
Pathogenesis and Clinical Management of Metabolic Dysfunction-Associated Steatotic Liver Disease
.jpg)
 ,
,  ,
,  ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. MASLD Pathogenesis
2.1. Physiopathology of Liver Fibrosis
2.2. MASLD and the Disruption of Carbohydrate Metabolism
2.3. MASLD and the Disruption of Lipid Metabolism
2.4. MASLD and Thyroid Hormones
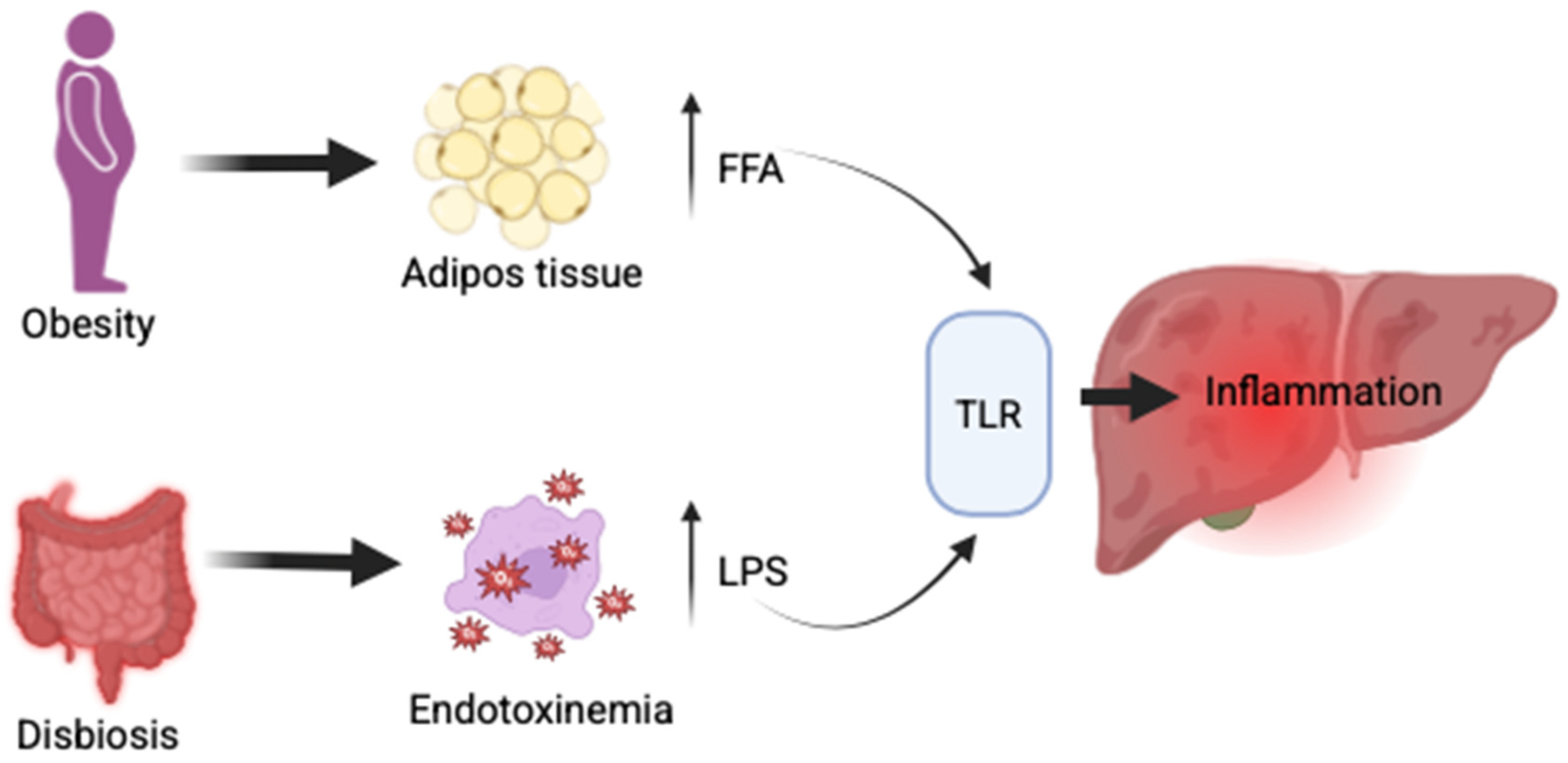
2.5. MASLD and Gut Dysbiosis
2.5.1. Mechanical Barrier Disruption in MASLD
2.5.2. Immunological Barrier Disruption in MASLD
2.5.3. Microbial Barrier Disruption in MASLD
2.5.4. Microbiome in MASLD
3. MASLD Diagnosis
3.1. Initial Clinical Evaluation
3.2. Imaging Assessment of Hepatic Steatosis
3.3. Liver Biopsy: Indications and Histology
3.4. Differential Diagnosis
3.5. Staging and Fibrosis Assessment
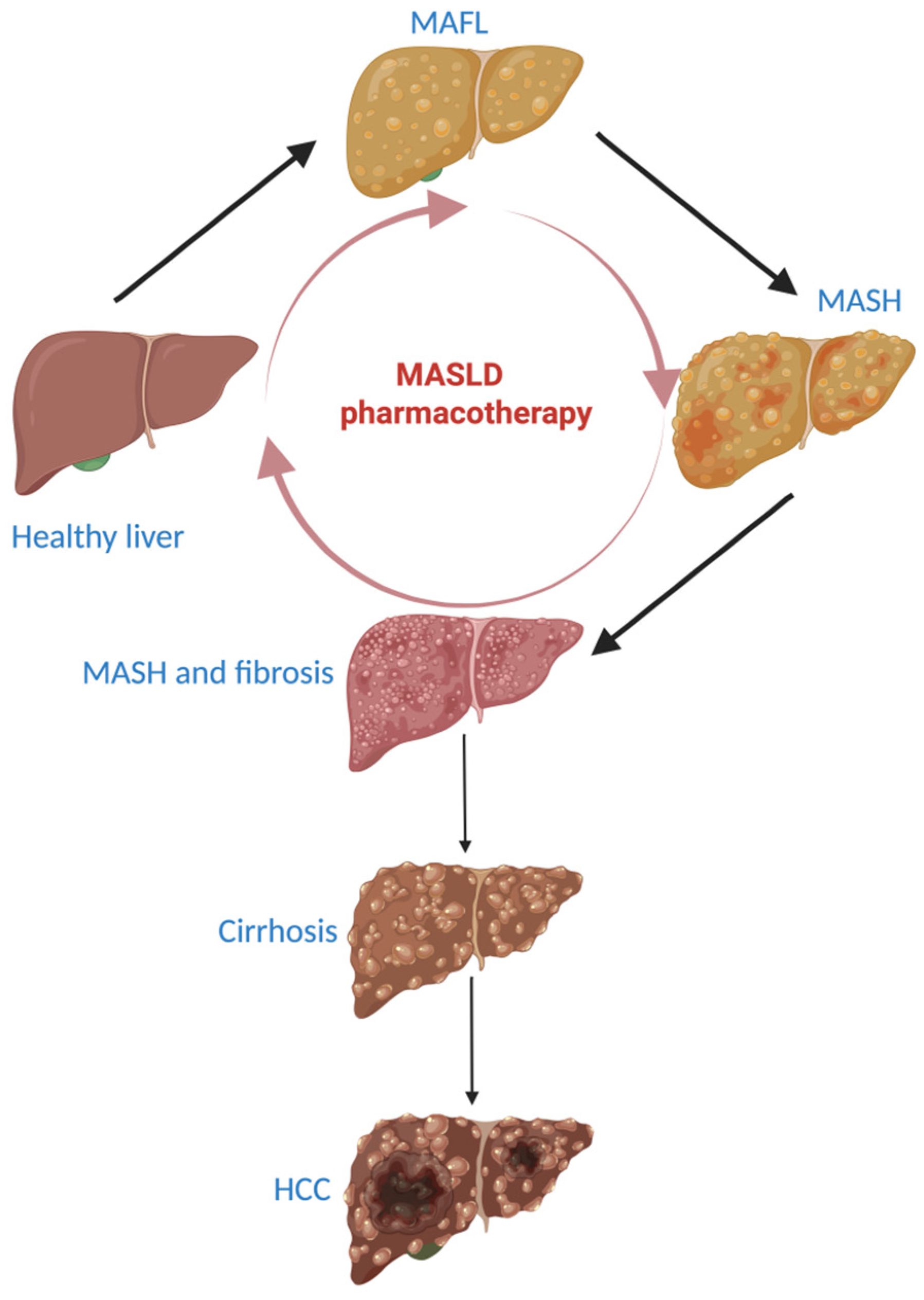
4. MASLD Therapeutic Options
- -
- 90–95% of patients will not progress to liver cirrhosis;
- -
- the progression of MASLD to liver cirrhosis lasts for 20 to 25 years;
- -
- MASLD has the possibility to regress. When steatosis is present, regression takes place in a short time, but when fibrosis is present and adiposopathy is mitigated, regression is still possible, but takes longer [102].
4.1. Healthy Lifestyle and MASLD
4.2. Drug Therapy in MASLD
4.2.1. Anti-Hyperglycemic Drugs
4.2.2. Ketohexokinase Inhibitors
4.2.3. Statins
4.2.4. Peroxisome Proliferator-Activator Receptor (PPAR) Agonists
4.2.5. Farnesoid-X Receptor (FXR) Agonists
4.2.6. Fibroblast Growth Factors (FGF) Agonists
4.2.7. Thyroid Hormone Receptor Beta (THR-β) Agonists and MASLD
4.2.8. Anti-Fibrotic and Anti-Inflammatory Agents
4.2.9. Microbiome Modulation in MASLD
4.3. Metabolic Surgery
4.3.1. Bariatric Surgery
4.3.2. Endoscopic Bariatric Interventions
4.4. Faecal Microbiota Transplant
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| ALD | alcohol-associated liver disease |
| ALT | alanine aminotransferase |
| AMPK | AMP-activated protein kinase |
| ApoB | apolipoprotein B |
| ARFI | acoustic radiation force impulse |
| ASK1 | apoptosis signal-regulating kinase-1 |
| AST | aspartate aminotransferase |
| BA | bile acids |
| BMI | body mass index |
| cAMP | cyclic adenosine monophosphate |
| CB1 | cannabinoid 1 receptor |
| CT | computed tomography |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| EVs | extracellular vesicles |
| FFAs | fatty acids |
| FGF15/19/21 | fibroblast growth factor 15/19/21 |
| FIB4 | fibrosis-4 index |
| FoxO1 | forkhead box protein O1 |
| FXR | farnesoid X receptor |
| GGT | gamma-glutamyl transferase |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP-1 | glucagon-like peptide-1 |
| HbA1c | hemoglobin A1c |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HDAC | histone deacetylase |
| HFD | high-fat diet |
| HK2 | hexokinase 2 |
| HOMA-IR | homeostatic model assessment of insulin resistance |
| IL-6/17/22 | interleukine-6/17/22 |
| INR | international normalized ratio |
| JAM-A | junctional adhesion molecule A |
| LDL | low-density lipoprotein |
| LPS | lipopolysacharides |
| LXR | liver X receptor |
| MAFL | metabolic-associated fatty liver |
| MASH | metabolic dysfunction-associated steatohepatitis |
| MASLD | metabolic dysfunction-associated steatotic liver disease |
| mGPD | mitochondrial glycerophosphate dehydrogenase |
| MRI | magnetic resonance imaging |
| MRE | magnetic resonance elastography |
| MTTP | microsomal triglyceride transport protein |
| NAFL | nonalcoholic fatty liver |
| NAFLD | nonalcoholic fatty liver disease |
| NAS | NAFLD activity score |
| NASH | non-alcoholic steatohepatitis |
| NF-kb | nuclear transcription factor Kappa B |
| NLPR-3 | family pyrin domain-containing 3 |
| NOD | nucleotide oligomerisation domain |
| NR1A2 | nuclear receptor subfamily 1, group A, member 2 = TR-β |
| PAMPs | pathogen-associated molecular patterns |
| PDL1 | programmed death-ligand 1 |
| p-HSL | phosphohormone-sensitive lipase |
| PKM2 | pyruvate kinase isozyme type M2 |
| PPAR | peroxisome proliferator-activator receptor |
| p-PLIN1 | phospho-perilipin 1 |
| PV-1 | plasmalemma vesicle-associated protein-1 |
| RCT | randomized clinical trial |
| ROS | reactive oxygen species |
| SAF-A score | steatosis activity fibrosis score |
| SCs | stellate cells |
| SFL | simple fatty liver |
| SGLT-2 | sodium-glucose cotransporter 2 |
| sIgA | secretory immunoglobulin A |
| SREBP | sterol regulatory element-binding protein |
| T3 | triiodothyronine |
| T4 | thyroxine |
| T2DM | type 2 diabetes mellitus |
| TG | tryglycerides |
| TGF-β | transforming growth factor-β |
| THR (α and β) | thyroid hormone receptor isoforms (α and β) |
| TLRs | toll-like receptors |
| Tregs | regulatory T cells |
| TSH | thyroid-stimulating hormone |
| VCTE | vibration-controlled transient elastography |
| VLDL | very low-density lipoprotein |
| ZO-1 | zonula occludens-1 |
References
- Scorletti, E.; Carr, R.M. A new perspective on NAFLD: Focusing on lipid droplets. J. Hepatol. 2022, 76, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, T.; Zhang, Q.; Jia, P.; Song, M.; Zhang, Q.; Ruan, G.; Ge, Y.; Lin, S.; Wang, Z.; et al. New-onset age of nonalcoholic fatty liver disease and cancer risk. JAMA Netw. Open 2023, 6, e2335511. [Google Scholar] [CrossRef] [PubMed]
- American association for the study of liver disease; Latin american association for the study of the liver; European association for the study of the liver. A call for unity: The path towards a more precise and patient-centric nomenclature for NAFLD. Hepatology 2023, 78, 3–5. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Hutchison, A.L.; Tavaglione, F.; Romeo, S.; Charlton, M. Endocrine aspects of metaboloc dysfunction-associated steatotic liver disease (MASLD): Beyond insuline resistence. J. Hepatol. 2023, 79, 1524–1541. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Li, Y.; Yang, P.; Ye, J.; Xu, Q.; Wu, J.; Wang, Y. Updated mechanisms of MASLD pathogenesis. Lipids Health Dis. 2024, 23, 117. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Younossi, Y.; Golabi, P.; Mishra, A.; Rafiq, N.; Henry, L. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 2020, 69, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P. NAFLD, MAFLD, and beyond: One or several acronyms for better comprehension and patient care. Intern. Emerg. Med. 2023, 18, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Kendall, B.J.; El-Serag, H.B.; Thrift, A.P.; Macdonald, G.A. Hepatocellular and extrahepatic cancer risk in people with non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2024, 9, 159–169. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef]
- Ramirez-Mejia, M.M.; Jimenez-Gutierrez, C.; Eslam, M.; George, J.; Mendez-Sanchez, N. Breaking new ground: MASLD vs. MAFLD-which holds the key for risk stratification? Hepatol. Int. 2024, 18, 168–178. [Google Scholar] [CrossRef]
- Njei, B.; Ameyaw, P.; Al-Ajlouni, Y.; Njei, L.P.; Boateng, S. Diagnosis and management of lean metabolic dysfunction associated steatotic liver disease (MASLD): A systematic review. Cureus 2024, 16, e71451. [Google Scholar] [CrossRef]
- Lai, J.C.T.; Liang, L.Y.; Wong, G.L.H. Noninvasive tests for liver fibrosis in 2024: Are there different scales for different diseases? Gastroenterol. Rep. 2024, 12, goae024. [Google Scholar] [CrossRef]
- Cathcart, J.; Barrett, R.; Bowness, J.S.; Mukhopadhya, A.; Lynch, R.; Dillon, J.F. Accuracy of non-invasive imaging techniques for the diagnosis of MASH in patients with MASLD: A systematic review. Liver Int. 2024, 45, e16127. [Google Scholar] [CrossRef]
- Gurjar, S.; Bhat, A.R.; Upadhya, R.; Shenoy, R.P. Extracellular vesicle-mediated approaches for the diagnosis and therapy of MASLD: Current advances and future prospective. Lipids Health Dis. 2025, 24, 5. [Google Scholar] [CrossRef]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD practice guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. NAFLD: Mechanisms, treatments, and biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V. MASLD treatment—A shift in the paradigm is imminent. Front. Med. 2023, 10, 1316284. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The intricate relationship between type 2 diabetes mellitus (T2DM), insulin resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Huang, X.; Chen, H.; Wen, S.; Dong, M.; Zhou, L.; Yuan, X. Therapeutic approaches for nonalcoholic fatty liver disease:established targets and drugs. Diabetes Metab. Syndr. Obes. 2023, 16, 1809–1819. [Google Scholar] [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwalder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease—Novel insights into cellular communication circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Qu, W.; Ma, T.; Cai, J.; Zhang, X.; Zhang, P.; She, Z.; Wan, F.; Li, H. Liver fibrosis and MAFLD: From molecular aspects to novel pharmacological strategies. Front. Med. 2021, 8, 761538. [Google Scholar] [CrossRef]
- Li, W.; Chang, N.; Li, L. Heterogeneity and function of Kupffer cells in liver injury. Front. Immunol. 2022, 13, 940867. [Google Scholar] [CrossRef]
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis in NAFLD/NASH: From pathophysiology towards diagnostic and therapeutic strategies. Mol. Aspects Med. 2024, 95, 101231. [Google Scholar] [CrossRef]
- Portincasa, P.; Khalil, M.; Mahdi, L.; Perniola, V.; Idone, V.; Graziani, A.; Baffy, G.; Di Ciaula, A. Metabolic dysfunction-associated steatotic liver disease: From pathogenesis to current therapeutic options. Int. J. Mol. Sci. 2024, 25, 5640. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, S.; Zhao, Y.; Sun, Q.; Zhang, J.; Shen, D.; Wu, J.; Shen, N.; Fu, X.; Sun, X.; et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J. Pathol. 2018, 246, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Passarella, S.; Schurr, A.; Portincasa, P. Mitochondrial transport in glycolysis and gluconeogenesis: Achievements and perspectives. Int. J. Mol. Sci. 2021, 22, 12620. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef]
- Beaven, S.W.; Matveyenko, A.; Wroblewski, K.; Chao, L.; Wilpitz, D.; Hsu, T.W.; Lentz, J.; Drew, B.; Hevener, A.L.; Tontonoz, P. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013, 18, 106–117. [Google Scholar] [CrossRef]
- Kamagate, A.; Qu, S.; Perdomo, G.; Su, D.; Kim, D.H.; Slusher, S.; Meseck, M.; Dong, H.H. FoxO1 mediates insulin-dependentregulation of hepatic VLDL production in mice. J. Clin. Investig. 2008, 118, 2347–2364. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Karin, M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021, 33, 2316–2328. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef]
- Yen, C.E.; Nelson, D.W.; Yen, M.I. Intestinal triacylglycerol synthesis in fat absorption and systemic energy metabolism. J. Lipid Res. 2015, 56, 489–501. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Colell, A.; Mari, M.; Morales, A.; Fernandez-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.P.; Collins, S. A compendium of G-protein-coupled receptors and cyclic nucleotide regulation of adipose tissue metabolism and energy expenditure. Clin. Sci. 2020, 134, 473–512. [Google Scholar] [CrossRef]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W.C. Progress and challenges of selective Farnesoid X receptor modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef]
- Jung, Y.; Koo, B.K.; Jang, S.Y.; Kim, D.; Lee, H.; Lee, D.H.; Joo, S.K.; Jung, Y.J.; Park, J.H.; Yoo, T.; et al. Association between circulating bile acid alterations and nonalcoholic steatohepatitis independent of obesity and diabetes mellitus. Liver Int. 2021, 41, 2892–2902. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, 4–14. [Google Scholar] [CrossRef]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic acid receptor beta stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPAR alpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jimenez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Selective agonists of thyroid hormone receptor beta for the treatment of NASH. N. Engl. J. Med. 2024, 390, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xie, H.; Shan, H.; Zheng, Z.; Li, G.; Li, M.; Hong, L. Development of thyroid hormones and synthetic thyromimetics in non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2022, 23, 1102. [Google Scholar] [CrossRef] [PubMed]
- Kizivat, T.; Maric, I.; Mudri, D.; Curcic, I.B.; Primorac, D.; Smolic, M. Hypothyroidism and nonalcoholic fatty liver disease: Pathophysiological associations and therapeutic implications. J. Clin. Transl. Hepatol. 2020, 8, 347–353. [Google Scholar] [CrossRef]
- Khaznadar, F.; Khaznadar, O.; Petrovic, A.; Hefer, M.; Gjoni, F.; Gjoni, S.; Steiner, J.; Smolic, M.; Bojanic, K. MAFLD pandemic: Updates in pharmacotherapeutic approach development. Curr. Issues Mol. Biol. 2024, 46, 6300–6314. [Google Scholar] [CrossRef]
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044. [Google Scholar] [CrossRef]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Portincasa, P. The role of microbiota in nonalcoholic fatty liver disease. Eur. J. Clin. Investig. 2022, 52, e13768. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; Angelis, M.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.; Di Ciaula, A. Intestinal barrier and permeability in health, obesity and NAFLD. Biomedicines 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Liver steatosis, gut-liver axis, microbiome and environmental factors. a never-ending bidirectional cross-talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Benedé-Ubieto, R.; Cubero, F.J.; Nevzorova, Y.A. Breaking the barriers: The role of gut homeostasis in metabolic-associated steatotic liver disease (MASLD). Gut Microbes 2024, 16, 2331460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019. [Google Scholar] [CrossRef]
- Liu, L.; Yin, M.; Gao, J.; Yu, C.; Lin, J.; Wu, A.; Zhu, J.; Xu, C.; Liu, X. Intestinal barrier function in the pathogenesis of nonalcoholic fatty liver disease. J. Clin. Transl. Hepatol. 2023, 11, 452–458. [Google Scholar] [CrossRef]
- Xin, D.; Zong-Shun, L.; Bang-Mao, W.; Lu, Z. Expression of intestinal tight junction proteins in patients with non-alcoholic fatty liver disease. Hepato-Gastroenterol. 2014, 61, 136–140. [Google Scholar]
- Brescia, P.; Rescigno, M. The gut vascular barrier: A new player in the gut–liver–brain axis. Trends Mol. Med. 2021, 27, 844–855. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 7, 1216–1228. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, Q.; Chang, R.; Zhou, X.; Xu, C. Intestinal barrier function–non-alcoholic fatty liver disease interactions and possible role of gut microbiota. J. Agric. Food Chem. 2019, 67, 2754–2762. [Google Scholar] [CrossRef]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
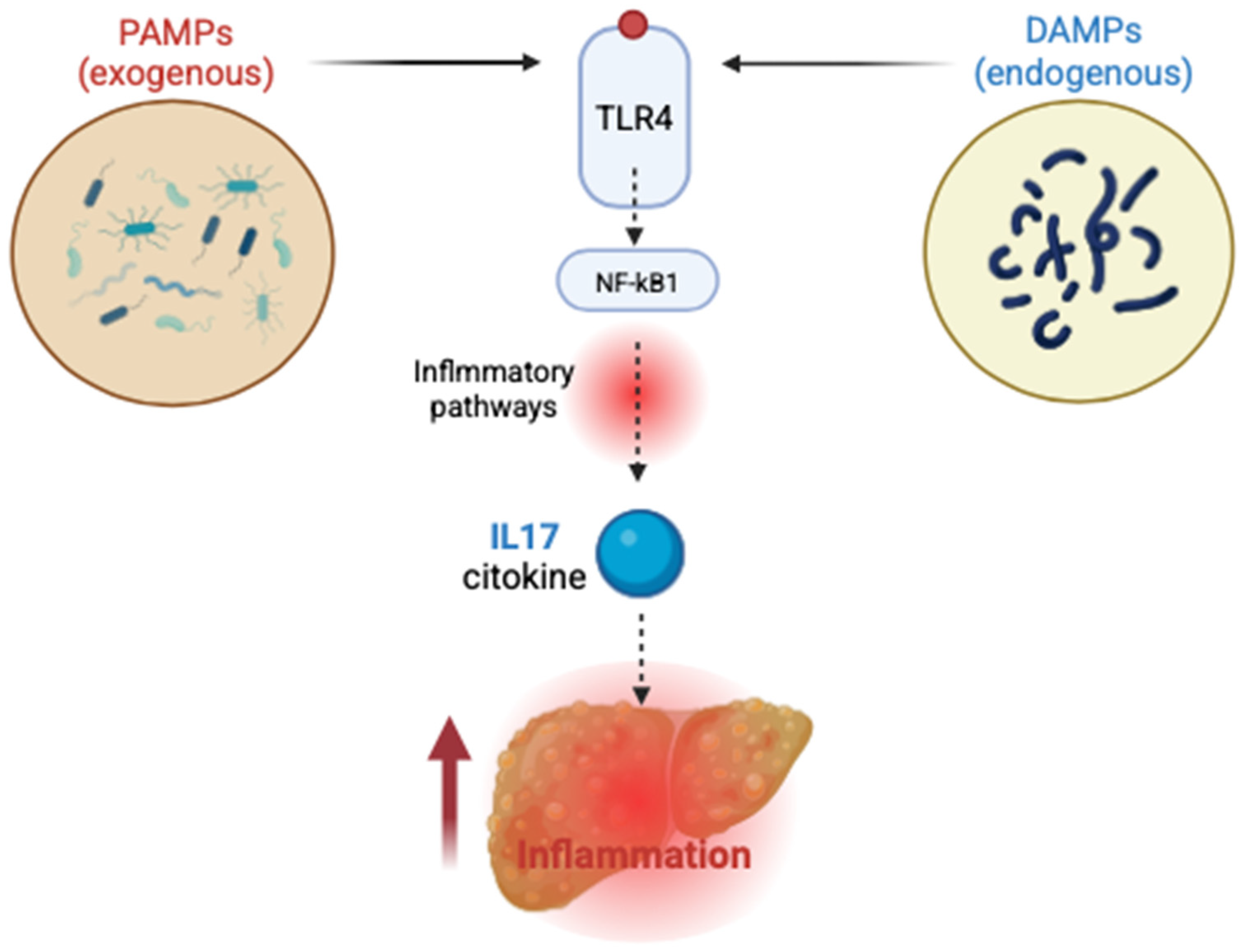
- Coste, S.C.; Orăsan, H.O.; Cozma, A.; Negrean, V.; Alexescu, T.G.; Perne, M.G.; Ciulei, G.; Hangan, A.C.; Lucaciu, R.L.; Iancu, M.; et al. Allelic, genotypic, and haplotypic analysis of cytokine IL17A, IL17F, and toll-like receptor TLR4 gene polymorphisms in metabolic dysfunction associated steatotic liver disease: Insights from an exploratory study. Life 2024, 14, 1327. [Google Scholar] [CrossRef] [PubMed]
- Coste, S.C.; Popovici, I.; Stefan, A.M.; Breaban, I.; Taut, A.S.; Sarlea, S.T.; Cozma, A.; Sampelean, D.; Orasan, O.H.; Negrean, V.; et al. Immune and inflammatory pathways in non-alcoholic steatohepatitis (NASH). An update. J. Mind Med. Sci. 2019, 6, 52–57. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Karl, M.; Hasselwander, S.; Zhou, Y.; Reifenberg, G.; Kim, Y.O.; Park, K.S.; Ridder, D.A.; Wang, X.; Seidel, E.; Hövelmeyer, N.; et al. Dual roles of B lymphocytes in mouse models of diet-induced nonalcoholic fatty liver disease. Hepatology 2022, 76, 1135–1149. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Wilcz-Villega, E.M.; McClean, S.; O’Sullivan, M.A. Mast cell tryptase reduces junctional adhesion molecule-A (JAM-A) expression in intestinal epithelial cells: Implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am. J. Gastroenterol. 2013, 108, 1140–1151. [Google Scholar] [CrossRef]
- McPherson, S.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 60, 1055–1062. [Google Scholar] [CrossRef]
- Coste, S.C.; Orăsan, O.H.; Cozma, A.; Negrean, V.; Sitar-Tăut, A.V.; Filip, G.A.; Hangan, A.C.; Lucaciu, R.L.; Iancu, M.; Procopciuc, L.M. Metabolic dysfunction associated steatotic liver disease: The associations between inflammatory markers, TLR4, and cytokines IL-17A/F, and their connections to the degree of steatosis and the risk of fibrosis. Biomedicines 2024, 12, 2144. [Google Scholar] [CrossRef]
- Quesada-Vazquez, S.; Bone, C.; Saha, S.; Triguero, I.; Colom-Pellicer, M.; Aragones, G.; Hildebrand, F.; Del Bas, J.M.; Caimari, A.; Beraza, N.; et al. Microbiota dysbiosis and gut barrier dysfunction associated with non-alcoholic fatty liver disease are modulated by a specific metabolic cofactors’ combination. Int. J. Mol. Sci. 2022, 23, 13675. [Google Scholar] [CrossRef]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of intestinal barrier function by microbial metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Chakaroun, R.M.; Massier, L.; Kovacs, P. Gut microbiome, intestinal permeability, and tissue bacteria in metabolic disease: Perpetrators or bystanders? Nutrients 2020, 12, 1082. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.M.B.; Stefano, J.T.; Oliveira, C.P. Microbiota and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH). Ann. Hepatol. 2019, 18, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Hernaez, R.; Lazo, M.; Bonekamp, S.; Kamel, I.; Brancati, F.L.; Guallar, E.; Clark, J.M. Diagnostic accuracy and reliability of ultrasonography for the detection of fatty liver: A meta-analysis. Hepatology 2011, 54, 1082–1090. [Google Scholar] [CrossRef]
- Mottin, C.C.; Moretto, M.; Padoin, A.V.; Swarowsky, A.M.; Toneto, M.G.; Glock, L.; Repetto, G. The role of ultrasound in the diagnosis of hepatic steatosis in morbidly obese patients. Obes. Surg. 2004, 14, 635–637. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Carpenter, D.H.; Rinella, M.; Harrison, S.A.; Loomba, R.; Younossi, Z.; Neuschwander-Tetri, B.A.; Sanyal, A.J. American association for the study of liver diseases NASH task force. NAFLD: Reporting histologic findings in clinical practice. Hepatology 2021, 73, 2028–2038. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathologic patterns and biopsy evaluation in clinical research. Semin. Liver Dis. 2012, 32, 3–13. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic steatohepatitis clinical research network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Ramrakhiani, S.; Cordes, B.G.; Neuschwander-Tetri, B.A.; Janney, C.G.; Bacon, B.R.; Di Bisceglie, A.M. Concurrence of histologic features of steatohepatitis with other forms of chronic liver disease. Mod. Pathol. 2003, 16, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.; Angulo, P.; Sanderson, S.; Jamil, L.H.; Stadheim, L.; Rosen, C.; Malinchoc, M.; Kamath, P.S.; Shah, V.H. Utility of a new model to diagnose an alcohol basis for steatohepatitis. Gastroenterology 2006, 131, 1057–1063. [Google Scholar] [CrossRef]
- Chan, A.; Patel, K.; Naggie, S. Genotype 3 infection: The last stand of hepatitis C Virus. Drugs 2017, 77, 131–144. [Google Scholar] [CrossRef]
- Jordan, T.; Popovič, P.; Rotovnik Kozjek, N. Liver steatosis in adult patients on home parenteral nutrition. Eur. J. Clin. Nutr. 2020, 74, 255–260. [Google Scholar] [CrossRef]
- López-Pascual, E.; Rienda, I.; Perez-Rojas, J.; Rapisarda, A.; Garcia-Llorens, G.; Jover, R.; Castell, J.V. Drug-induced fatty liver disease (DIFLD): A comprehensive analysis of clinical, biochemical, and histopathological data for mechanisms identification and consistency with current adverse outcome pathways. Int. J. Mol. Sci. 2024, 25, 5203. [Google Scholar] [CrossRef]
- Loomba, R.; Wolfson, T.; Ang, B.; Hooker, J.; Behling, C.; Peterson, M.; Valasek, M.; Lin, G.; Brenner, D.; Gamst, A.; et al. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: A prospective study. Hepatology 2014, 60, 1920–1928. [Google Scholar] [CrossRef]
- Selvaraj, E.A.; Mózes, F.E.; Jayaswal, A.N.A.; Zafarmand, M.H.; Vali, Y.; Lee, J.A.; Levick, C.K.; Young, L.A.J.; Palaniyappan, N.; Liu, C.H.; et al. LITMUS Investigators. Diagnostic accuracy of elastography and magnetic resonance imaging in patients with NAFLD: A systematic review and meta-analysis. J. Hepatol. 2021, 75, 770–785. [Google Scholar] [CrossRef]
- Hsu, C.; Caussy, C.; Imajo, K.; Chen, J.; Singh, S.; Kaulback, K.; Le, M.D.; Hooker, J.; Tu, X.; Bettencourt, R.; et al. Magnetic resonance vs transient elastography analysis of patients with nonalcoholic fatty liver disease: A systematic review and pooled analysis of individual participants. Clin. Gastroenterol. Hepatol. 2019, 17, 630–637.e8. [Google Scholar] [CrossRef]
- Vallet-Pichard, A.; Mallet, V.; Nalpas, B.; Verkarre, V.; Nalpas, A.; Dhalluin-Venier, V.; Fontaine, H.; Pol, S. FIB-4: An inexpensive and accurate marker of fibrosis in HCV infection. comparison with liver biopsy and fibrotest. Hepatology 2007, 46, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; Therneau, T.M.; Ahmed, O.T.; Gidener, T.; Mara, K.C.; Larson, J.J.; Canning, R.E.; Benson, J.T.; Kamath, P.S. Clinical course of non-alcoholic fatty liver disease and the implications for clinical trial design. J. Hepatol. 2022, 77, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Policarpo, S.; Coutinho, J.; Carvalhana, S.; Leitão, J.; Carvalho, A.; Silva, A.P.; Velasco, F.; Medeiros, I.; Alves, A.C.; et al. What is the role of the new index relative fat mass (RFM) in the assessment of nonalcoholic fatty liver disease (NAFLD)? Obes. Surg. 2020, 30, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V. Nonalcoholic fatty liver disease in lean subjects: Is it all metabolic-associated fatty liver disease? Hepatoma Res. 2020, 6, 84. [Google Scholar] [CrossRef]
- Simon, T.G.; Roelstraete, B.; Khalili, H.; Hagström, H.; Ludvigsson, J.F. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: Results from a nationwide cohort. Gut 2021, 70, 1375–1382. [Google Scholar] [CrossRef]
- Ng, C.H.; Lim, W.H.; Lim, G.E.H.; Tan, D.J.H.; Syn, N.; Muthiah, M.D.; Huang, D.Q.; Loomba, R. Mortality outcomes by fibrosis stage in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2023, 21, 931–939.e5. [Google Scholar] [CrossRef]
- Allen, A.M.; Hicks, S.B.; Mara, K.C.; Larson, J.J.; Therneau, T.M. The risk of incident extrahepatic cancers is higher in non-alcoholic fatty liver disease than obesity—A longitudinal cohort study. J. Hepatol. 2019, 71, 1229–1236. [Google Scholar] [CrossRef]
- Roeb, E.; Geier, A. Nonalcoholic steatohepatitis (NASH)—Current treatment recommendations and future developments. Z. Gastroenterol. 2019, 57, 508–517. [Google Scholar] [CrossRef]
- Dong, Q.; Bao, H.; Wang, J.; Shi, W.; Zou, X.; Sheng, J.; Gao, J.; Guan, C.; Xia, H.; Li, J.; et al. Liver fibrosis and MAFLD: The exploration of multi-drug combination therapy strategies. Front. Med. 2023, 10, 1120621. [Google Scholar] [CrossRef]
- Sato-Espinoza, K.; Chotiprasidhi, P.; Huaman, M.R.; Díaz-Ferrer, J. Update in lean metabolic dysfunction-associated steatotic liver disease. World J. Hepatol. 2024, 16, 452–464. [Google Scholar] [CrossRef]
- Alonso-Pena, M.; Del Barrio, M.; Peleteiro-Vigil, A.; Jimenez-Gonzalez, C.; Santos-Laso, A.; Arias-Loste, M.T.; Iruzubieta, P.; Crespo, J. Innovative therapeutic approaches in non-alcoholic fatty liver disease: When knowing your patient is key. Int. J. Mol. Sci. 2023, 24, 10718. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Nephew, L.D.; Vuppalanchi, R.; Gawrieh, S.; Mladenovic, A.; Pike, F.; Samala, N.; Chalasani, N. High-quality diet, physical activity, and college education are associated with low risk of NAFLD among the US population. Hepatology 2022, 75, 1491–1506. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V. What should we advise MAFLD patients to eat and drink? Metab. Target Organ Damage 2021, 1, 9. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Zhao, L.; Deng, C.; Lin, Z.; Giovannucci, E.; Zhang, X. Dietary fats, serum cholesterol and liver cancer risk: A systematic review and meta-analysis of prospective studies. Cancers 2021, 13, 1580. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Ivancovsky-Wajcman, D.; Isakov, N.F.; Webb, M.; Orenstein, D.; Shibolet, O.; Kariv, R. High red and processed meat consumption is associated with non-alcoholic fatty liver disease and insulin resistance. J. Hepatol. 2018, 68, 1239–1246. [Google Scholar] [CrossRef]
- Kouvari, M.; Boutari, C.; Chrysohoou, C.; Fragkopoulou, E.; Antonopoulou, S.; Tousoulis, D.; Pitsavos, C.; Panagiotakos, D.B.; Mantzoros, C.S. Mediterranean diet is inversely associated with steatosis and fibrosis and decreases ten-year diabetes and cardiovascular risk in NAFLD subjects: Results from the ATTICA prospective cohort study. Clin. Nutr. 2021, 40, 3314–3324. [Google Scholar] [CrossRef]
- Lange, M.; Nadkarni, D.; Martin, L.; Newberry, C.; Kumar, S.; Kushner, T. Intermittent fasting improves hepatic end points in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Hepatol. Commun. 2023, 7, e0212. [Google Scholar] [CrossRef]
- Guo, X.; Yin, X.; Liu, Z.; Wang, J. Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural products for prevention and treatment. Int. J. Mol. Sci. 2022, 23, 15489. [Google Scholar] [CrossRef]
- Chen, Y.P.; Lu, F.B.; Hu, Y.B.; Xu, L.M.; Zheng, M.H.; Hu, E.D. A systematic review and a dose-response meta-analysis of coffee dose and nonalcoholic fatty liver disease. Clin. Nutr. 2019, 38, 2552–2557. [Google Scholar] [CrossRef]
- Kennedy, O.J.; Roderick, P.; Buchanan, R.; Fallowfield, J.A.; Hayes, P.C.; Parkes, J. Coffee, including caffeinated and decaffeinated coffee, and the risk of hepatocellular carcinoma: A systematic review and dose-response meta-analysis. BMJ Open 2017, 7, e013739. [Google Scholar] [CrossRef] [PubMed]
- Åberg, F.; Byrne, C.D.; Pirola, C.J.; Männistö, V.; Sookoian, S. Alcohol consumption and metabolic syndrome: Clinical and epidemiological impact on liver disease. J. Hepatol. 2023, 78, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, H.; O’Keefe, H.; Craig, D.; Stow, D.; Hanratty, B.; Anstee, Q.M. Does moderate alcohol consumption accelerate the progression of liver disease in NAFLD? A systematic review and narrative synthesis. BMJ Open 2022, 12, e049767. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V. Aerobic exercise in the management of metabolic dysfunction associated fatty liver disease. Diabetes Metab. Syndr. Obes. 2021, 14, 627–645. [Google Scholar] [CrossRef]
- Oh, S.; So, R.; Shida, T.; Matsuo, T.; Kim, B.; Akiyama, K.; Isobe, T.; Okamoto, Y.; Tanaka, K.; Shoda, J. High-intensity aerobic exercise improves both hepatic fat content and stiffness in sedentary obese men with nonalcoholic fatty liver disease. Sci. Rep. 2017, 7, 43029. [Google Scholar] [CrossRef]
- Malespin, M.H.; Barritt, A.S.; Watkins, S.E.; Schoen, C.; Tincopa, M.A.; Corbin, K.D.; Mospan, A.R.; Munoz, B.; Trinh, H.N.; Weiss, M.; et al. Weight loss and weight regain in usual clinical practice: Results from the TARGET-NASH observational cohort. Clin. Gastroenterol. Hepatol. 2020, 20, 2393–2395. [Google Scholar] [CrossRef]
- Sohail, M.U.; Yassine, H.M.; Sohail, A.; Thani, A.A.A. Impact of physical exercise on gut microbiome, inflammation, and the pathobiology of metabolic disorders. Rev. Diabet. Stud. 2019, 15, 35–48. [Google Scholar] [CrossRef]
- Sangro, P.; de la Torre Alaez, M.; Sangro, B.; D’Avola, D. Metabolic dysfunction-associated fatty liver disease (MAFLD): An update of the recent advances in pharmacological treatment. J. Physiol. Biochem. 2023, 79, 869–879. [Google Scholar] [CrossRef]
- Cusi, K.; Isaacs, S.; Barb, D.; Basu, R.; Caprio, S.; Garvey, W.T.; Kashyap, S.; Mechanick, J.I.; Mouzaki, M.; Nadolsky, K.; et al. American Association of Clinical Endocrinology clinical practice guideline for the diagnosis and management of nonalcoholic fatty liver disease in primary care and endocrinology clinical settings: Co-sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr. Pract. 2022, 28, 528–562. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhao, Y.; Xu, X.; Ling, S. Advance of Metformin in liver disease. Curr. Med. Chem. 2024, 32, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.L.; Chow, E.; Chan, J.C. Cardiorenal diseases in type 2 diabetes mellitus: Clinical trials and real-world practice. Nat. Rev. Endocrinol. 2023, 19, 151–163. [Google Scholar] [CrossRef]
- Cariou, B. The metabolic triad of non-alcoholic fatty liver disease, visceral adiposity and type 2 diabetes: Implications for treatment. Diabetes Obes. Metab. 2022, 24 (Suppl. S2), 15–27. [Google Scholar] [CrossRef]
- Kansara, A.; Mubeen, F.; Shakil, J. SGLT2 Inhibitors in patients with chronic kidney disease and heart disease: A literature review. Methodist DeBakey Cardiovasc. J. 2022, 18, 62–72. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Maleki, M.; Jamialahmadi, T.; Moallem, S.A.; Sahebkar, A. Hepatic benefits of sodium-glucose cotransporter 2 inhibitors in liver disorders. EXCLI J. 2023, 22, 403–414. [Google Scholar] [CrossRef]
- Jasleen, B.; Vishal, G.K.; Sameera, M.; Fahad, M.; Brendan, O.; Deion, S.; Pemminati, S. Sodium-glucose cotransporter 2 (SGLT2) inhibitors: Benefits versus risk. Cureus 2023, 15, e33939. [Google Scholar] [CrossRef]
- Sun, L.; Deng, C.; Gu, Y.; He, Y.; Yang, L.; Shi, J. Effects of dapagliflozin in patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis of randomized controlled trials. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101876. [Google Scholar] [CrossRef]
- Liarakos, A.L.; Koliaki, C. Novel dual incretin receptor agonists in the spectrum of metabolic diseases with a focus on Tirzepatide: Real game-changers or great expectations? A narrative review. Biomedicines 2023, 11, 1875. [Google Scholar] [CrossRef]
- Yabut, J.M.; Drucker, D.J. Glucagon-like Peptide-1 receptor-based therapeutics for metabolic liver disease. Endocr. Rev. 2023, 44, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Zachou, M.; Flevari, P.; Nasiri-Ansari, N.; Varytimiadis, C.; Kalaitzakis, E.; Kassi, E.; Androutsakos, T. The role of anti-diabetic drugs in NAFLD. Have we found the Holy Grail? A narrative review. Eur. J. Clin. Pharmacol. 2024, 80, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Bergenstal, R.; Bode, B.; Kushner, R.F.; Lewin, A.; Skjøth, T.V.; Andreasen, A.H.; Jensen, C.B.; deFronzo, R.A. Efficacy of Liraglutide for weight loss among patients with type 2 diabetes: The SCALE diabetes randomized clinical trial. JAMA 2015, 314, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Alderslet, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Xie, Z.; Yang, S.; Deng, W.; Li, J.; Chen, J. Efficacy and safety of Liraglutide and Semaglutide on weight loss in people with obesity or overweight: A systematic review. Clin. Epidemiol. 2022, 14, 1463–1476. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A. A placebo-controlled trial of subcutaneous Semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Loomba, R.; Abdelmalek, M.F.; Armstrong, M.J.; Jara, M.; Kjær, M.S.; Krarup, N.; Lawitz, E.; Ratziu, V.; Sanyal, A.J.; Schattenberg, J.M.; et al. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2023, 8, 511–522. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef]
- Sfikas, G.; Psallas, M.; Koumaras, C.; Imprialos, K.; Perdikakis, E.; Doumas, M.; Giouleme, O.; Karagiannis, A.; Athyros, V.G. Prevalence, diagnosis, and treatment with 3 different statins of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis in military personnel. Do genetics play a role? Curr. Vasc. Pharmacol. 2021, 19, 572–581. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Zhang, J.; Zeng, F.Y.; Zhu, Y.Z. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol. Res. 2023, 192, 106786. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Xu, J.; Targher, G.; Byrne, C.D.; Zheng, M.H. Old and new classes of glucose-lowering agents as treatments for non-alcoholic fatty liver disease: A narrative review. Clin. Mol. Hepatol. 2022, 28, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldeli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, I.J.; Yasir, M.; Sarfraz, S.; Shlaghya, G.; Narayana, S.H.; Mushtaq, U.; Shaman Ameen, B.; Nie, C.; Nechi, D.; Penumetcha, S.S. Vitamin E and Pioglitazone: A comprehensive systematic review of their efficacy in non-alcoholic fatty liver disease. Cureus 2023, 15, e43635. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current clinical trial status and future prospects of PPAR-targeted drugs for treating nonalcoholic fatty liver disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef]
- Tidwell, J.; Balassiano, N.; Shaikh, A.; Nassar, M. Emerging therapeutic options for non-alcoholic fatty liver disease: A systematic review. World J. Hepatol. 2023, 15, 1001–1012. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: A randomized controlled double-blind phase 2 trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Idowu, M.O.; Parmar, D.; Borg, B.B.; Denham, D.; Loo, N.M.; Lazas, D.; Younes, Z.; Sanyal, A.J. A Phase 2 double blinded, randomized controlled trial of Saroglitazar in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2021, 19, 2670–2672. [Google Scholar] [CrossRef]
- Staels, B.; Butruille, L.; Francque, S. Treating NASH by targeting peroxisome proliferator-activated receptors. J. Hepatol. 2023, 79, 1302–1316. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A randomized, controlled trial of the Pan-PPAR agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L.; Trauner, M. Miniseries: Established pharmacological targets for NASHFXR agonists in NASH treatment. J. Hepatol. 2023, 79, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the treatment of NASH-two sides of the same coin? Differential and overlapping effects of FGF19 and FGF21 from mice to human. Front. Endocrinol. 2020, 11, 601349. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Lucas, K.J.; Francque, S.; Abdelmalek, M.F.; Sanyal, A.J.; Ratziu, V.; Gadano, A.C.; Rinella, M.E.; Charlton, M.; Loomba, R.; et al. Tropifexor plus cenicriviroc combination versus monotherapy in adult nonalcoholic steatohepatitis: Results from the phase 2b TANDEM study. Hepatology 2023, 78, 1223–1239. [Google Scholar] [CrossRef]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, L.; Liang, L.; Koral, K.; Zhang, Q.; Zhao, L.; Lu, S.; Tao, J. The role of fibroblast growth factor 19 in hepatocellular carcinoma. Am. J. Pathol. 2021, 191, 1180–1192. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Taub, R.; Neff, G.; Moussa, S.; Alkhouri, N.; Bashir, M. Primary data analyses of MAESTRO-NAFLD-1 a 52 week double-blind placebo-controlled phase 3 clinical trial of Resmetirom in patients with NAFLD. J. Hepatol. 2022, 77, S14. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Neff, G.; Gunn, N.; Guy, C.D.; Alkhouri, N.; Bashir, M.R.; Freilich, B.; Kohli, A.; Khazanchi, A.; et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol. Hepatol. 2022, 7, 603–616. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. A dozen years of discovery: Insights into the physiology and pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Muraca, E.; Vergani, M.; Invernizzi, P.; Perseghin, G. Advancements in pharmacological treatment of NAFLD/MASLD: A focus on metabolic and liver-targeted interventions. Gastroenterol. Rep. 2024, 12, goae029. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Bays, H.E.; Miller, M.; Cain, J.E.; Wasilewska, K.; Andrawis, N.S.; Parli, T.; Feng, S.; Sterling, L.; Tseng, L.; et al. The FGF21 analog pegozafermin in severe hypertriglyceridemia: A randomized phase 2 trial. Nat. Med. 2023, 29, 1782–1792. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J.; Kowdley, K.V.; Bhatt, D.L.; Alkhouri, N.; Frias, J.P.; Bedossa, P.; Harrison, S.A.; Lazas, D.; Barish, R.; et al. Randomized, controlled trial of the FGF21 analogue Pegozafermin in NASH. N. Engl. J. Med. 2023, 389, 998–1008. [Google Scholar] [CrossRef]
- Kounatidis, D.; Vallianou, N.G.; Geladari, E.; Panoilia, M.P.; Daskou, A.; Stratigou, T.; Karampela, I.; Tsilingiris, D.; Dalamaga, M. NAFLD in the 21st century: Current knowledge regarding its pathogenesis, diagnosis and therapeutics. Biomedicines 2024, 12, 826. [Google Scholar] [CrossRef]
- Amatya, R.; Lee, D.; Min, K.A.; Shin, M.C. pharmaceutical strategies to improve druggability of potential drug candidates in nonalcoholic fatty liver disease therapy. Pharmaceutics 2023, 15, 1963. [Google Scholar] [CrossRef]
- Harrison, S.A.; Frias, J.P.; Neff, G.; Abrams, G.A.; Lucas, K.J.; Sanchez, W.; Gogia, S.; Sheikh, M.Y.; Behling, C.; Bedossa, P.; et al. Safety and efficacy of once-weekly efruxifermin versus placebo in non-alcoholic steatohepatitis (HARMONY): A multicentre, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol. Hepatol. 2023, 8, 1080–1093. [Google Scholar] [CrossRef]
- Brennan, P.N.; Elsharkawy, A.M.; Kendall, T.J.; Loomba, R.; Mann, D.A.; Fallowfield, J.A. Antifibrotic therapy in nonalcoholic steatohepatitis: Time for a human-centric approach. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 679–688. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Treatment for Patients with Liver Scarring Due to Fatty Liver Disease. 2024. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-patients-liver-scarring-due-fatty-liver-disease (accessed on 28 April 2025).
- Karim, G.; Bansal, M.B. Resmetirom: An orally administered, small molecule, liver-directed, β-selective THR agonist for the treatment of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. touchREV Endocrinol. 2023, 19, 60–70. [Google Scholar] [CrossRef] [PubMed]
- A Study to Assess the Efficacy and Safety of VK2809 for 52 Weeks in Subjects with Biopsy Proven NASH (VOYAGE). 2024. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04173065 (accessed on 28 April 2025).
- VK2809 Selective Thyroid Receptor-β Agonist. 2024. Available online: https://www.prnewswire.com/news-releases/viking-therapeutics-presents-results-from-phase-2b-voyage-study-of-vk2809-in-biopsy-confirmed-nashmash-at-the-75th-liver-meeting-2024-302310271.html (accessed on 28 April 2025).
- Anstee, Q.M.; Neuschwander-Tetri, B.A.; Wong, V.W.; Abdelmalek, M.F.; Younossi, Z.M.; Yuan, J.; Pecoraro, M.L.; Seyedkazemi, S.; Fischer, L.; Bedossa, P.; et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA phase 3 study design. Contemp. Clin. Trials 2020, 89, 105922. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination oftropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef]
- Bilal, M.; Ashraf, S.; Zhao, X. Dietary component-induced inflammation and its amelioration by prebiotics, probiotics, and synbiotics. Front. Nutr. 2022, 9, 931458. [Google Scholar] [CrossRef]
- Gou, H.Z.; Zhang, Y.L.; Ren, L.F.; Li, Z.J.; Zhang, L. How do intestinal probiotics restore the intestinal barrier? Front. Microbiol. 2022, 13, 929346. [Google Scholar] [CrossRef]
- Xing, W.; Gao, W.; Lv, X.; Zhao, Z.; Mao, G.; Dong, X.; Zhang, Z. The effects of supplementation of probiotics, prebiotics, or synbiotics on patients with non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1024678. [Google Scholar] [CrossRef]
- Horvath, A.; Leber, B.; Feldbacher, N.; Tripolt, N.; Rainer, F.; Blesl, A.; Trieb, M.; Marsche, G.; Sourij, H.; Stadlbauer, V. Effects of a multispecies synbiotic on glucose metabolism, lipid marker, gut microbiome composition, gut permeability, and quality of life in diabesity: A randomized, double-blind, placebo-controlled pilot study. Eur. J. Nutr. 2020, 59, 2969–2983. [Google Scholar] [CrossRef]
- Zhang, C.; Bjornson, E.; Arif, M.; Tebani, A.; Lovric, A.; Benfeitas, R.; Ozcan, M.; Juszczak, K.; Kim, W.; Kim, J.T.; et al. The acute effect of metabolic cofactor supplementation: A potential therapeutic strategy against non-alcoholic fatty liver disease. Mol. Syst. Biol. 2020, 16, e9495. [Google Scholar] [CrossRef] [PubMed]
- Sindhughosa, D.A.; Wibawa, I.D.; Mariadi, I.K.; Somayana, G. Additional treatment of vitamin D for improvement of insulin resistance in non-alcoholic fatty liver disease patients: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 7716. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlen, M.; Boren, J. The potential use of metabolic cofactors in treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef]
- Hegazi, O.E.; Alalalmeh, S.O.; Shahwan, M.; Jairoun, A.A.; Alourfi, M.M.; Bokhari, G.A.; Alkhattabi, A.; Alsharif, S.; Aljehani, M.A.; Alsabban, A.M.; et al. Exploring promising therapies for non-alcoholic fatty liver disease: A ClinicalTrials.gov analysis. Diabetes Metab. Syndr. Obes. 2024, 17, 545–561. [Google Scholar] [CrossRef]
- Yeoh, A.; Wong, R.; Singal, A.K. The role bariatric surgery and endobariatric therapies in nonalcoholic steatohepatitis. Clin. Liver Dis. 2023, 27, 413–427. [Google Scholar] [CrossRef]
- Seeberg, K.A.; Borgeraas, H.; Hofsø, D.; Småstuen, M.C.; Kvan, N.P.; Grimnes, J.O.; Lindberg, M.; Fatima, F.; Seeberg, L.T.; Sandbu, R.; et al. Gastric bypass versus sleeve gastrectomy in type 2 diabetes: Effects on hepatic steatosis and fibrosis: A randomized controlled trial. Ann. Intern. Med. 2022, 175, 74–83. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V.; et al. Bariatric surgery provides Long-term resolution of NASH and regression of fibrosis. Gastroenterology 2020, 159, 1290–1301.e5. [Google Scholar] [CrossRef]
- Verrastro, O.; Panunzi, S.; Castagneto-Gissey, L.; De Gaetano, A.; Lembo, E.; Capristo, E.; Guidone, C.; Angelini, C.; Pennetri, F.; Sessa, L. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023, 401, 1786–1797. [Google Scholar] [CrossRef]
- Lee, Y.; Doumouras, A.G.; Yu, J.; Brar, K.; Banfield, L.; Gmora, S.; Anvari, M.; Hong, D. Complete resolution of nonalcoholic fatty liver disease after bariatric surgery: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1040–1060.e11. [Google Scholar] [CrossRef]
- Elsaid, M.I.; Li, Y.; Bridges, J.F.P.; Brock, G.; Minacapelli, C.D.; Rustgi, V.K. Association of bariatric surgery with cardiovascular outcomes in adults with severe obesity and nonalcoholic fatty liver disease. JAMA Netw. Open 2022, 5, e2235003. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Jia, Z.; Chen, Y.; Li, Y.; Zheng, S.; Duan, Z. Bariatric surgery is effective and safe for obese patients with compensated cirrhosis: A systematic review and meta-analysis. World J. Surg. 2022, 46, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, T.N.; Swain, J.M.; Kendrick, M.L.; Charlton, M.R.; Schroeder, B.J.; Lee, R.E.C.; Narr, B.J.; Ribeiro, T.C.R.; Schroeder, D.R.; Sprung, J. Nonalcoholic steatohepatitis (NASH) does not increase complications after laparoscopic bariatric surgery. Obes. Surg. 2021, 21, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Jirapinyo, P.; McCarty, T.R.; Dolan, R.D.; Shah, R.; Thompson, C.C. Effect of endoscopic bariatric and metabolic therapies on NAFLD: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 511–524. [Google Scholar] [CrossRef]
- Chandan, S.; Mohan, B.P.; Khan, S.R.; Facciorusso, A.; Ramai, D.; Kassab, L.L.; Bhogal, N.; Asokkumar, R.; Lopez-Nava, G.; McDonough, S.; et al. Efficacy and safety of Intragastric balloon in NAFLD: A comprehensive review and meta-analysis. Obes. Surg. 2021, 31, 1271–1279. [Google Scholar] [CrossRef]
- Hajifathalian, K.; Mehta, A.; Ang, B.; Skaf, D.; Shah, S.L.; Saumoy, M.; Dawod, Q.; Dawod, E.; Shukla, A.; Aronne, L.; et al. Improvement in insulin resistance and estimated hepatic steatosis and fibrosis after endoscopic sleeve gastroplasty. Gastrointest. Endosc. 2021, 93, 1110–1118. [Google Scholar] [CrossRef]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef]
- Ianiro, G.; Bibbo, S.; Porcari, S.; Settanni, C.R.; Giambo, F.; Curta, A.R.; Quaranta, G.; Scaldaferri, F.; Masucci, L.; Sanguinetti, M.; et al. Fecal microbiota transplantation for recurrent C. difficile infection in patients with inflammatory bowel disease: Experience of a large-volume European FMT center. Gut Microbes 2021, 13, 1994834. [Google Scholar] [CrossRef]
- Leong, K.S.W.; Jayasinghe, T.N.; Wilson, B.C.; Derraik, J.G.B.; Albert, B.B.; Chiavaroli, V.; Svirskis, D.M.; Beck, K.L.; Conlon, C.A.; Jiang, Y.; et al. Effects of fecal microbiome transfer in adolescents with obesity: The gut bugs randomized controlled trial. JAMA Netw. Open 2020, 3, e2030415. [Google Scholar] [CrossRef]
- Xue, L.; Deng, Z.; Luo, W.; He, X.; Chen, Y. Effect of fecal microbiota transplantation on non-alcoholic fatty liver disease: A randomized clinical trial. Front. Cell. Infect. Microbiol. 2022, 12, 759306. [Google Scholar] [CrossRef]
- Zhang, B.; Feng, Y.; Lu, J. Progress in the treatment of metabolic-related fatty liver disease. Altern. Ther. Health Med. 2023, 29, 86–93. [Google Scholar] [PubMed]
- Ratziu, V.; Charlton, M. Rational combination therapy for NASH: Insights from clinical trials and error. J. Hepatol. 2023, 78, 1073–1079. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucaciu, R.L.; Coste, S.C.; Hangan, A.C.; Iancu, M.; Orășan, O.H.; Cozma, A.; Gog Bogdan, S.; Procopciuc, L.M. Pathogenesis and Clinical Management of Metabolic Dysfunction-Associated Steatotic Liver Disease. Int. J. Mol. Sci. 2025, 26, 5717. https://doi.org/10.3390/ijms26125717
Lucaciu RL, Coste SC, Hangan AC, Iancu M, Orășan OH, Cozma A, Gog Bogdan S, Procopciuc LM. Pathogenesis and Clinical Management of Metabolic Dysfunction-Associated Steatotic Liver Disease. International Journal of Molecular Sciences. 2025; 26(12):5717. https://doi.org/10.3390/ijms26125717
Chicago/Turabian StyleLucaciu, Roxana Liana, Sorina Cezara Coste, Adriana Corina Hangan, Mihaela Iancu, Olga Hilda Orășan, Angela Cozma, Sidonia Gog Bogdan, and Lucia Maria Procopciuc. 2025. "Pathogenesis and Clinical Management of Metabolic Dysfunction-Associated Steatotic Liver Disease" International Journal of Molecular Sciences 26, no. 12: 5717. https://doi.org/10.3390/ijms26125717
APA StyleLucaciu, R. L., Coste, S. C., Hangan, A. C., Iancu, M., Orășan, O. H., Cozma, A., Gog Bogdan, S., & Procopciuc, L. M. (2025). Pathogenesis and Clinical Management of Metabolic Dysfunction-Associated Steatotic Liver Disease. International Journal of Molecular Sciences, 26(12), 5717. https://doi.org/10.3390/ijms26125717