
Molecular Insight into the Recognition of DNA by the DndCDE Complex in DNA Phosphorothioation

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
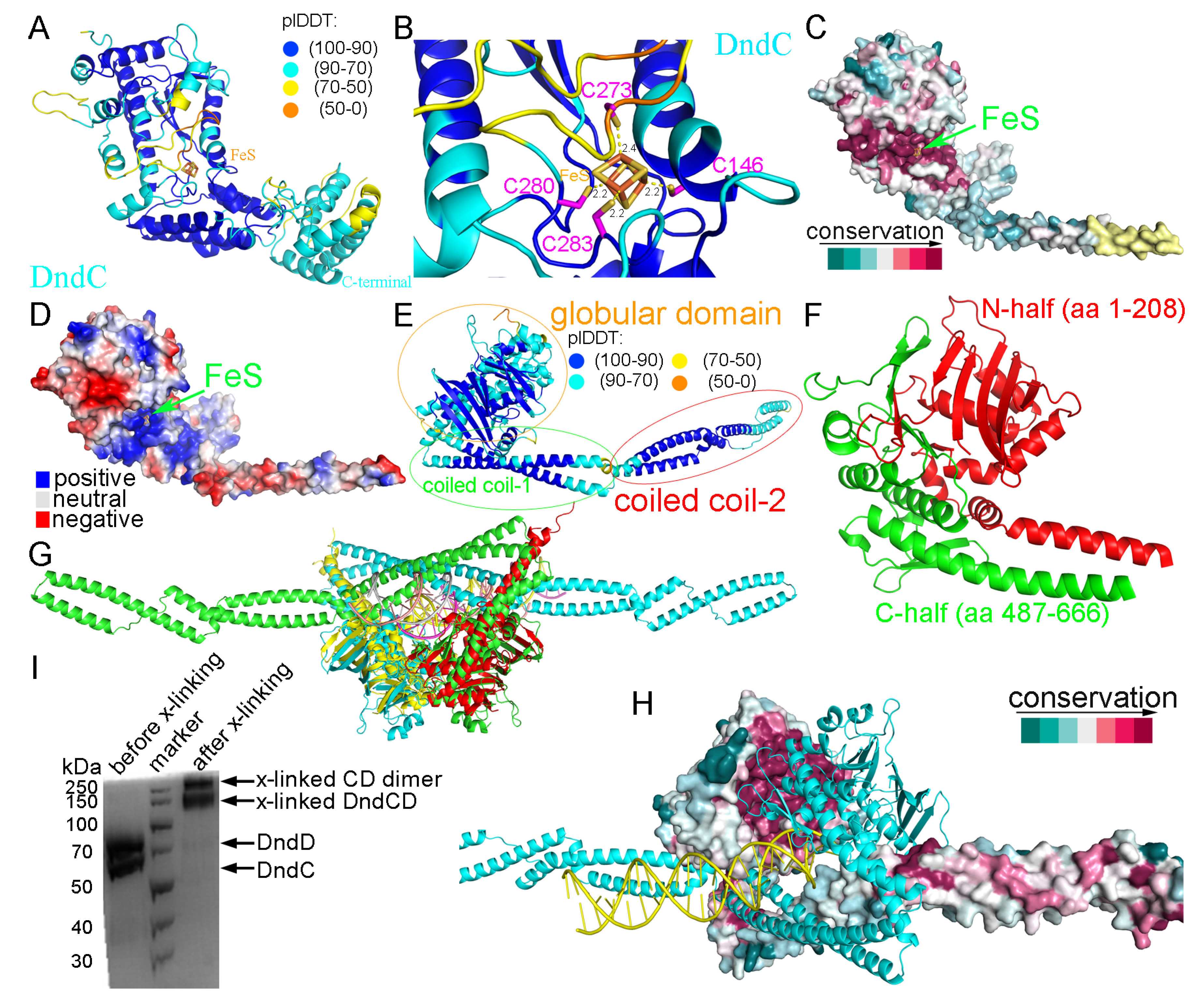
2.1. Structural Prediction of DndC
2.2. Structural Prediction of DndD
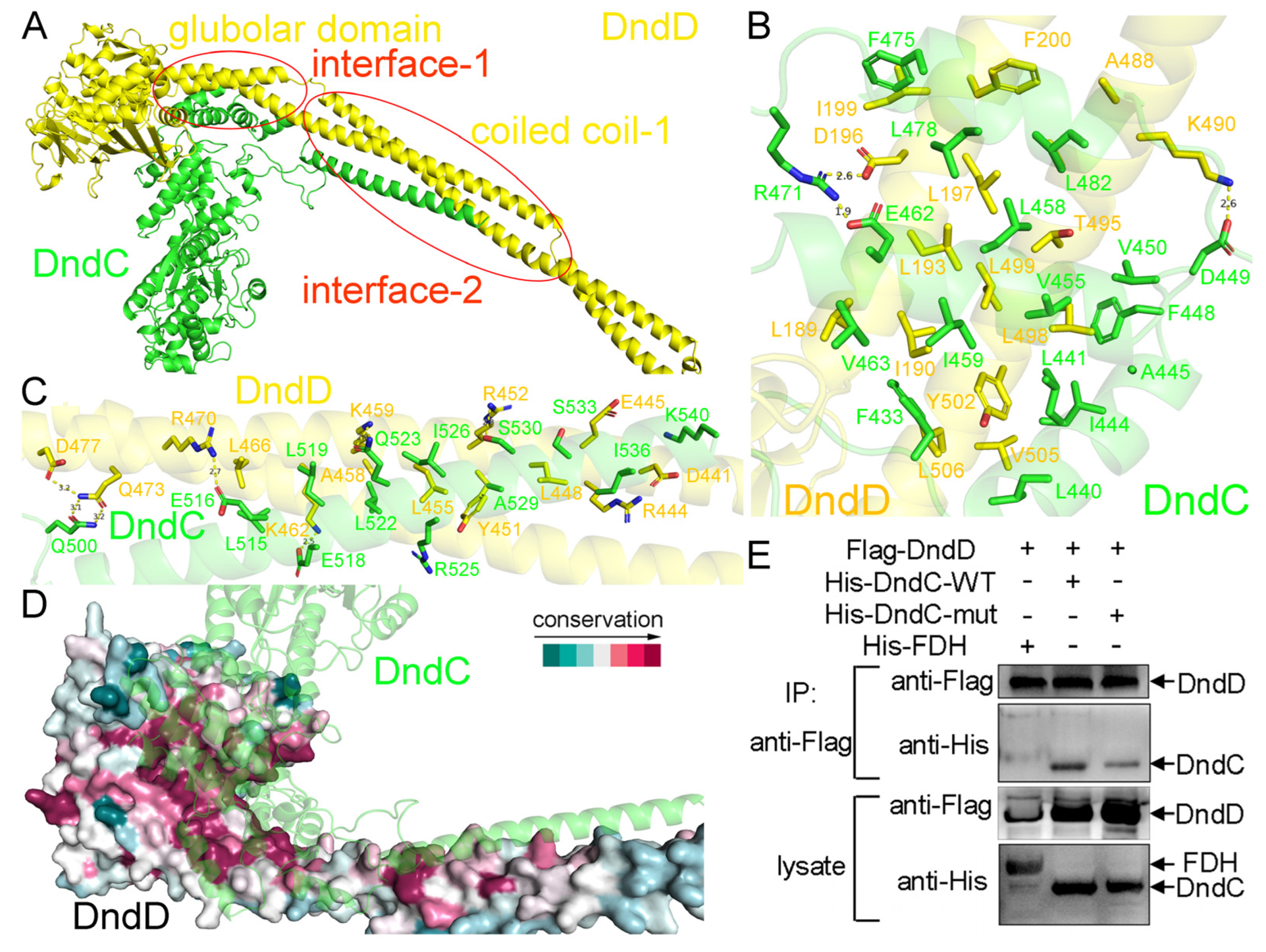
2.3. The Recognition Mechanism Between DndC and DndD
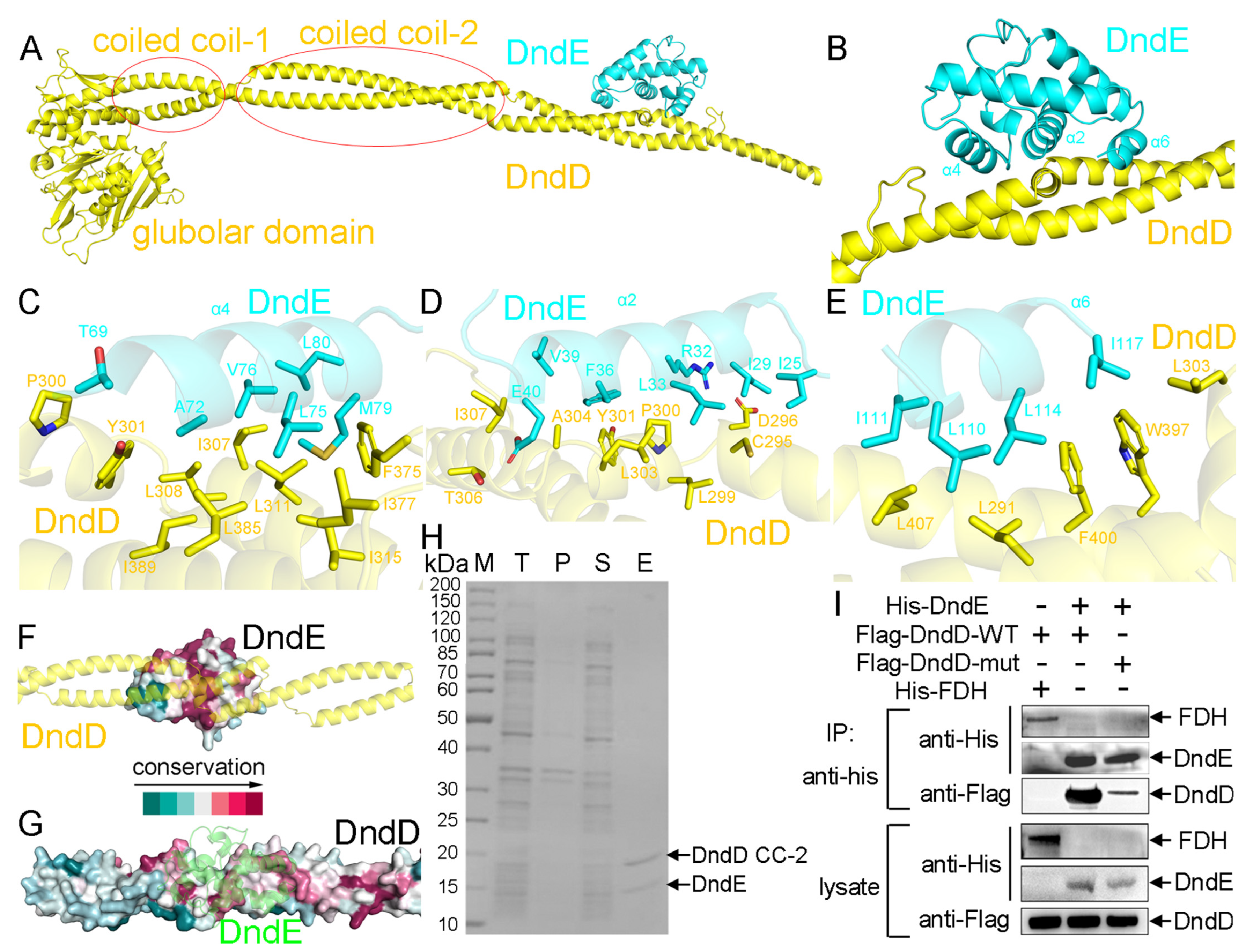
2.4. The Recognition Mechanism Between DndD and DndE
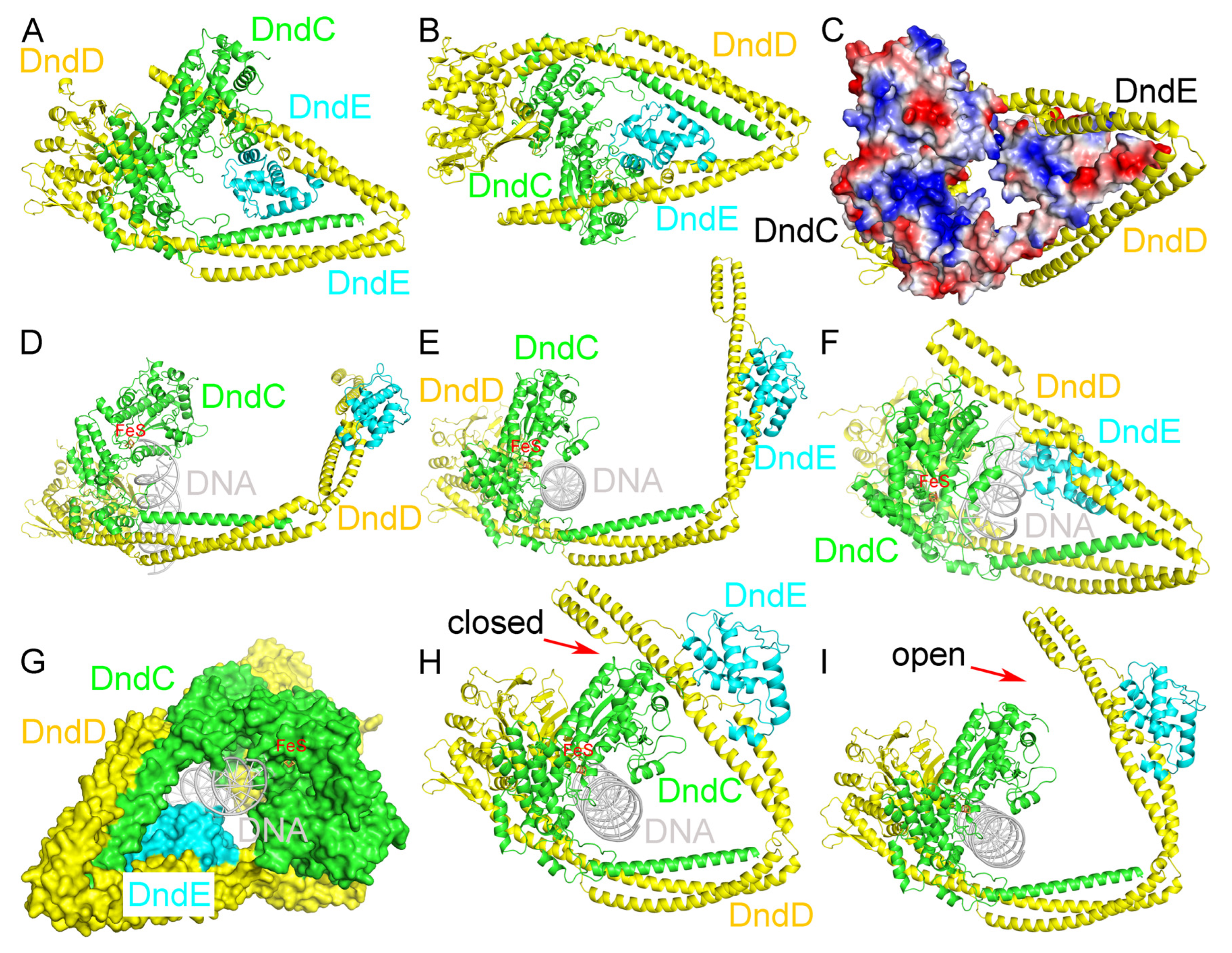
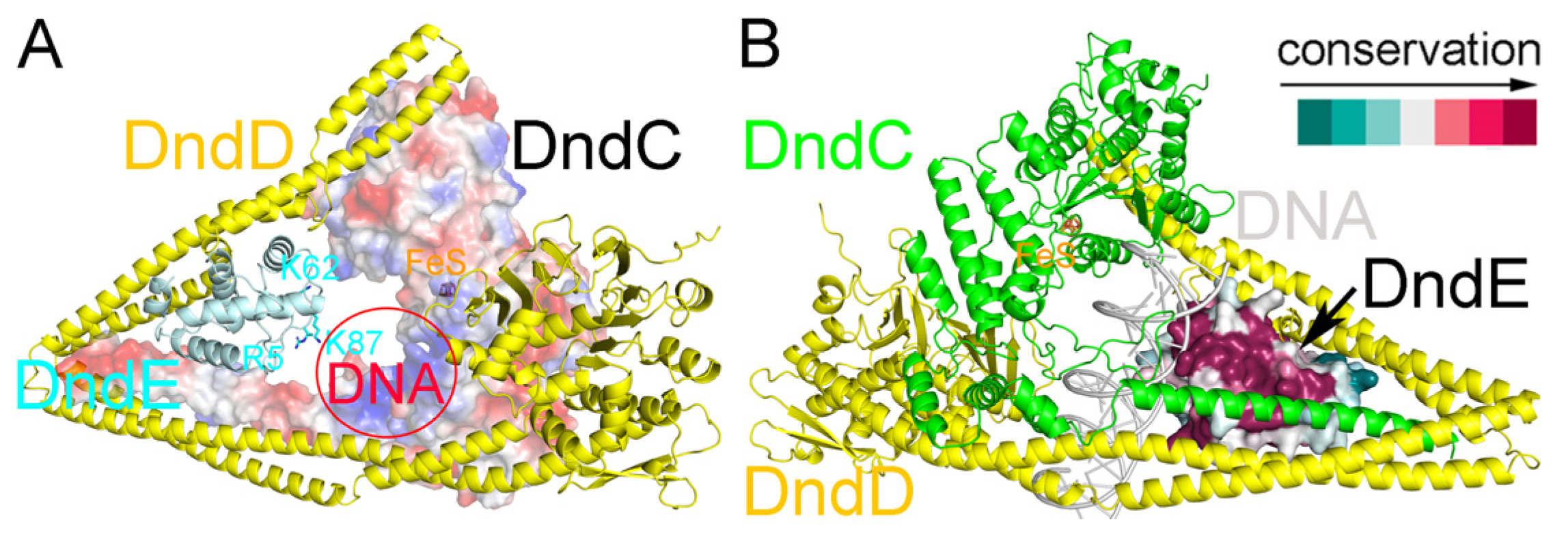
2.5. Recognition of DNA by the DndCDE Complex
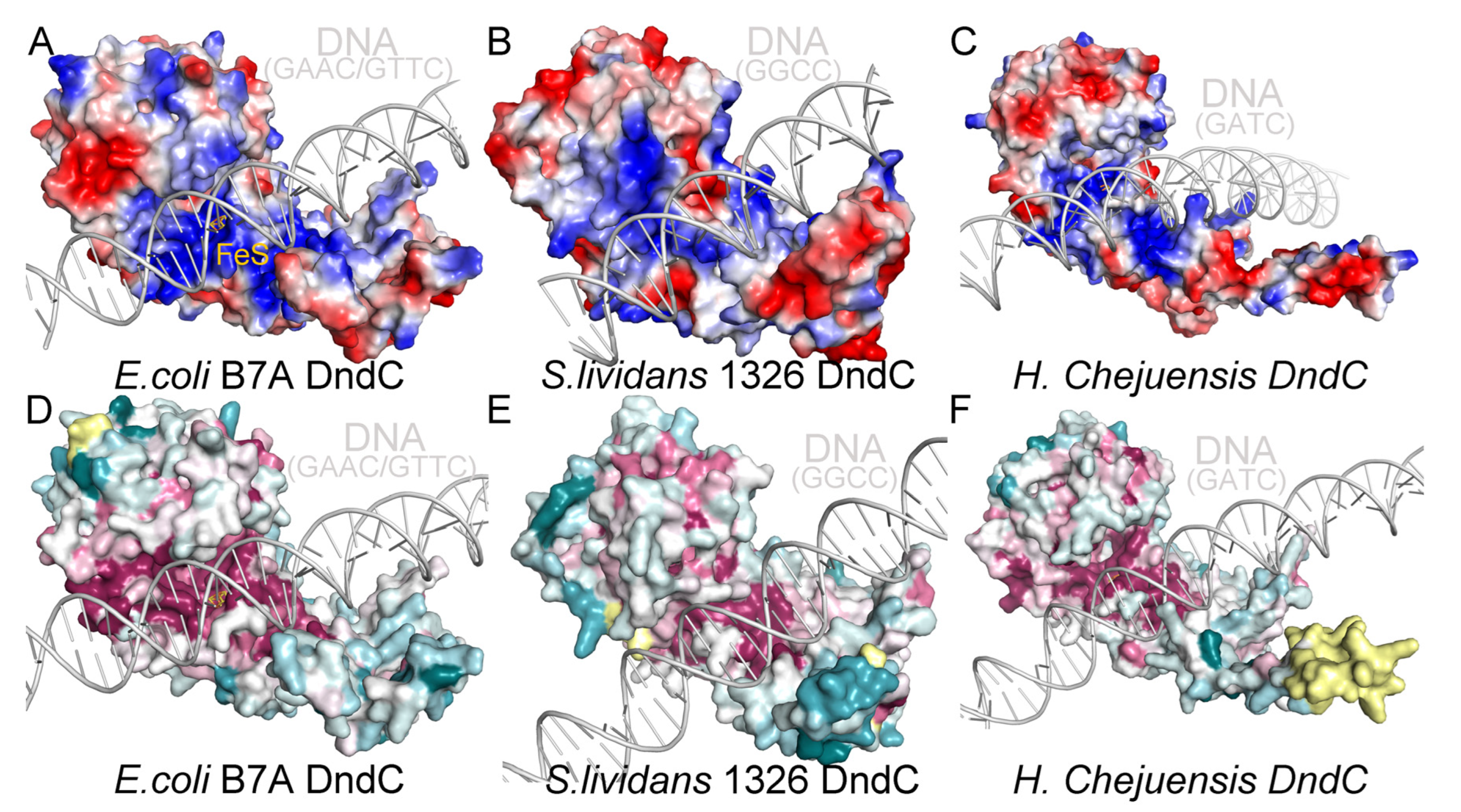
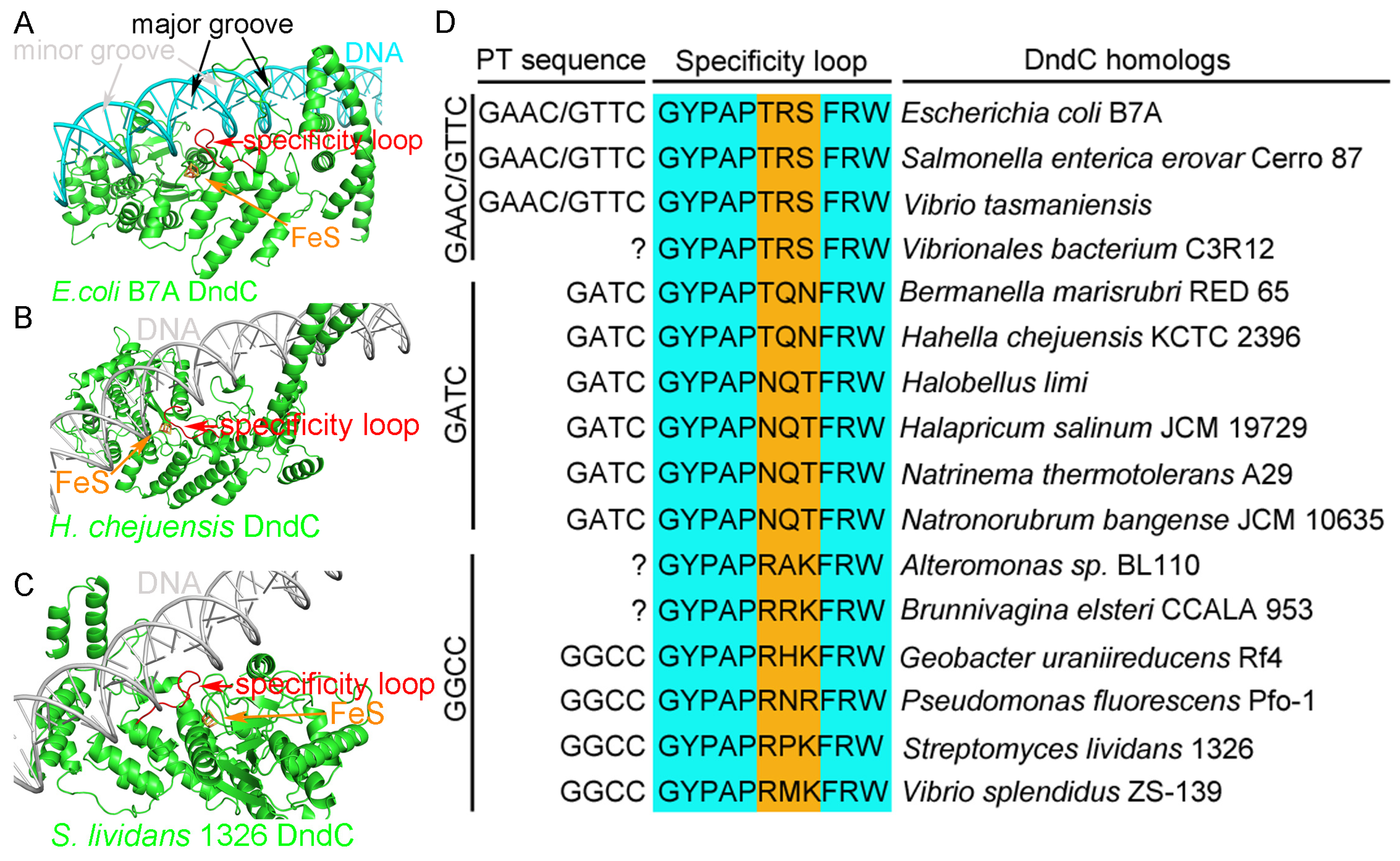
2.6. Recognition of the Core Consensus Phosphorothioation Motif by DndC
2.7. Recognition of DNA by DndE
2.8. Microbiological Assays Showing the Importance of the Specificity Loop in DNA PT
3. Discussion
4. Materials and Methods
4.1. Protein Structure Prediction
4.2. Protein Expression and Purification
4.3. Electron Microscopy
4.4. Conjugative Transfer [44]
4.5. Using LC-MS to Examine Bacterial DNA PT Modification [21]
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Jiang, S.; Deng, Z.; Dedon, P.C.; Chen, S. DNA phosphorothioate modification–A new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev. 2019, 43, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, N.A.; Chen, Y.; Needham, D.M.; Parada, A.E.; Sachdeva, R.; Trinh, V.; Chen, T.; Fuhrman, J.A. Genome and epigenome of a novel marine Thaumarchaeota strain suggest viral infection, phosphorothioation DNA modification and multiple restriction systems. Environ. Microbiol. 2017, 19, 2434–2452. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Lunazzi, A.; Fujiwara-Nagata, E.; Avendaño-Herrera, R.; Bernardet, J.F.; Touchon, M.; Duchaud, E. From the Flavobacterium genus to the phylum Bacteroidetes: Genomic analysis of dnd gene clusters. FEMS Microbiol. Lett. 2013, 348, 26–35. [Google Scholar] [CrossRef]
- Liu, G.; Fu, W.; Zhang, Z.; He, Y.; Yu, H.; Wang, Y.; Wang, X.; Zhao, Y.L.; Deng, Z.; Wu, G.; et al. Structural basis for the recognition of sulfur in phosphorothioated DNA. Nat. Commun. 2018, 9, 4689. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Xu, T.; Taghizadeh, K.; Wishnok, J.S.; Zhou, X.; You, D.; Deng, Z.; Dedon, P.C. Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 2007, 3, 709–710. [Google Scholar] [CrossRef]
- Zhou, X.; Deng, Z.; Firmin, J.L.; Hopwood, D.A.; Kieser, T. Site-specific degradation of Streptomyces lividans DNA during electrophoresis in buffers contaminated with ferrous iron. Nucleic Acids Res. 1988, 16, 4341–4352. [Google Scholar] [CrossRef]
- Xiong, W.; Zhao, G.; Yu, H.; He, X. Interactions of Dnd proteins involved in bacterial DNA phosphorothioate modification. Front. Microbiol. 2015, 6, 1139. [Google Scholar] [CrossRef] [PubMed]
- Pu, T.; Liang, J.; Mei, Z.; Yang, Y.; Wang, J.; Zhang, W.; Liang, W.J.; Zhou, X.; Deng, Z.; Wang, Z. Phosphorothioated DNA is shielded from oxidative damage. Appl. Environ. Microbiol. 2019, 85, e00104-19. [Google Scholar] [CrossRef]
- Tong, T.; Chen, S.; Wang, L.; Tang, Y.; Ryu, J.Y.; Jiang, S.; Wu, X.; Chen, C.; Luo, J.; Deng, Z.; et al. Occurrence, evolution, and functions of DNA phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E2988–E2996. [Google Scholar] [CrossRef]
- Yao, F.; Xu, T.; Zhou, X.; Deng, Z.; You, D. Functional analysis of spfD gene involved in DNA phosphorothioation in Pseudomonas fluorescens Pf0-1. FEBS Lett. 2009, 583, 729–733. [Google Scholar] [CrossRef]
- Xu, T.; Liang, J.; Chen, S.; Wang, L.; He, X.; You, D.; Wang, Z.; Li, A.; Xu, Z.; Zhou, X.; et al. DNA phosphorothioation in Streptomyces lividans: Mutational analysis of the dnd locus. BMC Microbiol. 2009, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; He, X.; Liang, J.; Li, A.; Xu, T.; Kieser, T.; Helmann, J.D.; Deng, Z. A novel DNA modification by sulphur. Mol. Microbiol. 2005, 57, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gao, H.; Wu, D.; Jiang, S.; Huang, W.; Chen, C.; Deng, Z.; Xiong, L.; Wu, G.; Wang, L. Structural and functional analysis of DndE involved in DNA phosphorothioation in the haloalkaliphilic archaea Natronorubrum bangense JCM10635. mBio 2022, 14, e00716-22. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, C.; Liang, J.; Zhang, T.; Hu, Z.; Wang, Z.; Lan, W.; Li, F.; Wu, H.; Ding, J.; et al. Structural insights into DndE from Escherichia coli B7A involved in DNA phosphorothioation modification. Cell Res. 2012, 22, 1203–1206. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Vergin, K.L.; Giovannoni, S.J.; Chan, S.W.; DeMott, M.S.; Taghizadeh, K.; Cordero, O.X.; Cutler, M.; Timberlake, S.; et al. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl. Acad. Sci. USA 2011, 108, 2963–2968. [Google Scholar] [CrossRef]
- You, D.; Wang, L.; Yao, F.; Zhou, X.; Deng, Z. A novel DNA modification by sulfur: DndA is a NifS-like cysteine desulfurase capable of assembling DndC as an iron-sulfur cluster protein in Streptomyces lividans. Biochemistry 2007, 46, 6126–6133. [Google Scholar] [CrossRef]
- An, X.; Xiong, W.; Yang, Y.; Li, F.; Zhou, X.; Wang, Z.; Deng, Z.; Liang, J. A novel target of IscS in Escherichia coli: Participating in DNA phosphorothioation. PLoS ONE 2014, 7, e51265. [Google Scholar] [CrossRef]
- Cupp-Vickery, J.R.; Urbina, H.; Vickery, L.E. Crystal structure of IscS, a cysteine desulfurase from Escherichia coli. J. Mol. Biol. 2003, 330, 1049–1059. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Z.; Lin, K.; Qian, T.; Zhang, Y.; You, D.; He, X.; Wang, Z.; Liang, J.; Deng, Z.; et al. Crystal structure of the cysteine desulfurase DndA from Streptomyces lividans which is involved in DNA phosphorothioation. PLoS ONE 2012, 7, e36635. [Google Scholar] [CrossRef]
- Kaiser, J.T.; Clausen, T.; Bourenkow, G.P.; Bartunik, H.D.; Steinbacher, S.; Huber, R. Crystal structure of a NifS-like protein from Thermotoga maritima: Implications for iron sulphur cluster assembly. J. Mol. Biol. 2000, 297, 451–464. [Google Scholar] [CrossRef]
- Shi, R.; Proteau, A.; Villarroya, M.; Moukadiri, I.; Zhang, L.; Trempe, J.F.; Matte, A.; Armengod, M.E.; Cygler, M. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol. 2010, 8, e1000354. [Google Scholar] [CrossRef] [PubMed]
- Tirupati, B.; Vey, J.L.; Drennan, C.L.; Bollinger, J.M. Kinetic and structural characterization of Slr0077/SufS, the essential cysteine desulfurase from Synechocystis sp. PCC 6803. Biochemistry 2004, 43, 12210–12219. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Wu, X.; He, W.; Liu, Z.; Wu, S.; Chen, C.; Chen, S.; Xiang, Q.; Deng, Z.; Liang, D.; et al. DNA phosphorothioate modifications influence the global transcriptional response and protect DNA from double-stranded breaks. Sci. Rep. 2014, 4, 6642. [Google Scholar] [CrossRef]
- Wu, D.; Tang, Y.Q.; Chen, S.W.; He, Y.; Chang, X.F.; Zheng, W.Z.; Deng, Z.X.; Li, Z.Q.; Wang, L.R.; Wu, G.; et al. The functional coupling between restriction and DNA phosphorothioate modification systems underlying the DndFGH restriction complex. Nat. Catal. 2022, 5, 1131–1144. [Google Scholar] [CrossRef]
- Xiong, X.; Wu, G.; Wei, Y.; Liu, L.; Zhang, Y.; Su, R.; Jiang, X.; Li, M.; Gao, H.; Tian, X.; et al. SspABCD-SspE is a phosphorothioation-sensing bacterial defence system with broad anti-phage activities. Nat. Microbiol. 2020, 5, 917–928. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, S.; Xing, M.; Chen, C.; Lai, C.; Li, N.; Liu, G.; Wu, D.; Gao, H.; Hong, L.; et al. Structural analysis of an L-cysteine desulfurase from an Ssp DNA phosphorothioation system. mBio 2020, 11, e00488-20. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- He, X.H.; Li, J.R.; Shen, S.Y.; Xu, H.E. AlphaFold3 versus experimental structures: Assessment of the accuracy in ligand-bound G protein-coupled receptors. Acta Pharmacol. Sin. 2025, 46, 1111–1122. [Google Scholar] [CrossRef]
- Coskuner-Weber, O. Structures prediction and replica exchange molecular dynamics simulations of α-synuclein: A case study for intrinsically disordered proteins. Int. J. Biol. Macromol. 2024, 276, 133813. [Google Scholar] [CrossRef]
- Aniana, A.; Nashed, N.T.; Ghirlando, R.; Drago, V.N.; Kovalevsky, A.; Louis, J.M. Characterization of alternate encounter assemblies of SARS-CoV-2 main protease. J. Biol. Chem. 2024, 300, 107675. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Y.X.; Gao, J.N.; Chen, J.; Huang, J.; Qiao, X. Multiple Mutations in the β1 Subunit of the Nicotinic Acetylcholine Receptor Confer Resistance to Neonicotinoids. J. Agric. Food Chem. 2025, 73, 12176–12183. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, T.; Chen, R.; Xu, Y. Discovery of CMNPD31124 as a novel marine-derived PKMYT1 inhibitor for pancreatic ductal adenocarcinoma therapy: Computational and biological insights. Front. Pharmacol. 2025, 16, 1569765. [Google Scholar] [CrossRef]
- Medvedev, K.E.; Schaeffer, R.D.; Grishin, N.V. Leveraging AI to explore structural contexts of post-translational modifications in drug binding. J. Cheminform. 2025, 17, 67. [Google Scholar] [CrossRef]
- Pu, T.; Mei, Z.; Zhang, W.; Liang, W.J.; Zhou, X.; Liang, J.; Deng, Z.; Wang, Z. An in vitro DNA phosphorothioate modification reaction. Mol. Microbiol. 2020, 113, 452–463. [Google Scholar] [CrossRef]
- Liu, Y.; Sung, S.; Kim, Y.; Li, F.; Gwon, G.; Jo, A.; Kim, A.K.; Kim, T.; Song, O.K.; Lee, S.E.; et al. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 2016, 35, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.; Polzer, P.; Diederichs, K.; Welte, W.; Dimroth, P. Structure of the rotor ring of F-Type Na+-ATPase from Ilyobacter tartaricus. Science 2005, 308, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Postic, G.; Ghannay, S.; Tahi, F. Has AlphaFold3 achieved success for RNA? Biol. Crystallogr. 2025, 81, 49–62. [Google Scholar] [CrossRef]
- Dürr, S.L.; Rothlisberger, U. Predicting metal-protein interactions using cofolding methods: Status quo. bioRxiv 2024. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Koumadorakis, D.E.; Lazaros, K.; Ivantsik, O.; Exarchos, T.P.; Vrahatis, A.G.; Kotsiantis, S.; Vlamos, P. AlphaFold3: An Overview of Applications and Performance Insights. Int. J. Mol. Sci. 2025, 26, 3671. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Elliott, L.G.; Simpkin, A.J.; Rigden, D.J. ABCFold: Easier running and comparison of AlphaFold 2025, 3, Boltz-1 and Chai-1. bioRxiv 2025. [Google Scholar] [CrossRef]
- Wlodawer, A.; Dauter, Z.; Porebski, P.J.; Minor, W.; Stanfield, R.; Jaskolski, M.; Pozharski, E.; Weichenberger, C.X.; Rupp, B. Detect, correct, retract: How to manage incorrect structural models. FEBS J. 2018, 285, 444–466. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ou, H.Y.; Wang, T.; Li, L.; Tan, H.; Zhou, X.; Rajakumar, K.; Deng, Z.; He, X. Cleavage of phosphorothioated DNA and methylated DNA by the type IV restriction endonuclease ScoMcrA. PLoS Genet. 2010, 6, e1001253. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, W.; Wang, Y.; Ge, Y.; Gao, H.; Sun, X.; Deng, Z.; Wang, L.; Chen, S.; He, X.; Wu, G. Molecular Insight into the Recognition of DNA by the DndCDE Complex in DNA Phosphorothioation. Int. J. Mol. Sci. 2025, 26, 5765. https://doi.org/10.3390/ijms26125765
Fu W, Wang Y, Ge Y, Gao H, Sun X, Deng Z, Wang L, Chen S, He X, Wu G. Molecular Insight into the Recognition of DNA by the DndCDE Complex in DNA Phosphorothioation. International Journal of Molecular Sciences. 2025; 26(12):5765. https://doi.org/10.3390/ijms26125765
Chicago/Turabian StyleFu, Wencheng, Yuli Wang, Yashi Ge, Haiyan Gao, Xuan Sun, Zixin Deng, Lianrong Wang, Shi Chen, Xinyi He, and Geng Wu. 2025. "Molecular Insight into the Recognition of DNA by the DndCDE Complex in DNA Phosphorothioation" International Journal of Molecular Sciences 26, no. 12: 5765. https://doi.org/10.3390/ijms26125765
APA StyleFu, W., Wang, Y., Ge, Y., Gao, H., Sun, X., Deng, Z., Wang, L., Chen, S., He, X., & Wu, G. (2025). Molecular Insight into the Recognition of DNA by the DndCDE Complex in DNA Phosphorothioation. International Journal of Molecular Sciences, 26(12), 5765. https://doi.org/10.3390/ijms26125765