [Google Scholar] [CrossRef]
- Sun, X.Y.; Jin, Y.Z.; Li, F.N.; Li, G.; Chai, K.Y.; Quan, Z.S. Synthesis of 8-alkoxy-4,5-dihydro-[1,2,4]triazole [4,3-a]quinoline-1-ones and evaluation of their anticonvulsant properties. Arch. Pharm. Res. 2006, 29, 1080–1085. [Google Scholar] [CrossRef]
- Vasudevan, A.; Wodka, D.; Verzal, M.K.; Souers, A.J.; Gao, J.; Brodjian, S.; Fry, D.; Dayton, B.; Marsh, K.C.; Hernandez, L.E.; et al. Synthesis and evaluation of 2-amino-8-alkoxy quinolines as MCHr1 antagonists. Part 2. Bioorg. Med. Chem. Lett. 2004, 14, 4879–4882. [Google Scholar] [CrossRef]
- Saadeh, H.A.; Sweidan, K.A.; Mubarak, M.S. Recent Advances in the Synthesis and Biological Activity of 8-Hydroxyquinolines. Molecules 2020, 25, 4321. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision A.03. Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wolinski, K.; Hinton, J.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, F.; Zhong, A.; Dugu, J.; Li, X. Determination of a new chromone from Aurantii Fructus Immaturus by DFT/GIAO method. Nat. Prod. Res. 2016, 30, 69. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef]
- Novak, U.; Grdadolnik, J. Infrared spectra of hydrogen bond network in lamellar perfluorocarboxylic acid monohydrates. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 253, 119551. [Google Scholar] [CrossRef]
- Novak, U.; Golobič, A.; Klančnik, N.; Mohaček-Grošev, V.; Stare, J.; Grdadolnik, J. Strong Hydrogen Bonds in Acetylenedicarboxylic Acid Dihydrate. Int. J. Mol. Sci. 2022, 23, 6164. [Google Scholar] [CrossRef]
- Socrates, G. Infrared Characteristic Group Frequencies; John Wiley & Sons, Inc.: Hobocken, NJ, USA, 1994. [Google Scholar]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS ONE 2021, 16, 0260853. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, T.; Mieszkowska, A.; Kempińska-Kupczyk, D.; Kot-Wasik, A.; Namieśnik, J.; Mazerska, Z. The impact of lipophilicity on environmental processes, drug delivery and bioavailability of food components. Microchem. J. 2019, 146, 393–406. [Google Scholar] [CrossRef]
- pkCSM. Available online: http://biosig.unimelb.edu.au/pkcsm/prediction# (accessed on 5 April 2025).
- SwissADME. Available online: http://www.swissadme.ch/index.php (accessed on 5 April 2025).
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef]
- Rezvan, V.H. Molecular structure, HOMO-LUMO, and NLO studies of some quinoxaline 1,4-dioxide derivatives: Computational (HF and DFT) analysis. Results Chem. 2024, 7, 101437. [Google Scholar] [CrossRef]
- Parr, R.; Pearson, R. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Jumabaev, A.; Khudaykulov, B.; Holikulov, U.; Norkulov, A.; Subbiah, J.; Al-Dossary, O.M.; Hushvaktov, H.; Absanov, A.; Issaoui, N. Molecular structure, vibrational spectral assignments, MEP, HOMO-LUMO, AIM, NCI, RDG, ELF, LOL properties of acetophenone and for its solutions based on DFT calculations. Opt. Mater. 2025, 159, 116683. [Google Scholar] [CrossRef]
- Shafieyoon, P.; Khalili, S.; Mehdipour, E.; Khorasani, S.N. Computational analysis and biological investigation of cellulose acetate: PED, HOMO–LUMO, MEP and molecular docking. Results Chem. 2024, 10, 101709. [Google Scholar] [CrossRef]
- Khan, A.; Arutla, V.; Srivenugopal, K. Human NQO1 as a Selective Target for Anticancer Therapeutics and Tumor Imaging. Cells 2024, 13, 1272. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Park, H. Implications of NQO1 in cancer therapy. BMB Rep. 2015, 48, 609. [Google Scholar] [CrossRef]
- Wu, L.Q.; Ma, X.; Zhang, C.; Liu, Z.P. Design, synthesis, and biological evaluation of 4-substituted-3,4-dihydrobenzo[h]quinoline-2,5,6(1H)-triones as NQO1-directed antitumor agents. Eur. J. Med. Chem. 2020, 198, 112396. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M. Design, Synthesis, Physicochemical Properties, and Biological Activity of Thymidine Compounds Attached to 5,8-Quinolinedione Derivatives as Potent DT-Diaphorase Substrates. Int. J. Mol. Sci. 2024, 25, 11211. [Google Scholar] [CrossRef]
- Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 5 April 2025).
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, 2169. [Google Scholar] [CrossRef]
- The Exact Mass Calculator, Single Isotope Version. Available online: http://www.sisweb.com/referenc/tools/exactmass.htm (accessed on 5 April 2025).
- Ceylan, Ü.; Hacıyusufoğlu, M.E.; Sönmez, M.; Yalçın, Ş.P.; Özdemir, N. Experimental and theoretical studies of (E)-2-(2-hydroxystyryl)-6-(4-methoxybenzoyl)-5-(4-methoxyphenyl)-1,2,4-triazin-3(2H)-one. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 141, 307. [Google Scholar] [CrossRef]
- Foresman, J.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian, Inc.: Pittsburg, PA, USA, 2015. [Google Scholar]
- Henschel, H.; Andersson, A.T.; Jespers, W.; Mehdi Ghahremanpour, M.; van der Spoel, D. Theoretical Infrared Spectra: Quantitative Similarity Measures and Force Fields. J. Chem. Theory Comput. 2020, 16, 3307. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 4, 42717. [Google Scholar] [CrossRef]
- Pires, D.; Blundell, T.; Ascher, D. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Li, X.; Bian, J.; Wang, N.; Qian, X.; Gu, J.; Mu, T.; Fan, J.; Yang, X.; Li, S.; Yang, T.; et al. Novel naphtho [2,1-d]oxazole-4,5-diones as NQO1 substrates with improved aqueous solubility: Design, synthesis, and in vivo antitumor evaluation. Bioorg. Med. Chem. 2016, 24, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Deng, B.; Xu, L.; Xu, X.; Wang, N.; Hu, T.; Yao, Z.; Du, J.; Yang, L.; Lei, Y.; et al. 2-Substituted 3-methylnaphtho [1,2-b]furan-4,5-diones as novel L-shaped ortho-quinone substrates for NAD(P)H:quinone oxidoreductase (NQO1). Eur. J. Med. Chem. 2014, 82, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Winski, S.; Hargreaves, R.; Moody, C.J.; Hudnott, A.R.; Ross, D.; Amzel, L.M. Structure-based development of anticancer drugs: Complexes of NAD(P)H:quinone oxidoreductase 1 with chemotherapeutic quinones. Structure 2001, 9, 659. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dessault Systemes BIOVIA. Available online: https://www.3ds.com/products/biovia/biosciences (accessed on 5 April 2025).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Proton | 1H NMR [ppm] | 1H NMRcalc [ppm] | ROESY | HSQC | Carbon | 13C NMR [ppm] | 13C NMRcalc [ppm] | HMBC |
| OH | 10.24 | 8.90 | ||||||
| H2′ | 8.89 | 8.94 | H2′(8.89)-H3′(7.53) | H2′(8.89)-C2′(148.7) | C2′ | 148.7 | 149.2 | H2′(8.89)-C3′(122.4) H2′(8.89)-C4′(135.3) H2′(8.89)-C8A′(138.5) |
| H4′ | 8.29 | 8.11 | H4′(8.29)-H3′(7.53) | H4′(8.28)-C4′(135.3) | C4′ | 135.3 | 140.2 | H4′(8.89)-C3′(122.4) H4′(8.89)-C8A′(138.5) H4′(8.89)-C2′(148.7) H4′(8.89)-C5′(127.8) H4′(8.89)-C8′(155.1) |
| H5 H8 | 8.08 | 8.46 8.42 | H8(8.08)-H6(7.93) | H5/H8(8.08)-C5(126.0) H5/H8(8.08)-C5(127.0) | C5 C8 | 126.0 127.0 | 135.7 136.3 | H5/H8(8.08)-C6(134.6) H5/H8(8.08)-C10(132.4) |
| H6 H7 | 7.93 | 7.93 7.89 | H7(7.93)-H8(8.08) | H6/H7(7.93)-C6/C7(134.6) | C6 C7 | 134.6 | 132.0 130.1 | H6/H7(7.93)-C8(127.0) H6/H7(7.93)-C5(126.0) H6/H7(7.93)-C10(132.4) H6/H7(7.93)-C9(132.7) |
| H3′ | 7.53 | 7.48 | H3′(7.53)-H4′(8.28) H3′(7.53)-H2′(8.89) | H3′(7.53)-C3′(122.4) | C3′ | 122.4 | 124.8 | H3′(7.53)-C2′(148.7) H3′(7.53)-C5′(127.8) H3′(7.53)-C8′(155.1) |
| H6′ | 7.50 | 7.71 | H6′(7.50)-H7′(7.17) | H6′(7.50)-C6′(130.0) | C6′ | 130.0 | 135.5 | H6′(7.50)-C5′(127.8) H6′(7.50)-C2(148.0) H6′(7.50)-C8′(155.1) H6′(7.50)-C8A′(138.5) |
| H7′ | 7.17 | 7.28 | H7′(7.17)-H6′(7.50) | H7′(7.17)-C7′(111.1) | C7′ | 111.1 | 113.5 | H7′(7.17)-122.6 H7′(7.17)-155.1 H7′(7.17)-138.5 |
| H3 | 7.12 | 6.95 | - | H3(7.12)-C3(138.0) | C3 | 138.0 | 140.1 | H3(7.12)-C4A′(122.6) H3(7.12)-C10(132.4) H3(7.12)-C2(148.0) H3(7.12)-C4(184.7) |
| C4A′ | 122.6 | 133.1 | C4A′(122.6)-H3(7.12) C4A′(122.6)-H7′(7.17) | |||||
| C5′ | 127.8 | 129.0 | C5′(127.8)-H4′(8.29) C5′(127.8)-H3′(7.53) C5′(127.8)-H6′(7.50) | |||||
| C10 | 132.4 | 136.1 | C10(132.4)-H7′(7.12) C10(132.4)-H6/H7(7.93) C10(132.4)-H5/H8(8.08) | |||||
| C9 | 132.7 | 136.6 | C9(132.7)-H6/H7(7.93) | |||||
| C8A′ | 138.5 | 142.2 | C8A′(138.5)-H2′(8.89) C8A′(138.5)-H4′(8.29) C8A′(138.5)-H6′(7.50) C8A′(138.5)-H7′(7.17) | |||||
| C2 | 148.0 | 154.9 | C2(148.0)-H6′(7.50) C2(148.0)-H3(7.12) | |||||
| C8′ | 155.1 | 159.3 | C8′(155.1)-H4′(8.29) C8′(155.1)-H3′(7.53) C8′(155.1)-H6′(7.50) C8′(155.1)-H7′(7.17) | |||||
| C4 | 184.7 | 188.6 | C4(184.7)-H3(7.12) | |||||
| C1 | 185.1 | 189.9 | - | |||||
| Band | Wavenumber [cm−1] | |||||
|---|---|---|---|---|---|---|
| 5 | 6 | 7 | ||||
| Exp. | Calc. | Exp. | Calc. | Exp. | Calc. | |
| v O-H | 3298 | 3463 | 3330 | 3455 | 3340 | 3483 |
| v C-H | 2953–2853 | 3108–3054 | 3072–2853 | 3108–2940 | 3077–2855 | 3118–2893 |
| vas C=O | 1662 | 1689 | 1664 | 1689 | 1665 | 1687 |
| Vsyn C=O | 1649 | 1673 | 1650 | 1673 | 1652 | 1669 |
| v C-C naph | 1591–1566 | 1588 | 1590–1565 | 1589 | 1590 | 1583 |
| δ C-H quinone | 1514–1477 | 1496–1465 | 1510–1479 | 1496–1472 | 1515–1480 | 1496–1470 |
| δ C-H quinone | 1419 | 1411 | 1412 | 1403 | 1428 | 1418 |
| δ C-C quinone | 1369 | 1350 | 1374 | 1357 | 1385–1379 | 1357 |
| δ C-C naphtho | 1294 | 1280 | 1307 | 1280 | 1265 | 1270 |
| δ C-C quinone | 1254–1240 | 1242 | 1260–1220 | 1248–1233 | 1246–1220 | 1253–1227 |
| δ C-H naphto | 1211–1167 | 1195–1141 | 1180–1154 | 1206 | 1186–1169 | 1201 |
| δ C-C nap | 1118 | 1103 | 1118 | 1102 | 1118–1114 | 1114 |
| δ C-C quinone | 1092 | 1072 | 1088 | 1072 | 1084 | 1066 |
| δ C-H quinone | 843–781 | 825–802 | 902–893 | 902 | 867–816 | 865–821 |
| δ C-C | 719–708 | 717–702 | 719–698 | 717–697 | 729–715 | 715 |
| δ O-H | 680–638 | 663 | 680–662 | 670 | 639 | 629 |
| δ C-C | 569–529 | 563–524 | 552–534 | 544–524 | 550–530 | 530 |
| Parameter | 5 | 6 | 7 |
|---|---|---|---|
| logS | −4.05 | −4.36 | −4.45 |
| logP | 2.21 | 2.49 | 2.71 |
| M | 301.3 | 315.32 | 386.4 |
| TPSA | 64.26 | 67.26 | 79.73 |
| nHA | 4 | 4 | 5 |
| nHD | 1 | 1 | 1 |
| nRT | 1 | 1 | 2 |
| logPapp | 1.250 | 1.214 | 1.342 |
| HIA [%] | 98 | 97 | 99 |
| logKp | −2.738 | −2.747 | −2.766 |
| logBB | −0.126 | −0.143 | −0.499 |
| logPS | −2.000 | −1.926 | −2.231 |
| logVDss | 0.086 | 0.076 | 0.166 |
| CYP3A4 substrate | Yes | Yes | Yes |
| CYP3A4 inhibitor | No | No | No |
| Descriptor | 5 | 6 | 7 |
|---|---|---|---|
| EHOMO [eV] | −5.855 | −5.772 | −5.496 |
| ELUMO [eV] | −3.113 | −3.069 | −3.004 |
| ΔE [eV] | 2.743 | 2.704 | 2.491 |
| I [eV] | 5.855 | 5.772 | 5.496 |
| A [eV] | 3.113 | 3.069 | 3.004 |
| η [eV] | 1.371 | 1.352 | 1.246 |
| µ [eV] | −4.484 | −4.421 | −4.250 |
| χ [eV] | 4.484 | 4.421 | 4.250 |
| ω [eV] | 7.331 | 7.227 | 7.249 |
| Ligand | Enzymatic Conversion Rate of NQO1 [µmol NADPH/µmol NQO1/min] |
|---|---|
| 1 | 595 ± 50 |
| 5 | 782 ± 42 |
| 6 | 1026 ± 69 |
| 7 | 851 ± 54 |
| β-Lap | 985 ± 45 |
| Compounds | Cell Lines/IC50 ± SD [µM] | |||
|---|---|---|---|---|
| A549 | MCF-7 | C-32 | Colo-829 | |
| 1 | 2.33 ± 0.28 | 26.34 ± 1.51 | 7.45 ± 0.08 | 86.93 ± 2.07 |
| 5 | 1.08 ± 0.24 | 14.08 ± 0.88 | 3.57 ± 0.02 | 23.69 ± 2.90 |
| 6 | 0.89 ± 0.05 | 11.69 ± 1.71 | 2.52 ± 0.05 | 19.56 ± 2.31 |
| 7 | 1.64 ± 0.16 | 18.64 ± 1.22 | 6.85 ± 0.31 | 29.73 ± 2.78 |
| β-Lap | 4.50 ± 0.32 | 5.85 ± 0.62 | 4.89 ± 0.36 | 8.12 ± 0.51 |
| Cisplatin | 5.96 ± 0.60 | 20.91 ± 1.78 | 2.92 ± 0.01 | 20.43 ± 1.12 |
| Ligand | ΔG [kcal/mol] |
|---|---|
| 1 | −8.1 |
| 5 | −9.5 |
| 6 | −10.0 |
| 7 | −9.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokal, A.; Wrzalik, R.; Latocha, M.; Kadela-Tomanek, M. The 8-Hydroxyquinoline Derivatives of 1,4-Naphthoquinone: Synthesis, Computational Analysis, and Anticancer Activity. Int. J. Mol. Sci. 2025, 26, 5331. https://doi.org/10.3390/ijms26115331
Sokal A, Wrzalik R, Latocha M, Kadela-Tomanek M. The 8-Hydroxyquinoline Derivatives of 1,4-Naphthoquinone: Synthesis, Computational Analysis, and Anticancer Activity. International Journal of Molecular Sciences. 2025; 26(11):5331. https://doi.org/10.3390/ijms26115331
Chicago/Turabian StyleSokal, Arkadiusz, Roman Wrzalik, Małgorzata Latocha, and Monika Kadela-Tomanek. 2025. "The 8-Hydroxyquinoline Derivatives of 1,4-Naphthoquinone: Synthesis, Computational Analysis, and Anticancer Activity" International Journal of Molecular Sciences 26, no. 11: 5331. https://doi.org/10.3390/ijms26115331
APA StyleSokal, A., Wrzalik, R., Latocha, M., & Kadela-Tomanek, M. (2025). The 8-Hydroxyquinoline Derivatives of 1,4-Naphthoquinone: Synthesis, Computational Analysis, and Anticancer Activity. International Journal of Molecular Sciences, 26(11), 5331. https://doi.org/10.3390/ijms26115331