
Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2



Abstract

1. Introduction
2. Results and Discussion
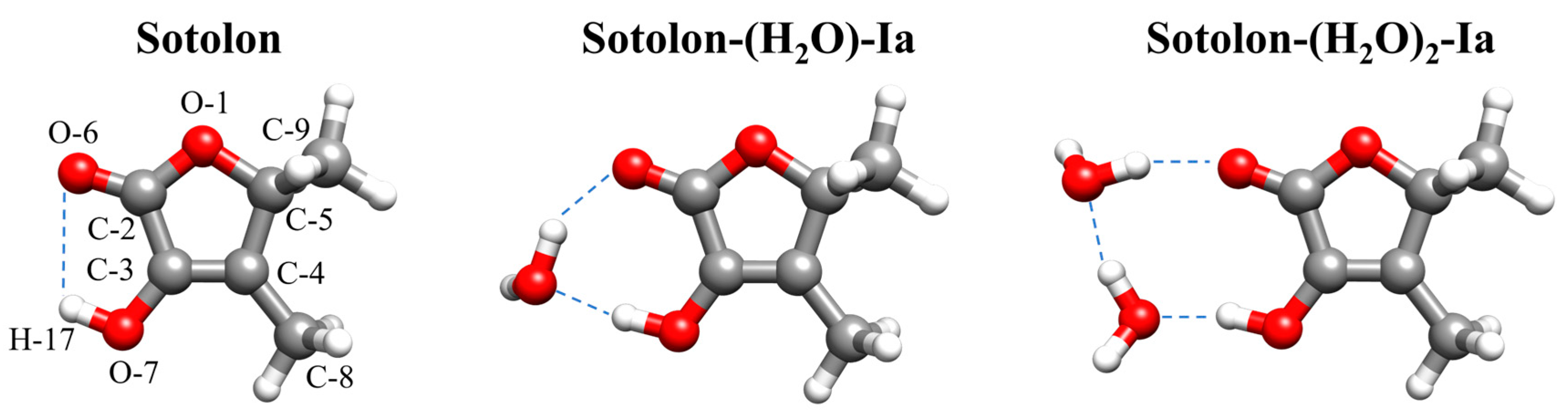
2.1. Conformational Panorama
2.2. Rotational Spectrum
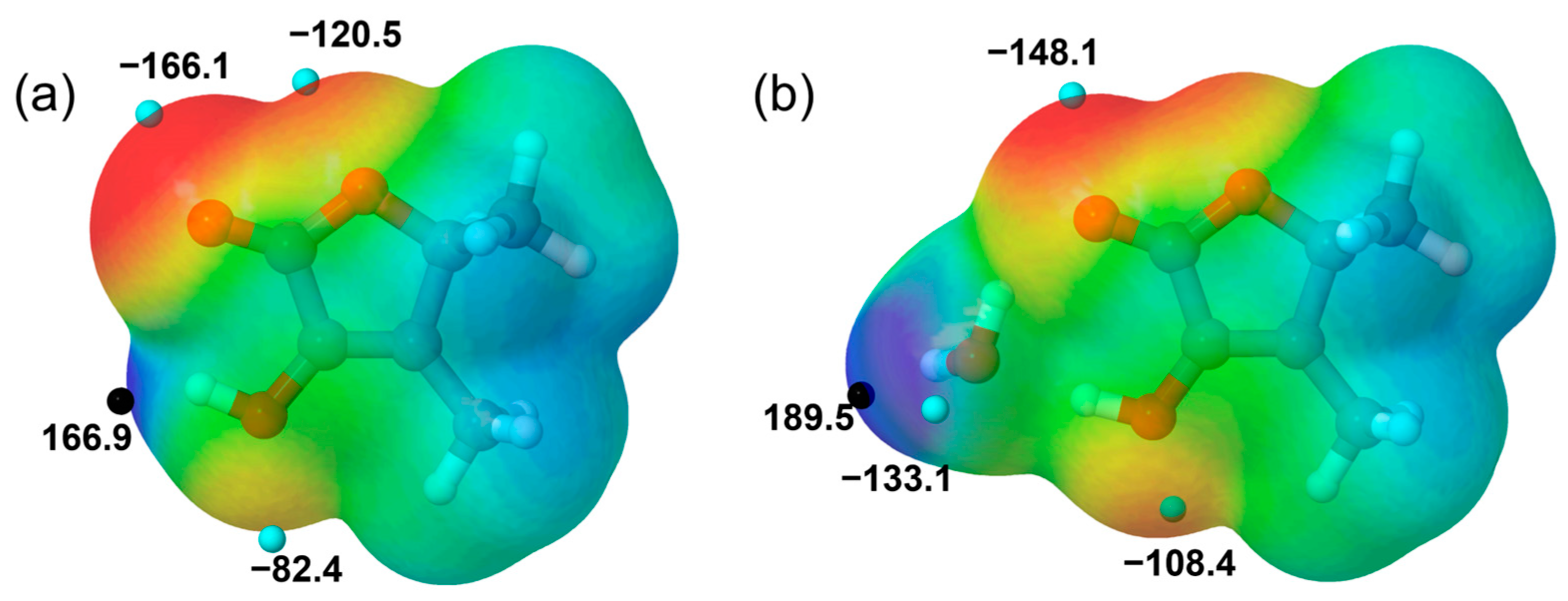
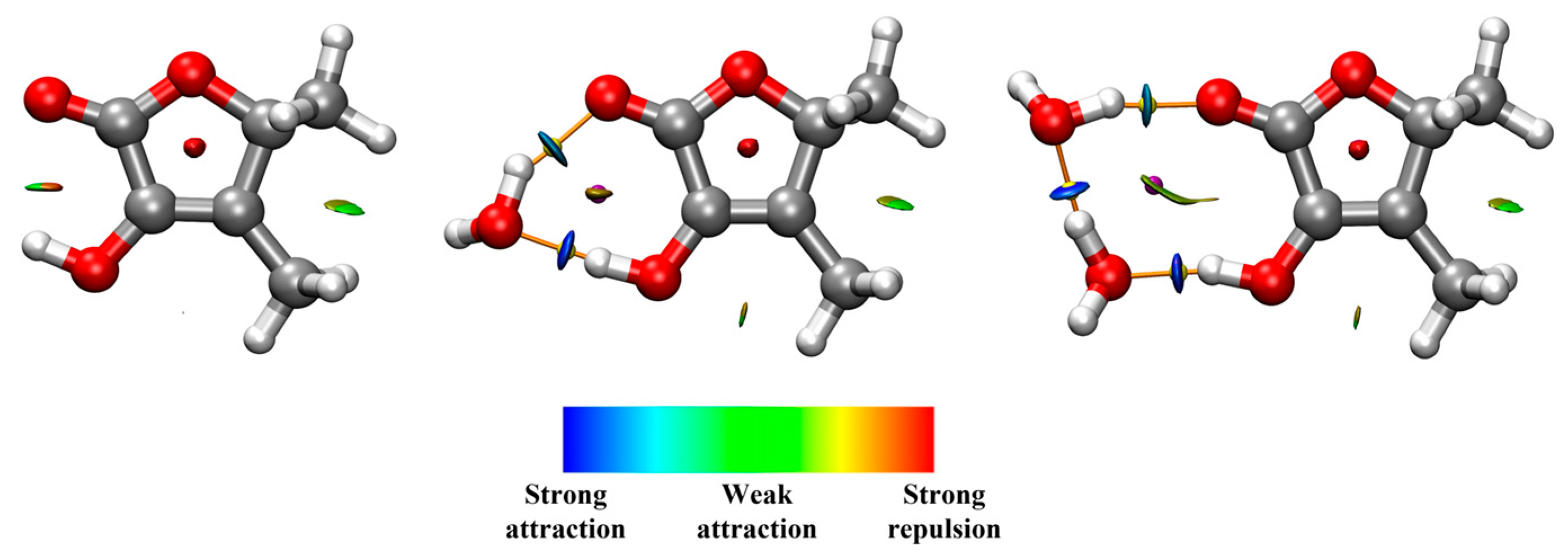
2.3. Molecular Structure and Molecular Interactions
2.4. Methyl Internal Rotation
3. Materials and Methods
3.1. Experimental Details
3.2. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Martín-Fernández, C.; Montero-Campillo, M.M.; Alkorta, I. Hydrogen Bonds Are Never of an “Anti-Electrostatic” Nature: A Brief Tour of a Misleading Nomenclature. J. Phys. Chem. Lett. 2024, 15, 4105–4110. [Google Scholar] [CrossRef]
- Lehn, J.-M. Toward Self-Organization and Complex Matter. Science 2002, 295, 2400–2403. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Bruce, C.D.; Redfield, C.; Lorenz, C.D.; McLain, S.E. Water Mediation Is Essential to Nucleation of β-Turn Formation in Peptide Folding Motifs. Angew. Chem. Int. Ed. 2013, 52, 13091–13095. [Google Scholar] [CrossRef] [PubMed]
- Hazrah, A.S.; Insausti, A.; Ma, J.; Al-Jabiri, M.H.; Jäger, W.; Xu, Y. Wetting vs. Droplet Aggregation: A Broadband Rotational Spectroscopic Study of 3-Methylcatechol⋅⋅⋅Water Clusters. Angew. Chem. Int. Ed. 2023, 62, e202310610. [Google Scholar] [CrossRef] [PubMed]
- Burevschi, E.; Chrayteh, M.; Murugachandran, S.I.; Loru, D.; Dréan, P.; Sanz, M.E. Water Arrangements upon Interaction with a Rigid Solute: Multiconfigurational Fenchone-(H2O)4–7 Hydrates. J. Am. Chem. Soc. 2024, 146, 10925–10933. [Google Scholar] [CrossRef]
- Macario, A.; López, J.C.; Blanco, S. Molecular Structure of Salicylic Acid and Its Hydrates: A Rotational Spectroscopy Study. Int. J. Mol. Sci. 2024, 25, 4074. [Google Scholar] [CrossRef]
- Steber, A.L.; Temelso, B.; Kisiel, Z.; Schnell, M.; Pérez, C. Rotational Dive into the Water Clusters on a Simple Sugar Substrate. Proc. Natl. Acad. Sci. USA 2023, 120, e2214970120. [Google Scholar] [CrossRef]
- Pérez, C.; López, J.C.; Blanco, S.; Schnell, M. Water-Induced Structural Changes in Crown Ethers from Broadband Rotational Spectroscopy. J. Phys. Chem. Lett. 2016, 7, 4053–4058. [Google Scholar] [CrossRef]
- Blanco, S.; Pinacho, P.; López, J.C. Structure and Dynamics in Formamide–(H2O)3: A Water Pentamer Analogue. J. Phys. Chem. Lett. 2017, 8, 6060–6066. [Google Scholar] [CrossRef]
- Krin, A.; Pérez, C.; Pinacho, P.; Quesada-Moreno, M.M.; López-González, J.J.; Avilés-Moreno, J.R.; Blanco, S.; López, J.C.; Schnell, M. Structure Determination, Conformational Flexibility, Internal Dynamics, and Chiral Analysis of Pulegone and Its Complex with Water. Chem. A Eur. J. 2018, 24, 721–729. [Google Scholar] [CrossRef]
- Domingos, S.R.; Pérez, C.; Marshall, M.D.; Leung, H.O.; Schnell, M. Assessing the Performance of Rotational Spectroscopy in Chiral Analysis. Chem. Sci. 2020, 11, 10863–10870. [Google Scholar] [CrossRef]
- Mills, M.D.; Sonstrom, R.E.; Vang, Z.P.; Neill, J.L.; Scolati, H.N.; West, C.T.; Pate, B.H.; Clark, J.R. Enantioselective Synthesis of Enantioisotopomers with Quantitative Chiral Analysis by Chiral Tag Rotational Spectroscopy. Angew. Chem. Int. Ed. 2022, 61, e202207275. [Google Scholar] [CrossRef] [PubMed]
- Pinacho, P.; López, J.C.; Kisiel, Z.; Blanco, S. The Effect of Microsolvation on the Structure, Nuclear Quadrupole Coupling, and Internal Rotation: The Methyl Carbamate⋯(H2O)1–3 Complexes. J. Chem. Phys. 2024, 160, 164315. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Sukhorukov, O.; Jäger, W.; Xu, Y. Direct Spectroscopic Detection of the Orientation of Free OH Groups in Methyl Lactate–(Water)1,2 Clusters: Hydration of a Chiral Hydroxy Ester. Angew. Chem. Int. Ed. 2014, 53, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Hazrah, A.S.; Al-Jabiri, M.H.; Jäger, W. Structure and Conformations of 3-Methylcatechol: A Rotational Spectroscopic and Theoretical Study. J. Mol. Spectrosc. 2022, 390, 111715. [Google Scholar] [CrossRef]
- Cummings, C.N.; Kleiner, I.; Walker, N.R. Noncovalent Interactions in the Molecular Geometries of 4-Methylthiazole···H2O and 5-Methylthiazole···H2O Revealed by Microwave Spectroscopy. J. Phys. Chem. A 2023, 127, 8133–8145. [Google Scholar] [CrossRef]
- Blank, I.; Sen, A.; Grosch, W. Potent Odorants of the Roasted Powder and Brew of Arabica Coffee. Z. Lebensm. Unters. Forsch. 1992, 195, 239–245. [Google Scholar] [CrossRef]
- Pons, A.; Lavigne, V.; Landais, Y.; Darriet, P.; Dubourdieu, D. Identification of a Sotolon Pathway in Dry White Wines. J. Agric. Food Chem. 2010, 58, 7273–7279. [Google Scholar] [CrossRef]
- Martin, B.; Etievant, P.X.; Le Quere, J.L.; Schlich, P. More Clues about Sensory Impact of Sotolon in Some Flor Sherry Wines. J. Agric. Food Chem. 1992, 40, 475–478. [Google Scholar] [CrossRef]
- Nakahashi, A.; Yaguchi, Y.; Miura, N.; Emura, M.; Monde, K. A Vibrational Circular Dichroism Approach to the Determination of the Absolute Configurations of Flavorous 5-Substituted-2(5H)-Furanones. J. Nat. Prod. 2011, 74, 707–711. [Google Scholar] [CrossRef]
- Pons, A.; Lavigne, V.; Landais, Y.; Darriet, P.; Dubourdieu, D. Distribution and Organoleptic Impact of Sotolon Enantiomers in Dry White Wines. J. Agric. Food Chem. 2008, 56, 1606–1610. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liang, M.-M.; Wang, H.-J.; Zhao, Q.-Q.; Zhu, H.-J.; Liu, L.; Pittman, C.U. Investigating Cyclic Sotolon, Maple Furanone and Their Dimers in Solution Using Optical Rotation, Electronic Circular Dichroism and Vibrational Circular Dichroism. Tetrahedron 2017, 73, 2432–2438. [Google Scholar] [CrossRef]
- Dindić, C.; Lüchow, A.; Vogt, N.; Demaison, J.; Nguyen, H.V.L. Equilibrium Structure in the Presence of Methyl Internal Rotation: Microwave Spectroscopy and Quantum Chemistry Study of the Two Conformers of 2-Acetylfuran. J. Phys. Chem. A 2021, 125, 4986–4997. [Google Scholar] [CrossRef]
- Van, V.; Stahl, W.; Nguyen, M.T.; Nguyen, H.V.L. The Smell of Coffee: The Carbon Atom Microwave Structure of Coffee Furanone Validated by Quantum Chemistry. Can. J. Phys. 2020, 98, 538–542. [Google Scholar] [CrossRef]
- Ray, B.S. Uber Die Eigenwerte Des Asymmetrischen Kreisels. Z. Phys. 1932, 78, 74–91. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure a Series of Advances; During, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 6. [Google Scholar]
- Pickett, H.M. The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra; Wiley-Interscience: New York, NY, USA, 1984. [Google Scholar]
- Gerhard, D.; Hellweg, A.; Merke, I.; Stahl, W.; Baudelet, M.; Petitprez, D.; Wlodarczak, G. Internal Rotation and Chlorine Nuclear Quadrupole Coupling of O-Chlorotoluene Studied by Microwave Spectroscopy and Ab Initio Calculations. J. Mol. Spectrosc. 2003, 220, 234–241. [Google Scholar] [CrossRef]
- Herschbach, D.R. Calculation of Energy Levels for Internal Torsion and Over-All Rotation. III. J. Chem. Phys. 1959, 31, 91–108. [Google Scholar] [CrossRef]
- Hartwig, H.; Dreizler, H. The Microwave Spectrum of Trans-2,3-Eimethyloxirane in Torsional Excited States. Z. Naturforschung A 1996, 51, 923–932. [Google Scholar] [CrossRef]
- Dyke, T.R.; Muenter, J.S. Microwave Spectrum and Structure of Hydrogen Bonded Water Dimer. J. Chem. Phys. 1974, 60, 2929–2930. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Simple, Efficient, and Universal Energy Decomposition Analysis Method Based on Dispersion-Corrected Density Functional Theory. J. Phys. Chem. A 2023, 127, 7023–7035. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, I.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J.; Trujillo, C. Evaluation of Electron Density Shifts in Noncovalent Interactions. J. Phys. Chem. A 2021, 125, 4741–4749. [Google Scholar] [CrossRef]
- Brown, G.G.; Dian, B.C.; Douglass, K.O.; Geyer, S.M.; Shipman, S.T.; Pate, B.H. A Broadband Fourier Transform Microwave Spectrometer Based on Chirped Pulse Excitation. Rev. Sci. Instrum. 2008, 79, 053103. [Google Scholar] [CrossRef]
- Pinacho, P.; Blanco, S.; López, J.C. The Complete Conformational Panorama of Formanilide–Water Complexes: The Role of Water as a Conformational Switch. Phys. Chem. Chem. Phys. 2019, 21, 2177–2185. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Pracht, P.; Grimme, S.; Bannwarth, C.; Bohle, F.; Ehlert, S.; Feldmann, G.; Gorges, J.; Müller, M.; Neudecker, T.; Plett, C.; et al. CREST—A Program for the Exploration of Low-Energy Molecular Chemical Space. J. Chem. Phys. 2024, 160, 114110. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Loru, D.; Vigorito, A.; Santos, A.F.M.; Tang, J.; Sanz, M.E. The Axial/Equatorial Conformational Landscape and Intramolecular Dispersion: New Insights from the Rotational Spectra of Monoterpenoids. Phys. Chem. Chem. Phys. 2019, 21, 26111–26116. [Google Scholar] [CrossRef]
- Verde, A.; López, J.C.; Blanco, S. The Role of the Transient Atropisomerism and Chirality of Flurbiprofen Unveiled by Laser-Ablation Rotational Spectroscopy. Chem. A Eur. J. 2023, 29, e202300064. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 16, Revision 16; Gaussian, Inc.: Wallingford, CT, USA, 2017. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction Methods for Noncovalent Interactions. WIREs Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An Open-source Ab Initio Electronic Structure Program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]

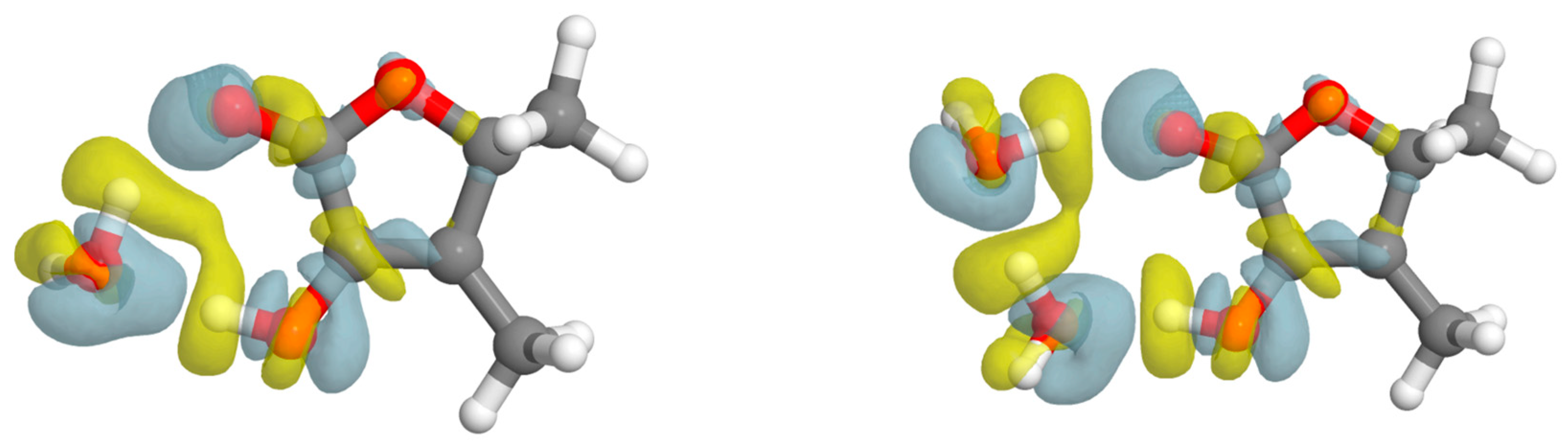

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sotolon | Sotolon-(H2O) | |||||
|---|---|---|---|---|---|---|
| SPFIT | XIAM | SPFIT | XIAM | |||
| Param. a | A | E | A | E | ||
| A/MHz | 2232.14921(35) b | 2232.0820(17) | 2232.09770(49) | 2113.48296(59) | 2113.43803(57) | 2113.44992(56) |
| B/MHz | 1683.79965(26) | 1683.73308(68) | 1683.76052(33) | 890.95221(48) | 890.91708(16) | 890.92982(20) |
| C/MHz | 1026.48763(23) | 1026.48750(42) | 1026.48327(28) | 659.47245(45) | 659.47015(14) | 659.46969(19) |
| ΔJ/kHz | - | - | - | 0.051(10) | - | - |
| ΔJK/kHz | - | 0.417(87) | - | - | - | - |
| ΔK/kHz | - | - | - | - | - | - |
| Da/MHz | - | −4.933(15) | - | - | −4.4371(23) | - |
| Db/MHz | - | −3.63(34) | - | - | −3.73(12) | - |
| Pa/uÅ2 | 283.035549(95) | 283.04151(25) | 283.03747(12) | 547.22591(45) | 547.23588(17) | 547.23277(31) |
| Pb/uÅ2 | 209.302678(95) | 209.29678(25) | 209.30285(12) | 219.11255(45) | 219.10525(17) | 219.10890(31) |
| Pc/uÅ2 | 17.106514(95) | 17.11242(25) | 17.11157(12) | 20.00889(45) | 20.02128(17) | 20.01629(31) |
| s | 30.73 | 29.02 | ||||
| V3/cm−1 | - | - | 372.38(43) | - | - | 348.78(24) |
| F/GHz | - | - | 161.45(derived) | - | - | 160.15 (derived) |
| F0/GHz | - | - | 159.521(93) | - | - | 158.973(54) |
| Iα/uÅ2 | - | - | 3.1681(18) | - | - | 3.1790(11) |
| ρ/deg | - | - | 0.012(calculated) | - | - | 0.0080(calculated) |
| β/deg | - | - | 0.7390(20) | - | - | 2.48(calculated) |
| γ | - | - | 0.0146(16) | - | - | 3.107(23) |
| µa/D | YES | YES | YES | YES | YES | YES |
| µb/D | YES | YES | YES | YES | YES | YES |
| µc/D | NO | NO | NO | NO | NO | NO |
| N | 30 | 30 | 60 | 48 | 39 | 82 |
| σ/kHz | 3.6 | 5.8 | 6.5 | 6.9 | 4.3 | 8.8 |
| Sotolon-(H2O)2 | ||||||
| SPFIT | XIAM | |||||
| Param. a | A | E | ||||
| A/MHz | 1680.068(25) | 1680.038(39) | 1680.094(21) | |||
| B/MHz | 565.40988(53) | 565.40074(58) | 565.40482(43) | |||
| C/MHz | 438.63777(62) | 438.63742(65) | 438.63699(49) | |||
| ΔJ/kHz | 0.0106(47) | 0.0465(53) | 0.0284(39) | |||
| ΔJK/kHz | 0.177(35) | - | 0.136(25) | |||
| ΔK/kHz | - | 2.23(67) | - | |||
| Da/MHz | - | −4.4339(41) | - | |||
| Db/MHz | - | −2.31(54) | - | |||
| Pa/uÅ2 | 872.5875(36) | 873.5925(48) | 872.5949(29) | |||
| Pb/uÅ2 | 279.5684(36) | 279.5643(48) | 279.5631(29) | |||
| Pc/uÅ2 | 21.2403(36) | 21.2498(48) | 21.2410(29) | |||
| s | 28.81 | |||||
| V3/cm−1 | 345.808(74) | |||||
| F/GHz | - | - | 159.93(derived) | |||
| F0/GHz | - | - | 158.99(derived) | |||
| Iα/uÅ2 | - | - | 3.18 | |||
| ρ/deg | - | - | 0.0068(calculated) | |||
| β/deg | - | - | 0.44(calculated) | |||
| γ | - | - | 0.054(calculated) | |||
| µa/D | YES | YES | YES | |||
| µb/D | NO | NO | NO | |||
| µc/D | NO | NO | NO | |||
| N | 33 | 31 | 68 | |||
| σ/kHz | 7.5 | 6.9 | 8.8 | |||
| Sotolon-(H2O) | Sotolon-(H2O)2 | |
|---|---|---|
| De/kJmol−1 | 56.74 | 100.00 |
| De/nHB/kJmol−1 | 28.37 | 33.33 |
| ΔEeles | ΔEex-rep | ΔEorb | ΔEdisp | ΔEint | |
|---|---|---|---|---|---|
| Sotolon-(H2O) | −107.6 | 136.6 | −55.4 | −31.5 | −57.9 |
| Sotolon-(H2O)2 | −161.5 | 202.4 | −90.6 | −50.7 | −100.4 |
| DFT | Experiment | |
|---|---|---|
| Sotolon | 326 | 372.38(43) |
| Sotolon-(H2O) | 294 | 348.78(24) |
| Sotolon-(H2O)2 | 298 | 345.808(74) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verde, A.; López, J.C.; Blanco, S. Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2. Int. J. Mol. Sci. 2025, 26, 5806. https://doi.org/10.3390/ijms26125806
Verde A, López JC, Blanco S. Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2. International Journal of Molecular Sciences. 2025; 26(12):5806. https://doi.org/10.3390/ijms26125806
Chicago/Turabian StyleVerde, Andrés, Juan Carlos López, and Susana Blanco. 2025. "Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2" International Journal of Molecular Sciences 26, no. 12: 5806. https://doi.org/10.3390/ijms26125806
APA StyleVerde, A., López, J. C., & Blanco, S. (2025). Probing Hydrogen-Bonding Preferences and Methyl Internal Rotation in Sotolon and Sotolon-(H2O)1,2. International Journal of Molecular Sciences, 26(12), 5806. https://doi.org/10.3390/ijms26125806