
Oxamate, an LDHA Inhibitor, Inhibits Stemness, Including EMT and High DNA Repair Ability, Induces Senescence, and Exhibits Radiosensitizing Effects in Glioblastoma Cells


Abstract
1. Introduction
2. Results
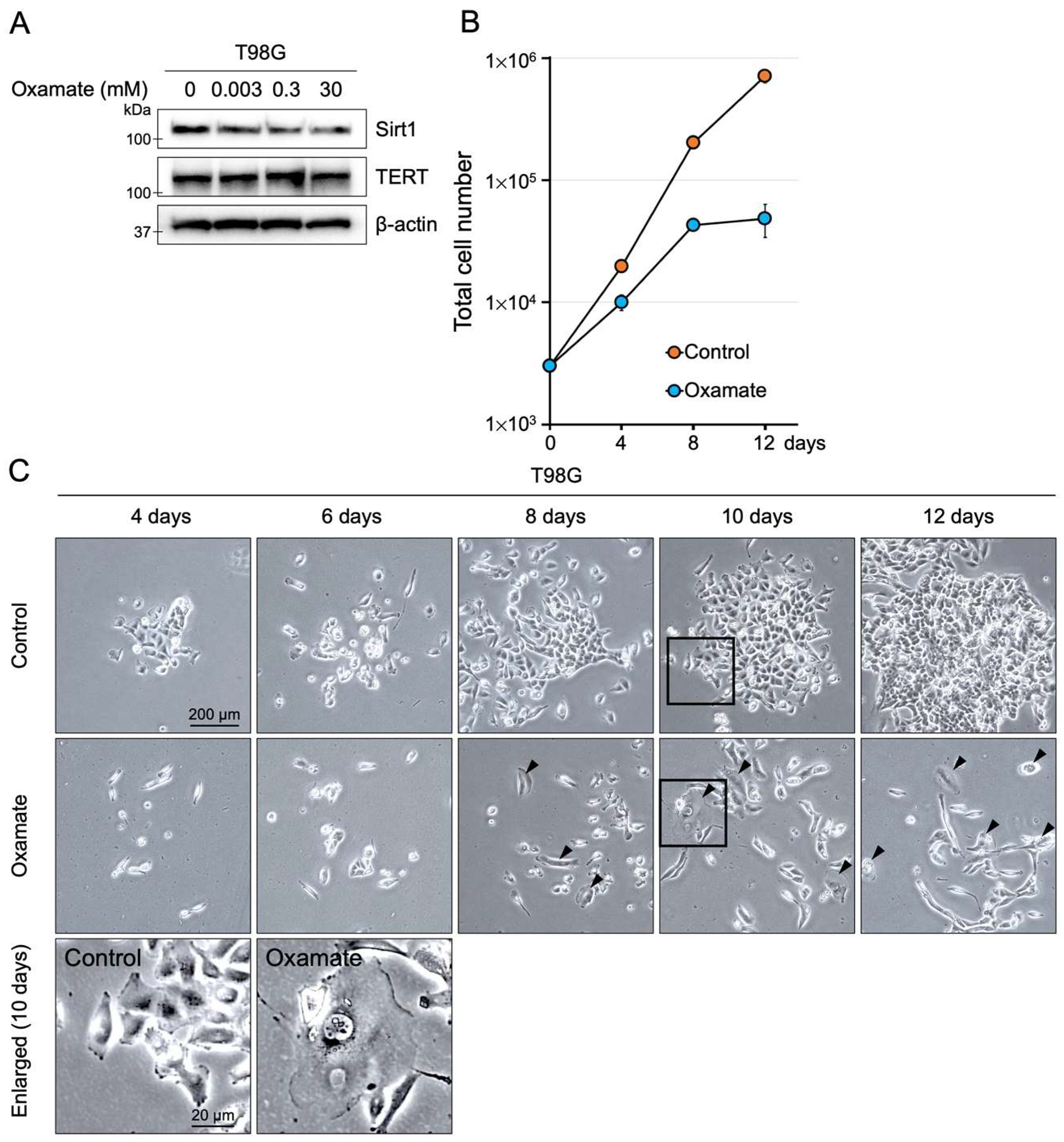
2.1. Effects of Oxamate on Proliferative Ability
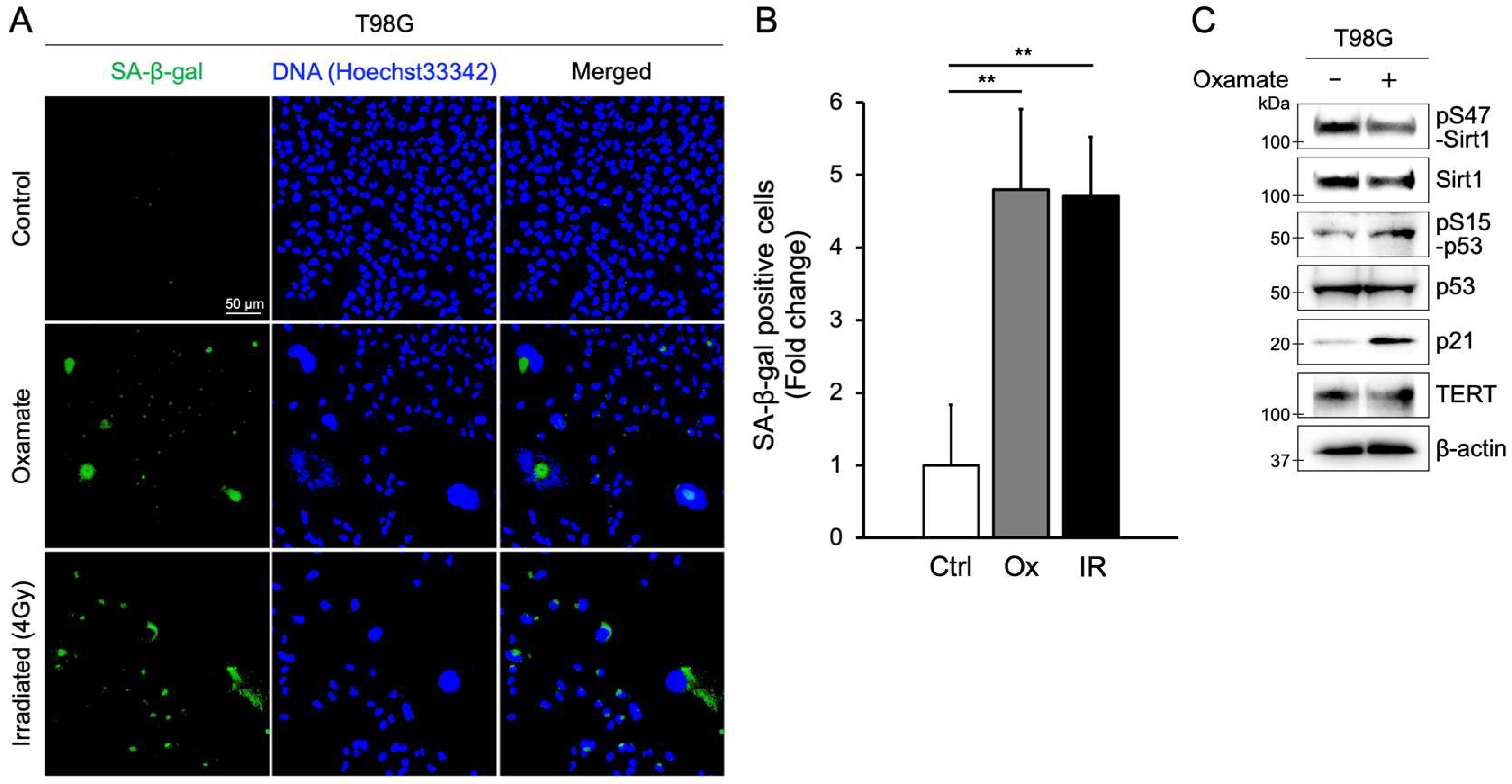
2.2. Oxamate Induces Cellular Senescence
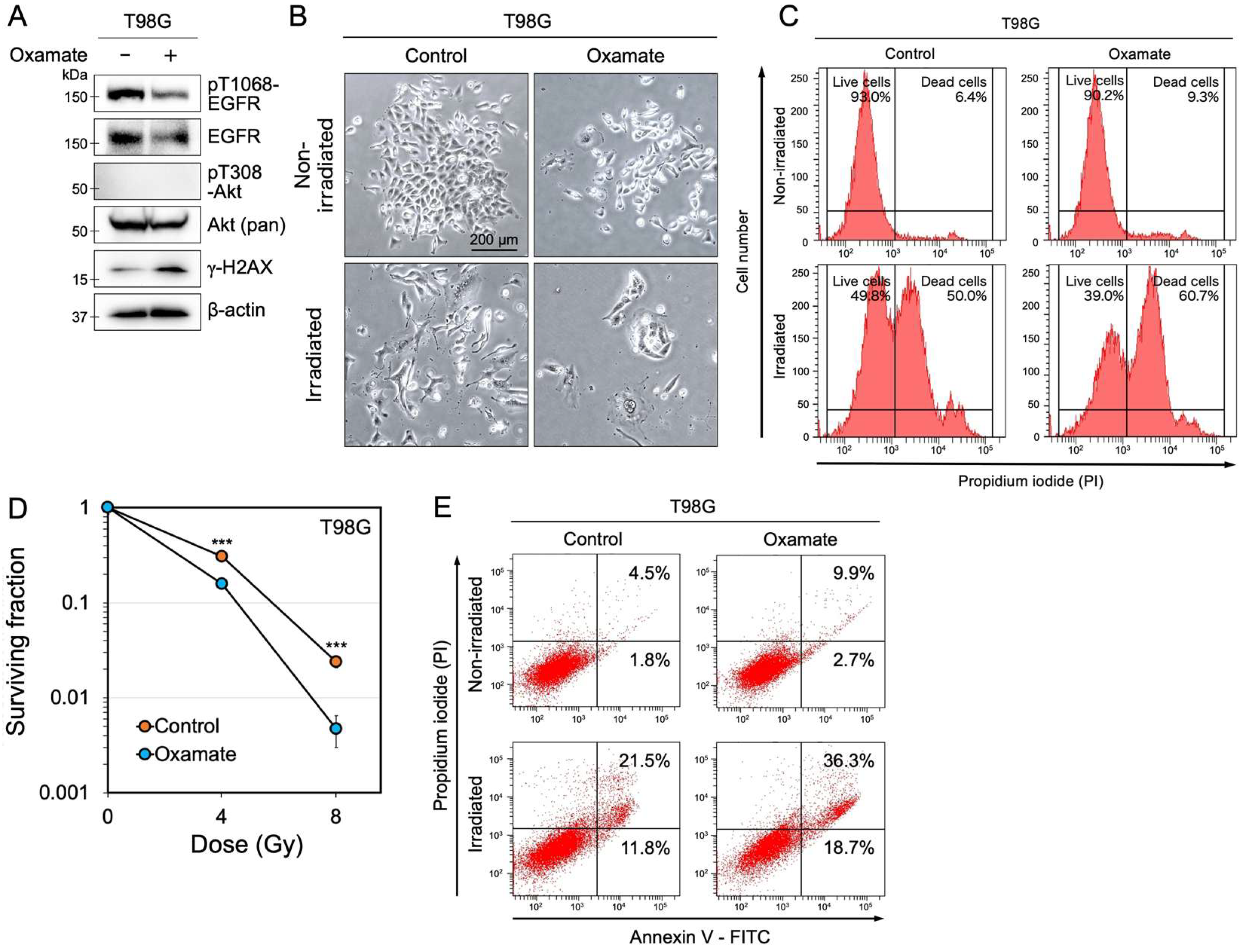
2.3. Oxamate Suppresses EGFR Expression and Increases Radiosensitivity
2.4. Oxamate Induces Apoptosis
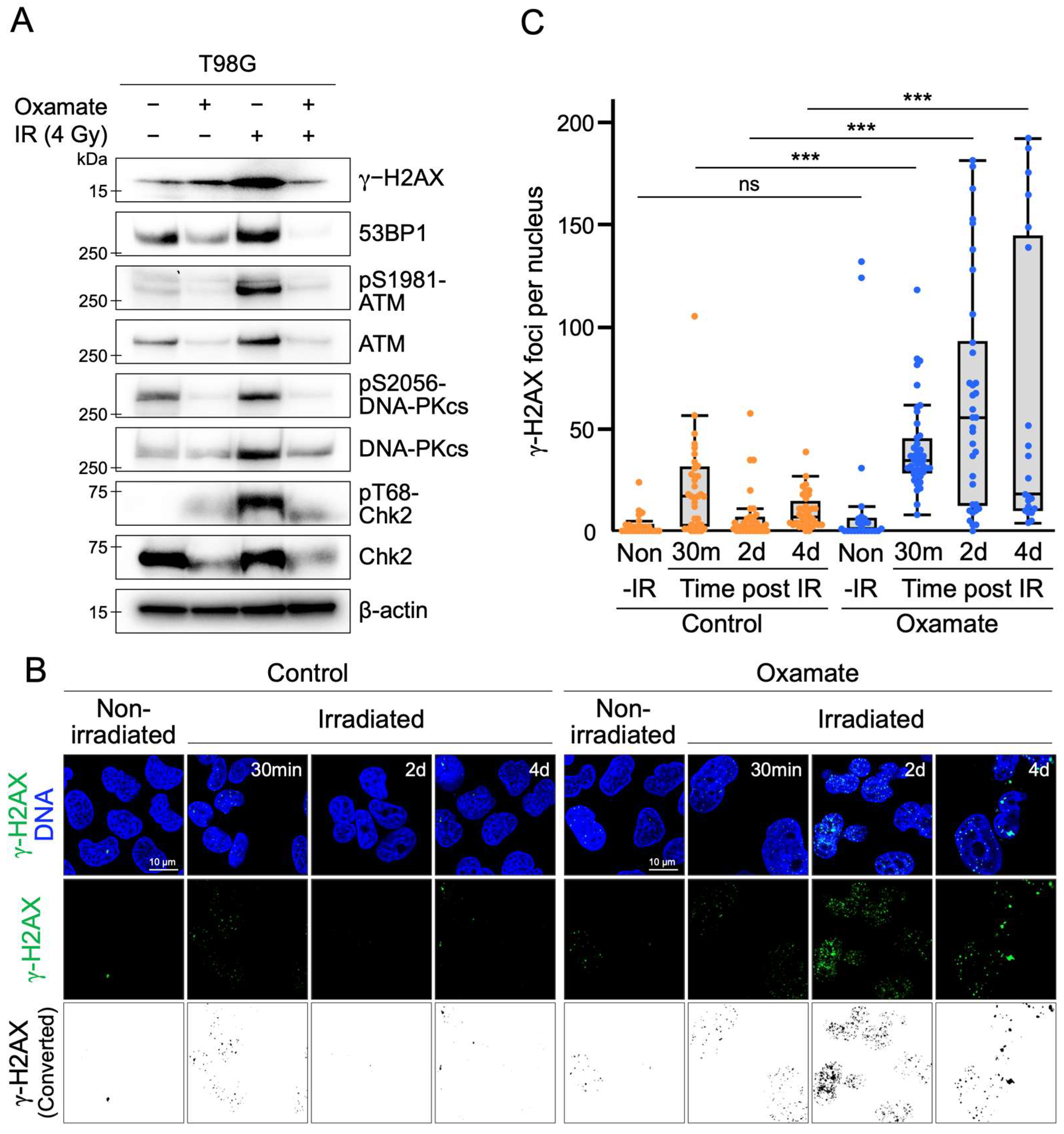
2.5. Oxamate Suppresses Radiation-Induced Expression and Activation of DNA Repair Factors
2.6. Oxamate Delays DNA Repair
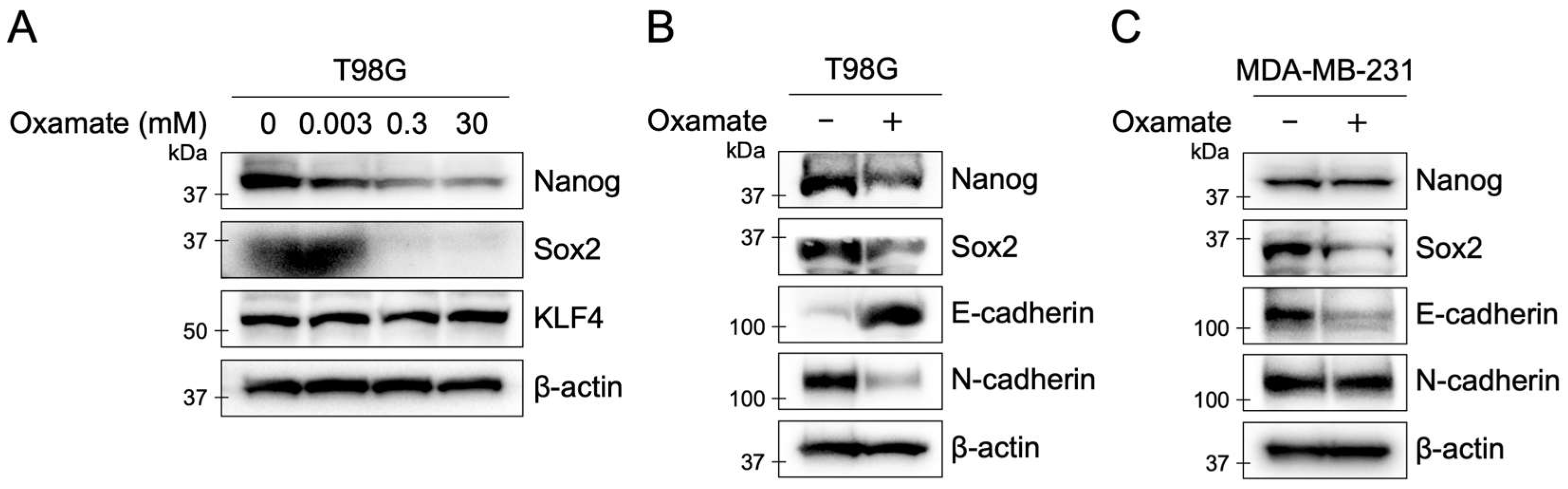
2.7. Oxamate Suppresses Stemness and EMT in T98G Cells
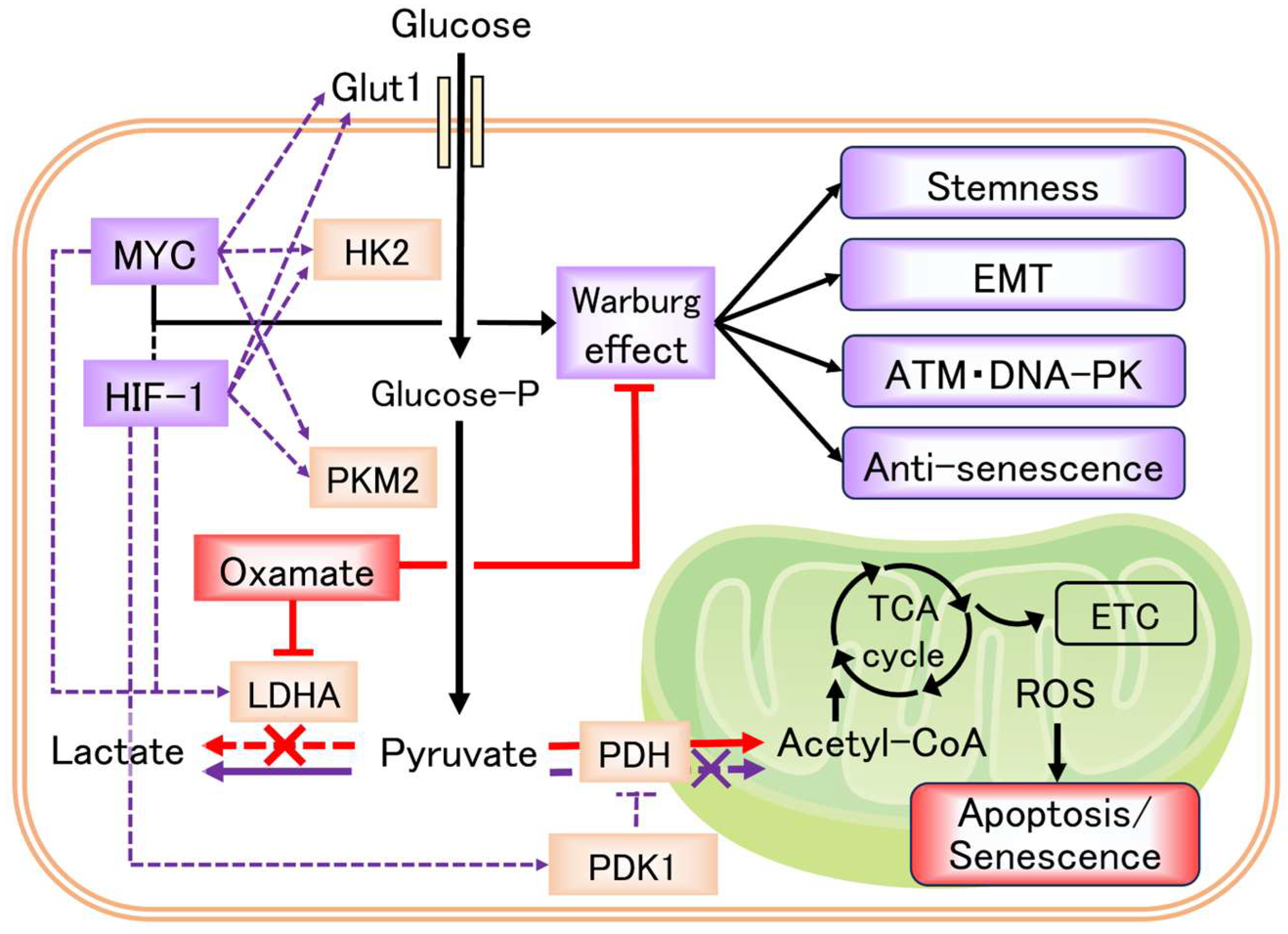
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemical
4.3. Proliferation Assay
4.4. X-Ray Irradiation
4.5. Colony Formation Assay
4.6. Western Blotting
4.7. Flow Cytometry
4.8. Detection of γH2AX Foci
4.9. Detection of Ki-67 Signals
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Girgis, H.; Masui, O.; White, N.M.; Scorilas, A.; Rotondo, F.; Seivwright, A.; Gabril, M.; Filter, E.R.; Girgis, A.H.; Bjarnason, G.A.; et al. Lactate Dehydrogenase A Is a Potential Prognostic Marker in Clear Cell Renal Cell Carcinoma. Mol. Cancer 2014, 13, 101. [Google Scholar] [CrossRef]
- Di, H.; Zhang, X.; Guo, Y.; Shi, Y.; Fang, C.; Yuan, Y.; Wang, J.; Shang, C.; Guo, W.; Li, C. Silencing LDHA Inhibits Proliferation, Induces Apoptosis and Increases Chemosensitivity to Temozolomide in Glioma Cells. Oncol. Lett. 2018, 15, 5131–5136. [Google Scholar] [CrossRef]
- Givechian, K.B.; Garner, C.; Benz, S.; Rabizadeh, S.; Soon-Shiong, P. Glycolytic Expression in Lower-Grade Glioma Reveals an Epigenetic Association between IDH Mutation Status and PDL1/2 Expression. Neurooncol. Adv. 2020, 3, vdaa162. [Google Scholar] [CrossRef]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate Dehydrogenase A in Cancer: A Promising Target for Diagnosis and Therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 632364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Han, F.; Yang, S.; Wu, J.; Zhan, W. Oxamate-Mediated Inhibition of Lactate Dehydrogenase Induces Protective Autophagy in Gastric Cancer Cells: Involvement of the Akt–mTOR Signaling Pathway. Cancer Lett. 2015, 358, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by Oxamate Induces G2/M Arrest, Apoptosis and Increases Radiosensitivity in Nasopharyngeal Carcinoma Cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Al-Salam, S.; Kandhan, K.; Sudhadevi, M. Down Regulation of Lactate Dehydrogenase Initiates Apoptosis in HeLa and MCF-7 Cancer Cells through Increased Voltage-Dependent Anion Channel Protein and Inhibition of BCL2. Oncotarget 2021, 12, 923–935. [Google Scholar] [CrossRef]
- Koukourakis, M.; Tsolou, A.; Pouliliou, S.; Lamprou, I.; Papadopoulou, M.; Ilemosoglou, M.; Kostoglou, G.; Ananiadou, D.; Sivridis, E.; Giatromanolaki, A. Blocking LDHA Glycolytic Pathway Sensitizes Glioblastoma Cells to Radiation and Temozolomide. Biochem. Biophys. Res. Commun. 2017, 491, 932–938. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to Improve Radiotherapy with Targeted Drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef]
- Chen, B.P.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double-strand breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; Golding, S.E.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin functions in health and disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef]
- Liu, P.; Wang, L.; Yu, H. Polyploid giant cancer cells: Origin, possible pathways of formation, characteristics, and mechanisms of regulation. Front. Cell Dev. Biol. 2024, 12, 1410637. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- McKenzie, P.P.; Guichard, S.M.; Middlemas, D.S.; Ashmun, R.A.; Danks, M.K.; Harris, L.C. Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clin. Cancer Res. 1999, 5, 4199–4207. [Google Scholar]
- Chinnaiyan, P.; Huang, S.; Vallabhaneni, G.; Armstrong, E.; Varambally, S.; Tomlins, S.A.; Chinnaiyan, A.M.; Harari, P.M. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res. 2005, 65, 3328–3335. [Google Scholar] [CrossRef]
- Liang, K.; Ang, K.K.; Milas, L.; Hunter, N.; Fan, Z. The epidermal growth factor receptor mediates radioresistance. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 246–254. [Google Scholar] [CrossRef]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Martin, O.A.; Parekh, P.R.; Weyemi, U.S.; Bonner, W.M. Recent developments in the use of γ-H2AX as a quantitative DNA double-strand break biomarker. Aging 2011, 3, 168–174. [Google Scholar] [CrossRef]
- Schwab, M.; Thunborg, K.; Azimzadeh, O.; von Toerne, C.; Werner, C.; Shevtsov, M.; Di Genio, T.; Zdralevic, M.; Pouyssegur, J.; Renner, K.; et al. Targeting cancer metabolism breaks radioresistance by impairing the stress response. Cancers 2021, 13, 3762. [Google Scholar] [CrossRef]
- Hollenberg, A.M.; Smith, C.O.; Shum, L.C.; Awad, H.; Eliseev, R. Lactate dehydrogenase inhibition with Oxamate exerts bone anabolic effect. J. Bone Miner. Res. 2020, 35, 2343–2356. [Google Scholar] [CrossRef]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef]
- Rose, G.P.; Dewar, A.J.; Stratford, I.J. A biochemical method for assessing the neurotoxic effects of misonidazole in the rat. Br. J. Cancer 1980, 42, 890–899. [Google Scholar] [CrossRef]
- Palayoor, S.T.; Bump, E.A.; Saroff, D.M.; Delfs, J.R.; Geula, C.; Menton-Brennan, L.; Hurwitz, S.J.; Coleman, C.N.; Stevenson, M.A. Effect of BSO and etanidazole on neurofilament degradation in neonatal rat spinal cord cultures. Br. J. Cancer Suppl. 1996, 27, S117–S121. [Google Scholar]
- Tassinari, I.D.; Rodrigues, F.D.S.; Bertram, C.; Mendes-da-Cruz, D.A.; Guedes, R.P.; Paz, A.H.; Bambini-Junior, V.; de Fraga, L.S. Lactate protects microglia and neurons from oxygen-glucose deprivation/reoxygenation. Neurochem. Res. 2024, 49, 1762–1781. [Google Scholar] [CrossRef]
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem. Biophys. Res. Commun. 2022, 590, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Urushihara, Y.; Hashimoto, T.; Fujishima, Y.; Hosoi, Y. AMPK/FOXO3a pathway increases activity and/or expression of ATM, DNA-PKcs, Src, EGFR, PDK1, and SOD2 and induces radioresistance under nutrient starvation. Int. J. Mol. Sci. 2023, 24, 12828. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Tsubota, K.; Hatabi, K.; Hosoi, Y. FDX1 Regulates the Phosphorylation of ATM, DNA-PKcs Akt, and EGFR and Affects Radioresistance Under Severe Hypoxia in the Glioblastoma Cell Line T98G. Int. J. Mol. Sci. 2025, 26, 3378. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Horikawa, D.D.; Saito, Y.; Kuwahara, H.; Kozuka-Hata, H.; Shin-I, T.; Minakuchi, Y.; Ohishi, K.; Motoyama, A.; Aizu, T.; et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nat. Commun. 2016, 7, 12808. [Google Scholar] [CrossRef]
- Cai, Z.; Vallis, K.A.; Reilly, R.M. Computational analysis of the number, area, and density of γ-H2AX foci in breast cancer cells exposed to 111In-DTPA-hEGF or γ-rays using Image-J software. Int. J. Radiat. Biol. 2009, 85, 262–271. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Finding | Experimental Assay | Figure Reference |
|---|---|---|
| Induction of senescence and apoptosis | Proliferation assay, MTT assay, SA-β-gal staining, Ki-67 staining, Annexin V/PI staining, Western blot (Sirt1, p53, p21) | Figure 1, Figure 2 and Figure 3E, Figures S1 and S2 |
| Delayed DNA repair and enhanced radiosensitivity | Colony formation assay, γ-H2AX foci counting, Western blot (53BP1, ATM, DNA-PKcs) | Figure 3 and Figure 4 |
| Downregulation of stemness markers | Western blot (Nanog, Sox2) | Figure 5A,B |
| Modulation of EMT proteins | Western blot (E-cadherin, N-cadherin) | Figure 5A,B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, T.; Ushikubo, G.; Arao, N.; Hatabi, K.; Tsubota, K.; Hosoi, Y. Oxamate, an LDHA Inhibitor, Inhibits Stemness, Including EMT and High DNA Repair Ability, Induces Senescence, and Exhibits Radiosensitizing Effects in Glioblastoma Cells. Int. J. Mol. Sci. 2025, 26, 5710. https://doi.org/10.3390/ijms26125710
Hashimoto T, Ushikubo G, Arao N, Hatabi K, Tsubota K, Hosoi Y. Oxamate, an LDHA Inhibitor, Inhibits Stemness, Including EMT and High DNA Repair Ability, Induces Senescence, and Exhibits Radiosensitizing Effects in Glioblastoma Cells. International Journal of Molecular Sciences. 2025; 26(12):5710. https://doi.org/10.3390/ijms26125710
Chicago/Turabian StyleHashimoto, Takuma, Go Ushikubo, Naoya Arao, Khaled Hatabi, Kazuki Tsubota, and Yoshio Hosoi. 2025. "Oxamate, an LDHA Inhibitor, Inhibits Stemness, Including EMT and High DNA Repair Ability, Induces Senescence, and Exhibits Radiosensitizing Effects in Glioblastoma Cells" International Journal of Molecular Sciences 26, no. 12: 5710. https://doi.org/10.3390/ijms26125710
APA StyleHashimoto, T., Ushikubo, G., Arao, N., Hatabi, K., Tsubota, K., & Hosoi, Y. (2025). Oxamate, an LDHA Inhibitor, Inhibits Stemness, Including EMT and High DNA Repair Ability, Induces Senescence, and Exhibits Radiosensitizing Effects in Glioblastoma Cells. International Journal of Molecular Sciences, 26(12), 5710. https://doi.org/10.3390/ijms26125710