
Baculovirus Variant Detection from Transient CRISPR-Cas9-Mediated Disruption of gp64 at Different Gene Locations

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Effect of gp64 Gene Disruption on Foreign Protein and Progeny Virus Production
2.2. Confirmation of CRISPR-Cas9-Mediated Gene Editing of gp64
2.3. Variant Generation or Conservation over Passages
2.4. Evaluation of Mutations Outside the Targeted gp64 Gene
3. Discussion
3.1. CRISPR-Cas9-Mediated Targeted Disruption of gp64 During Virus Propagation
3.2. Are CRISPR-Cas9 Off-Targets Observed in Our System?
3.3. Variant Conservation or Random Mutations upon Virus Propagation in Cell Culture
4. Materials and Methods
4.1. Cell Line and Maintenance
4.2. Plasmid Design and Construction
4.3. Baculovirus Amplification and Quantification
4.4. Transfection-Infection Assay (T-I Assay)
4.5. Flow Cytometry Analysis of GFP upon gp64 Gene Disruption
4.6. Total Baculovirus Quantification via Flow Cytometry
4.7. Tiled-Amplicon Sequencing Assay for rBEV Genomes
4.8. Bioinformatics Pipeline for Minor Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcMNPV | Autographa californica multiple nucleopolyhedrovirus |
| au | arbitrary units |
| BEVS | baculovirus expression vector system |
| DIPs | defective interfering particles |
| EPDA | end-point dilution assay |
| GFP | green fluorescent protein |
| HDR | homology-directed repair |
| hpi | hours post-infection |
| hpt | hours post-transfection |
| hrs | homologous repeat regions |
| IDT | integrated DNA technologies |
| IVT | infectious virus titer |
| MOI | multiplicity of infection |
| NHEJ | non-homologous end joining |
| NGS | next-generation sequencing |
| ORF | open reading frame |
| ori | origin of replication |
| PAM | protospacer adjacent motif |
| PBS | phosphate-buffered saline |
| pfu/mL | plaque-forming units per mL |
| rBEV | recombinant baculovirus expression vector |
| SFM | serum-free media |
| sgRNA | single-guide RNA |
| T-I assay | transfection-infection assay |
References
- Smith, G.E.; Summers, M.D.; Fraser, M.J. Production of Human Beta Interferon in Insect Cells Infected with a Baculovirus Expression Vector. J. Mol. Cell. Biol. 1983, 3, 2156–2165. [Google Scholar]
- Urabe, M.; Ding, C.; Kotin, R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002, 13, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Kost, T.A.; Condreay, J.P.; Jarvis, D.L. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 2005, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Airenne, K.J.; Hu, Y.C.; Kost, T.A.; Smith, R.H.; Kotin, R.M.; Ono, C.; Matsuura, Y.; Wang, S.; Ylä-Herttuala, S. Baculovirus: An insect-derived vector for diverse gene transfer applications. Mol. Ther. 2013, 21, 739–749. [Google Scholar] [CrossRef]
- Felberbaum, R.S. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol. J. 2015, 10, 702–714. [Google Scholar] [CrossRef]
- Van Oers, M.M.; Pijlman, G.P.; Vlak, J.M. Thirty years of baculovirus-insect cell protein expression: From dark horse to mainstream technology. J. Gen. Virol. 2015, 96, 6–23. [Google Scholar] [CrossRef]
- Yee, C.M.; Zak, A.J.; Hill, B.D.; Wen, F. The Coming Age of Insect Cells for Manufacturing and Development of Protein Therapeutics. Ind. Eng. Chem. Res. 2018, 57, 10061–10070. [Google Scholar] [CrossRef]
- Zhang, X.; He, A.; Zong, Y.; Tian, H.; Zhang, Z.; Zhao, K.; Xu, X.; Chen, H. Improvement of protein production in baculovirus expression vector system by removing a total of 10 kb of nonessential fragments from Autographa californica multiple nucleopolyhedrovirus genome. Front. Microbiol. 2023, 14, 1171500. [Google Scholar] [CrossRef]
- Rohrmann, G.F. The AcMNPV genome: Gene content, conservation, and function. In Baculovirus Molecular Biology; National Center for Biotechnology Information: Bethesda, MD, USA, 2019; pp. 201–275. [Google Scholar]
- Ayres, M.D.; Howard, S.C.; Kuzio, J.; Lopez-Ferber, M.; Possee, R.D. The Complete DNA Sequence of Autographa californica Nuclear Polyhedrosis Virus. Virology 1994, 202, 586–605. [Google Scholar] [CrossRef]
- Miele, S.A.B.; Garavaglia, M.J.; Belaich, M.N.; Ghiringhelli, P.D. Baculovirus: Molecular Insights on Their Diversity and Conservation. Int. J. Evol. Biol. 2011, 2011, 379424. [Google Scholar] [CrossRef]
- Wang, R.; Deng, F.; Hou, D.; Zhao, Y.; Guo, L.; Wang, H.; Hu, Z. Proteomics of the Autographa californica Nucleopolyhedrovirus Budded Virions. J. Virol. 2010, 84, 7233–7242. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.N.; Rohrmann, G.F. Transfer, Incorporation, and Substitution of Envelope Fusion Proteins among Members of the Baculoviridae, Orthomyxoviridae, and Metaviridae (Insect Retrovirus) Families. J. Virol. 2002, 76, 5301–5304. [Google Scholar] [CrossRef] [PubMed]
- Blissard, G.W.; Wenz, J.R. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J. Virol. 1992, 66, 6829–6835. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, S.; Bai, L.; Zhao, H.; Shang, W.; Zhong, Z.; Maimaiti, T.; Gao, X.; Ji, N.; Chao, Y.; et al. Structural transition of GP64 triggered by a pH-sensitive multi-histidine switch. Nat. Commun. 2024, 15, 7668. [Google Scholar] [CrossRef]
- Oomens, A.G.; Blissard, G.W. Requirement for GP64 to drive efficient budding of Autographa californica multicapsid nucleopolyhedrovirus. Virology 1999, 254, 297–314. [Google Scholar] [CrossRef]
- Monsma, S.A.; Oomens, A.G.; Blissard, G.W. The GP64 envelope fusion protein is an essential baculovirus protein required for cell-to-cell transmission of infection. J. Virol. 1996, 70, 4607–4616. [Google Scholar] [CrossRef]
- Marek, M.; Van Oers, M.M.; Devaraj, F.F.; Vlak, J.M.; Merten, O.W. Engineering of baculovirus vectors for the manufacture of virion-free biopharmaceuticals. Biotechnol. Bioeng. 2011, 108, 1056–1067. [Google Scholar] [CrossRef]
- Bruder, M.R.; Aucoin, M.G. Evaluation of Virus-Free Manufacture of Recombinant Proteins Using CRISPR-Mediated Gene Disruption in Baculovirus-Infected Insect Cells. Vaccines 2023, 11, 225. [Google Scholar] [CrossRef]
- Bruder, M.R.; Aucoin, M.G. A sensitive assay for scrutiny of Autographa californica multiple nucleopolyhedrovirus genes using CRISPR-Cas9. Appl. Microbiol. Biotechnol. 2023, 107, 4323–4335. [Google Scholar] [CrossRef]
- Chakraborty, M.; Nielsen, L.; Nash, D.; Nissimov, J.I.; Charles, T.C.; Aucoin, M.G. Adapting next-generation sequencing to in process CRISPR-Cas9 genome editing of recombinant AcMNPV vectors: From shotgun to tiled-amplicon sequencing. Viruses 2025, 17, 437. [Google Scholar] [CrossRef]
- Hausjell, C.S.; Klausberger, M.; Ernst, W.; Grabherr, R. Evaluation of an inducible knockout system in insect cells based on co-infection and CRISPR/Cas9. PLoS ONE 2023, 18, e0289178. [Google Scholar] [CrossRef] [PubMed]
- Bruder, M.R.; Walji, S.D.; Aucoin, M.G. Comparison of CRISPR-Cas9 Tools for Transcriptional Repression and Gene Disruption in the BEVS. Viruses 2021, 13, 1925. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Hong, M.; Li, T.; Xue, W.; Zhang, S.; Cui, L.; Wang, H.; Zhang, Y.; Zhou, L.; Gu, Y.; Xia, N.; et al. Genetic engineering of baculovirus-insect cell system to improve protein production. Front. Bioeng. Biotechnol. 2022, 10, 994743. [Google Scholar] [CrossRef]
- Hong, Q.; Liu, J.; Wei, Y.; Wei, X. Application of Baculovirus Expression Vector System (BEVS) in Vaccine Development. Vaccines 2023, 11, 1218. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA—Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–822. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Sari-Ak, D.; Alomari, O.; Shomali, R.A.; Lim, J.; Thimiri Govinda Raj, D.B. Advances in CRISPR-Cas9 for the Baculovirus Vector System: A Systematic Review. Viruses 2023, 15, 54. [Google Scholar] [CrossRef]
- Guo, T.; Feng, Y.L.; Xiao, J.J.; Liu, Q.; Sun, X.N.; Xiang, J.F.; Kong, N.; Liu, S.C.; Chen, G.Q.; Wang, Y.; et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018, 19, 170. [Google Scholar] [CrossRef]
- Hussain, S.S.; Majumdar, R.; Moore, G.M.; Narang, H.; Buechelmaier, E.S.; Bazil, M.J.; Ravindran, P.T.; Leeman, J.E.; Li, Y.; Jalan, M.; et al. Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell. Nucleic Acids Res. 2021, 49, e74. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Powichrowski, J.; Bruder, M.R.; Nielsen, L.; Sung, C.; Boegel, S.J.; Aucoin, M.G. Probing Baculovirus Vector Gene Essentiality for Foreign Gene Expression Using a CRISPR-Cas9 System. Methods Mol. Biol. 2024, 2829, 127–156. [Google Scholar]
- Oxford Expression Technologies. Baculovirus Gene Mutations and Protein Expression. Oxford Expression Technologies Blog. 2016. Available online: https://www.oetltd.com/post/baculovirus-gene-mutations-and-protein-expression (accessed on 8 May 2025).
- Pijlman, G.P.; Van Den Born, E.; Martens, D.E.; Vlak, J.M. Autographa californica baculoviruses with large genomic deletions are rapidly generated in infected insect cells. Virology 2001, 283, 132–138. [Google Scholar] [CrossRef]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [CrossRef]
- Boezen, D.; Ali, G.; Wang, M.; Wang, X.; van der Werf, W.; Vlak, J.M.; Zwart, M.P. Empirical estimates of the mutation rate for an alphabaculovirus. PLoS Genet. 2022, 18, e1009806. [Google Scholar] [CrossRef]
- Kool, M.; Voncken, J.W.; Van Lier, F.L.J.; Tramper, J.; Vlak, J.M. Detection and analysis of Autographa californica nuclear polyhedrosis virus mutants with defective interfering properties. Virology 1991, 183, 739–746. [Google Scholar] [CrossRef]
- Mabashi-Asazuma, H.; Jarvis, D.L. CRISPR-Cas9 vectors for genome editing and host engineering in the baculovirus–insect cell system. Proc. Natl. Acad. Sci. USA 2017, 114, 9068–9073. [Google Scholar] [CrossRef]
- Port, F.; Chen, H.M.; Lee, T.; Bullock, S.L. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E2967–E2976. [Google Scholar] [CrossRef]
- Claudi, B.; Spröte, P.; Chirkova, A.; Personnic, N.; Zankl, J.; Schürmann, N.; Schmidt, A.; Bumann, D. Phenotypic variation of salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 2014, 158, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method for estimating fifty per cent endpoints. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]
- O’Reilly, D.R.; Miller, L.K.; Luckow, V.A. Baculovirus Expression Vectors: A Laboratory Manual; W. H. Freeman and Company: New York, NY, USA, 1992. [Google Scholar]
- Shen, C.F.; Meghrous, J.; Kamen, A. Quantitation of baculo v irus particles by flow cytometry. J. Virol. Methods 2002, 105, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Chakraborty, M.; Nielsen, L.; Nash, D.; Bruder, M.R.; Nissimov, J.I.; Charles, T.C.; Aucoin, M.G. Tiled-amplicon sequencing and variant analysis of p6.9 rBEV genomes. Borealis 2025, V1. [Google Scholar] [CrossRef]


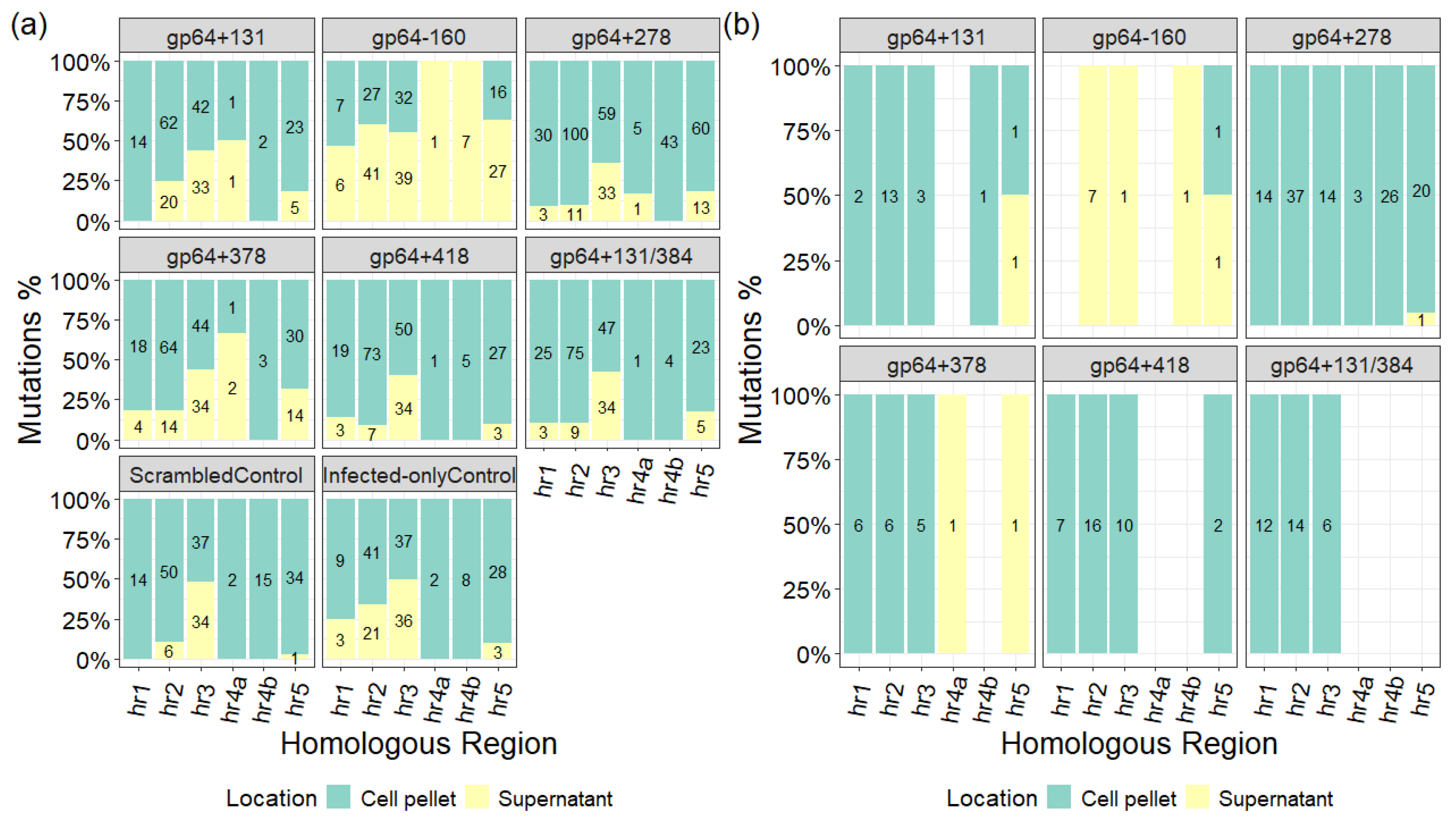
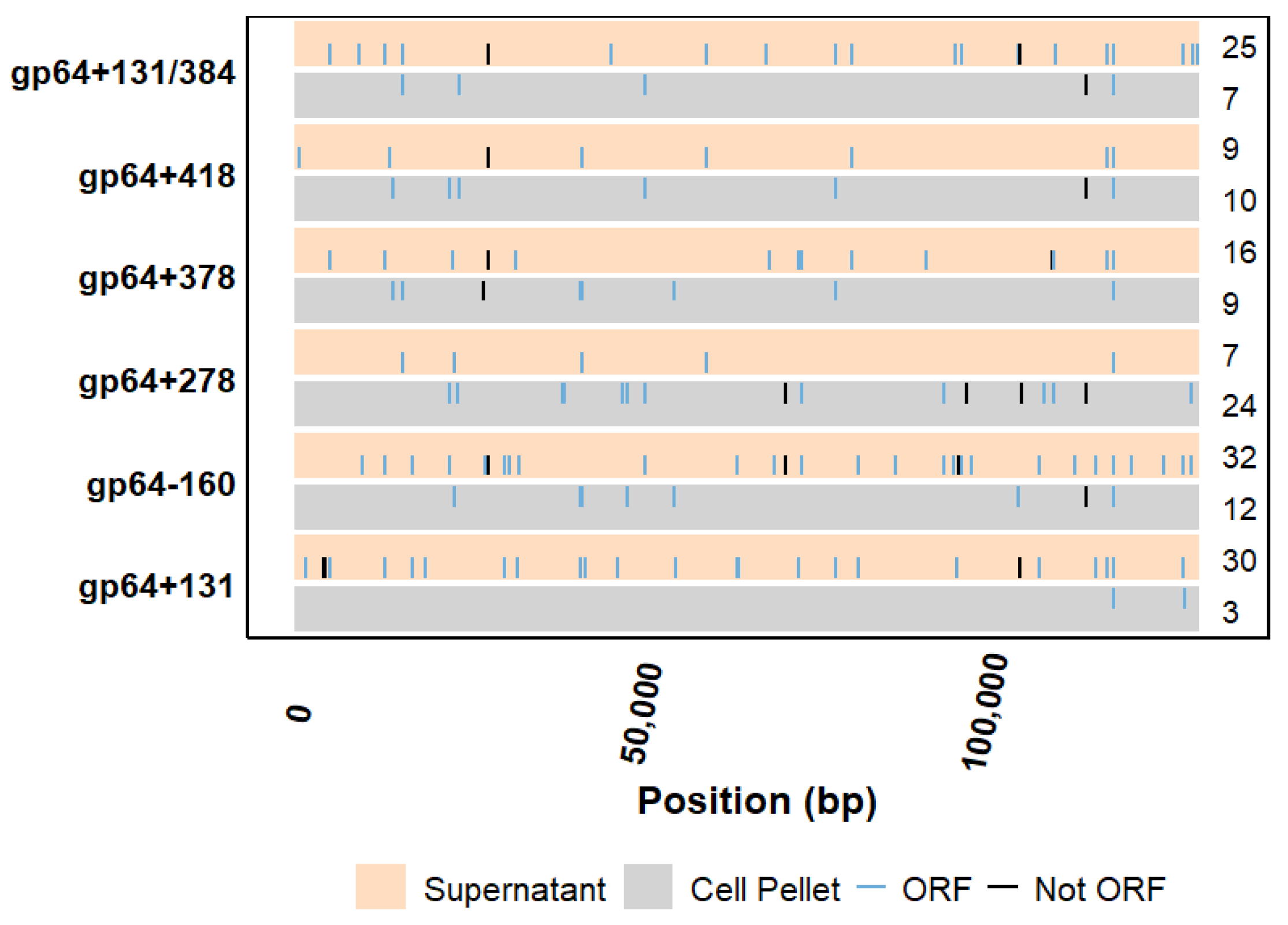

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target 1 | Estimated Position (bp) 2 | Observed Position (bp) [Mutation] (Frequency) 3 | |
|---|---|---|---|
| Cell Pellet | Supernatant | ||
| gp64+131 | 107,222–107,227 | 107,222 [−C] (6.62%) | Not observed |
| gp64−160 | 107,199–107,204 | 107,201 [−CCA] (6.30%) 107,202 [−CA] (8.47%) | 107,199 [−CTCCA] (3.05%) 107,200 [−TCCA] (3.48%) 107,201 [−CCA] (5.44%) 107,202 [−CA] (6.34%) 107,204 [−C] (1.43%) |
| gp64+278 | 107,075–107,080 | Not observed | Not observed |
| gp64+378 | 106,975–106,980 | 106,975 [−T] (4.51%) | 106,975 [−T] (4.04%) |
| gp64+418 | 106,935–106,940 | 106,935 [−CA] (4.24%) | 106,935 [−CA] (2.17%) |
| gp64+131/384 | 107,222–107,227 106,969–106,974 | Not observed | Not observed |
| Mutation Region | Mutation Type | Virus Stock Consensus/ Virus Stock Variant/ T-I Assay Variant 1 | Mutation Position |
|---|---|---|---|
| AcOrf-84 promoter | SNPs | G/T/T | 71,443 |
| fgf 3′ UTR | SNPs | A/G/G | 27,502 |
| fgf 3′ UTR | SNPs | G/A/A | 27,505 |
| fgf 3′ UTR | SNPs | G/A/A | 27,506 |
| fgf 3′ UTR | SNPs | G/A/A | 27,509 |
| AcOrf-603 | SNPs | C/T/T | 3960 |
| AcOrf-1629 | SNPs | G/A/A | 6375 |
| lef10 | SNPs | G/A/A | 45,761 |
| AcOrf-91 | SNPs | T/A/A | 78,627 |
| AcOrf-91 | SNPs | A/T/T | 78,666 |
| AcOrf-1629 | Insertion | -/GATC/GATC | 7304 |
| AcOrf-51 | Insertion | -/A/A | 44,073 |
| egt | Deletion | 2 CTAGAGA/-/- | 12,427 |
| AcOrf-91 | Deletion | TAT/-/- | 78,926 |
| Gene | Location | Spacer Sequence (5′-3′) | PAM | Strand |
|---|---|---|---|---|
| Scrambled control | N/A | CACCTTGAAGCGCATGAACT | N/A | N/A |
| gp64 | +131 | GGAAACGCTGCAAAAGGACG | TGG | Antisense |
| gp64 | −160 | GTTGTAGTCCGTCTCCACGA | TGG | Sense |
| gp64 | +278 | AACGCTGAATGTGGGCAAAG | AGG | Antisense |
| gp64 | +378 | GACTGTTTTCGCGACAACGA | GGG | Antisense |
| gp64 | +418 | AAGGCAAAGAGTTGGTGAAG | CGG | Antisense |
gp64 | +131/ 384 | GGAAACGCTGCAAAAGGACG/ TTTCGCGACAACGAGGGCCG | TGG/ CGG | Antisense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, M.; Nielsen, L.; Nash, D.; Bruder, M.R.; Nissimov, J.I.; Charles, T.C.; Aucoin, M.G. Baculovirus Variant Detection from Transient CRISPR-Cas9-Mediated Disruption of gp64 at Different Gene Locations. Int. J. Mol. Sci. 2025, 26, 5805. https://doi.org/10.3390/ijms26125805
Chakraborty M, Nielsen L, Nash D, Bruder MR, Nissimov JI, Charles TC, Aucoin MG. Baculovirus Variant Detection from Transient CRISPR-Cas9-Mediated Disruption of gp64 at Different Gene Locations. International Journal of Molecular Sciences. 2025; 26(12):5805. https://doi.org/10.3390/ijms26125805
Chicago/Turabian StyleChakraborty, Madhuja, Lisa Nielsen, Delaney Nash, Mark R. Bruder, Jozef I. Nissimov, Trevor C. Charles, and Marc G. Aucoin. 2025. "Baculovirus Variant Detection from Transient CRISPR-Cas9-Mediated Disruption of gp64 at Different Gene Locations" International Journal of Molecular Sciences 26, no. 12: 5805. https://doi.org/10.3390/ijms26125805
APA StyleChakraborty, M., Nielsen, L., Nash, D., Bruder, M. R., Nissimov, J. I., Charles, T. C., & Aucoin, M. G. (2025). Baculovirus Variant Detection from Transient CRISPR-Cas9-Mediated Disruption of gp64 at Different Gene Locations. International Journal of Molecular Sciences, 26(12), 5805. https://doi.org/10.3390/ijms26125805