
Low LDL-Cholesterol and Hemorrhagic Risk: Mechanistic Insights and Clinical Perspectives
 ,
,  ,
,  and
and
Abstract

1. Introduction
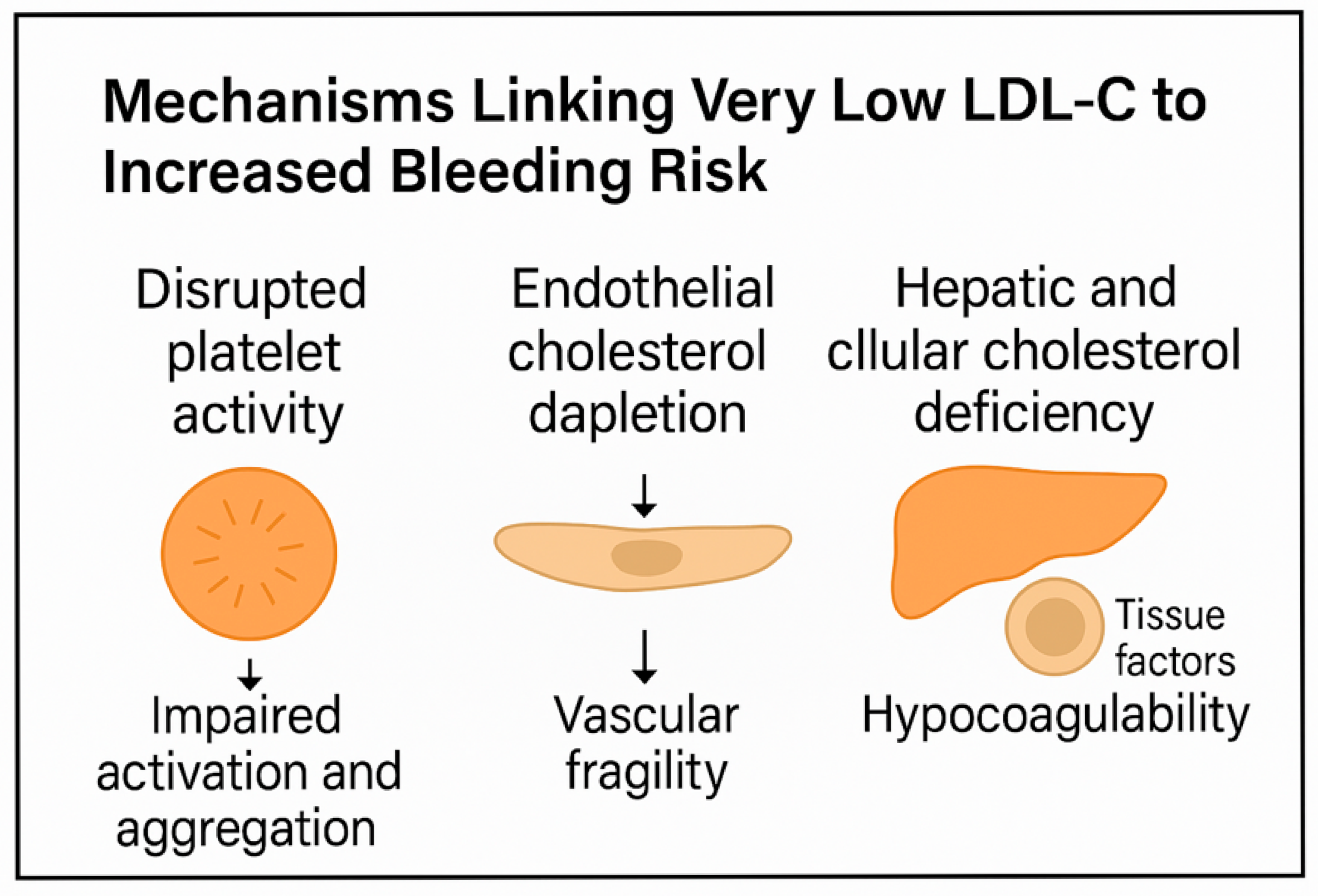
2. Cholesterol and Platelet Biology: From Lipid Rafts to Aggregation
3. Endothelial Integrity and LDL-C: Mechanisms of Microvascular Fragility
4. Clinical Evidence Linking Low LDL-C to Hemorrhagic Outcomes
4.1. Hemorrhagic Stroke
4.2. Gastrointestinal and Mucosal Bleeding
4.3. Meta-Analyses and Pooled Data
5. Implications for Clinical Practice and Future Research
5.1. Personalizing LDL-C Targets Based on Bleeding Risk
- A history of intracerebral hemorrhage or cerebral microbleeds on MRI;
- Advanced age (>75 years);
- Concurrent use of anticoagulants or dual antiplatelet therapy;
- A low body mass index (BMI < 20 kg/m2);
- A history of gastrointestinal bleeding or peptic ulcer disease;
5.2. Tailoring Lipid-Lowering Regimens and Co-Medications
- Discontinuing dual-antiplatelet therapy after 12 months, unless indicated as absolutely necessary;
- Co-prescribing proton pump inhibitors for gastroprotection;
- Using bleeding risk calculators (e.g., HAS-BLED, PRECISE-DAPT) to guide the intensity and duration of therapy [59].
5.3. Knowledge Gaps and Research Priorities
- What is the “threshold” LDL-C level at which bleeding risk significantly increases? While <50 mg/dL is commonly recognized as a general marker, individual susceptibility varies.
- How does the duration of exposure to very low LDL-C influence bleeding risk? Longitudinal cohort studies are needed to assess time-dependent effects.
- Are certain genetic variants associated with an increased risk of bleeding when LDL-C is low? Mendelian randomization and genome-wide association studies may offer valuable insights.
- Can we develop predictive models that integrate both ischemic and hemorrhagic risks for managing LDL-C? Such models could help personalize therapy beyond the current binary targets.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Licata, A.; Giammanco, A.; Minissale, M.G.; Pagano, S.; Petta, S.; Averna, M. Liver and Statins: A Critical Appraisal of the Evidence. Curr. Med. Chem. 2018, 25, 5835–5846. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in Drug and Herb Induced Liver Injury: The Update. Int. J. Mol. Sci. 2015, 17, 14. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Guo, Y.; Chang, L.; Zhang, G.; Gao, Z.; Lin, H.; Zhang, Y.; Hu, L.; Chen, S.; Fan, B.; Zhang, S.; et al. The role of Sphingomyelin synthase 2 (SMS2) in platelet activation and its clinical significance. Thromb. J. 2021, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Payrastre, B.; Missy, K.; Trumel, C.; Bodin, S.; Plantavid, M.; Chap, H. The integrin alpha IIb/beta 3 in human platelet signal transduction. Biochem. Pharmacol. 2000, 60, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Bodin, S.; Giuriato, S.; Ragab, J.; Humbel, B.M.; Viala, C.; Vieu, C.; Chap, H.; Payrastre, B. Production of phosphatidylinositol 3,4,5-trisphosphate and phosphatidic acid in platelet rafts: Evidence for a critical role of cholesterol-enriched domains in human platelet activation. Biochemistry 2001, 40, 15290–15299. [Google Scholar] [CrossRef]
- Andrews, R.K.; Lopez, J.A.; Berndt, M.C. Molecular mechanisms of platelet adhesion and activation. Int. J. Biochem. Cell Biol. 1997, 29, 91–105. [Google Scholar] [CrossRef]
- Safouris, A.; Magoufis, G.; Tsivgoulis, G. Emerging agents for the treatment and prevention of stroke: Progress in clinical trials. Expert. Opin. Investig. Drugs 2021, 30, 1025–1035. [Google Scholar] [CrossRef]
- Owens, A.P., 3rd; Mackman, N. The antithrombotic effects of statins. Annu. Rev. Med. 2014, 65, 433–445. [Google Scholar] [CrossRef]
- Siniscalchi, C.; Basaglia, M.; Riva, M.; Meschi, M.; Meschi, T.; Castaldo, G.; Di Micco, P. Statins Effects on Blood Clotting: A Review. Cells 2023, 12, 2719. [Google Scholar] [CrossRef] [PubMed]
- Siniscalchi, C.; Quintavalla, R.; Rocci, A.; Riera-Mestre, A.; Trujillo-Santos, J.; Surinach, J.M.; Jara-Palomares, L.; Bikdeli, B.; Moustafa, F.; Monreal, M.; et al. Statin and all-cause mortality in patients receiving anticoagulant therapy for venous thromboembolism. Data from the RIETE registry. Eur. J. Intern. Med. 2019, 68, 30–35. [Google Scholar] [CrossRef]
- Betrisey, S.; Haller, M.L.; Efthimiou, O.; Speierer, A.; Del Giovane, C.; Moutzouri, E.; Blum, M.R.; Aujesky, D.; Rodondi, N.; Gencer, B. Lipid-Lowering Therapy and Risk of Hemorrhagic Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2024, 13, e030714. [Google Scholar] [CrossRef]
- Li, W.; Wu, C.; Li, W.; Li, L. LDL-cholesterol lowering agents (statins and PCSK9 inhibitors) and the risk of intracerebral hemorrhage: A network meta-analysis. J. Stroke Cerebrovasc. Dis. 2025, 34, 108224. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Liu, W.; Li, H.; Fu, R.; Liu, X.; Xue, F.; Yang, R. Abnormal lipid rafts related ganglioside expression and signaling in T lymphocytes in immune thrombocytopenia patients. Autoimmunity 2016, 49, 58–68. [Google Scholar] [CrossRef]
- Siniscalchi, C.; Muriel, A.; Surinach Caralt, J.M.; Bikdeli, B.; Jimenez, D.; Lobo, J.L.; Amado, C.; Gil-Diaz, A.; Imbalzano, E.; Monreal, M.; et al. Statin use and 30-day mortality in patients with acute symptomatic pulmonary embolism. J. Thromb. Haemost. 2022, 20, 1839–1851. [Google Scholar] [CrossRef]
- Siniscalchi, C.; Bikdeli, B.; Jimenez, D.; Surinach, J.M.; Demelo-Rodriguez, P.; Moustafa, F.; Gil-Diaz, A.; Garcia-Ortega, A.; Bui, H.M.; Monreal, M.; et al. Statin use and mortality in patients with deep vein thrombosis. Data from the RIETE Registry. Thromb. Res. 2024, 236, 88–96. [Google Scholar] [CrossRef]
- Patel, H.H.; Insel, P.A. Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxid. Redox Signal. 2009, 11, 1357–1372. [Google Scholar] [CrossRef]
- Immanuel, J.; Yun, S. Vascular Inflammatory Diseases and Endothelial Phenotypes. Cells 2023, 12, 1640. [Google Scholar] [CrossRef] [PubMed]
- Gistera, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Drozd, M.; Bruns, A.F.; Yuldasheva, N.Y.; Maqbool, A.; Viswambharan, H.; Skromna, A.; Makava, N.; Cheng, C.W.; Sukumar, P.; Eades, L.; et al. Endothelial insulin-like growth factor-1 signaling regulates vascular barrier function and atherogenesis. Cardiovasc. Res. 2025, cvaf055. [Google Scholar] [CrossRef]
- Li, J.; Zhong, Z.; Yuan, J.; Chen, X.; Huang, Z.; Wu, Z. Resveratrol improves endothelial dysfunction and attenuates atherogenesis in apolipoprotein E-deficient mice. J. Nutr. Biochem. 2019, 67, 63–71. [Google Scholar] [CrossRef]
- Cao, C.; Shi, Y.; Zhang, X.; Li, Q.; Zhang, J.; Zhao, F.; Meng, Q.; Dai, W.; Liu, Z.; Yan, W.; et al. Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4. Nat. Commun. 2022, 13, 7139. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Sessa, W.C. Regulation of endothelial derived nitric oxide in health and disease. Mem. Inst. Oswaldo Cruz 2005, 100 (Suppl. S1), 15–18. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, A.; Wang, Y.; Tang, D.; Zhong, Y.; Liu, B.; Yan, M.; Jiu, Q.; Wu, W.; Wang, G. A study of lovastatin and L-arginine co-loaded PLGA nanomedicine for enhancing nitric oxide production and eNOS expression. J. Mater. Chem. B 2022, 10, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, Y.; Zhou, H.; Gong, X.; Luo, Z.; Li, J.; Sun, J.; Lou, M.; Yan, S. Low-density lipoprotein cholesterol, statin therapy, and cerebral microbleeds: The CIRCLE study. Neuroimage Clin. 2023, 39, 103502. [Google Scholar] [CrossRef]
- Kim, J.S. Role of Blood Lipid Levels and Lipid-Lowering Therapy in Stroke Patients with Different Levels of Cerebral Artery Diseases: Reconsidering Recent Stroke Guidelines. J. Stroke 2021, 23, 149–161. [Google Scholar] [CrossRef]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Naito, Y.; Ueda, S.; Takahashi, S.; Oyamada, H.; Yoneta, T.; Sugino, S.; Kondo, M. Ischemia-reperfusion injury and free radical involvement in gastric mucosal disorders. Adv. Exp. Med. Biol. 1990, 264, 401–410. [Google Scholar] [PubMed]
- Yu, Z.; Zhang, L.; Zhang, G.; Xia, K.; Yang, Q.; Huang, T.; Fan, D. Lipids, Apolipoproteins, Statins, and Intracerebral Hemorrhage: A Mendelian Randomization Study. Ann. Neurol. 2022, 92, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Siniscalchi, C.; Meschi, T.; Di Micco, P.; Imbalzano, E.; Hernandez-Blasco, L.; Mahe, I.; Fernandez-Reyes, J.L.; Garcia-Ortega, A.; Verhamme, P.; Alfonso-Megido, J.; et al. Low-Density Lipoprotein Cholesterol Levels and Bleeding Risk in Venous Thromboembolism. JAMA Netw. Open 2025, 8, e259467. [Google Scholar] [CrossRef] [PubMed]
- Deveci, B.; Gazi, E. Relation Between Globulin, Fibrinogen, and Albumin With the Presence and Severity of Coronary Artery Disease. Angiology 2021, 72, 174–180. [Google Scholar] [CrossRef]
- Schiano, C.; Trama, U.; Coscioni, E.; Infante, T.; Coppola, A.; de Nigris, F.; Napoli, C. Epitranscriptome: A Novel Regulatory Layer during Atherosclerosis Progression. Curr. Med. Chem. 2024, 32, 1234–1245. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.; Balbi, C.; Burrello, J.; Ruocco, A.; Infante, T.; Fiorito, C.; Panella, S.; Barile, L.; Mauro, C.; Vassalli, G.; et al. De novo DNA methylation induced by circulating extracellular vesicles from acute coronary syndrome patients. Atherosclerosis 2022, 354, 41–52. [Google Scholar] [CrossRef]
- Mandal, S.K.; Iakhiaev, A.; Pendurthi, U.R.; Rao, L.V. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: Modulation of tissue factor activity by membrane cholesterol. Blood 2005, 105, 153–160. [Google Scholar] [CrossRef]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef]
- Speer, T.; Dimmeler, S.; Schunk, S.J.; Fliser, D.; Ridker, P.M. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat. Rev. Nephrol. 2022, 18, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Zhang, C.; George, D.; Kotecha, S.; Abdelghaffar, M.; Forster, T.; Santos Rodrigues, P.D.; Reisinger, A.C.; White, D.; Hamilton, F.; et al. Low circulatory levels of total cholesterol, HDL-C and LDL-C are associated with death of patients with sepsis and critical illness: Systematic review, meta-analysis, and perspective of observational studies. eBioMedicine 2024, 100, 104981. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; Kim, T.J.; Yoon, B.W. Epidemiology, Risk Factors, and Clinical Features of Intracerebral Hemorrhage: An Update. J. Stroke 2017, 19, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Khalil, F.; Badshah, M.; Sathyanarayanan, S.P.; Amin, N. Statins Therapy and Intracranial Hemorrhage. South Dak. Med. 2023, 76, 170–173. [Google Scholar]
- Prado, Y.; Aravena, D.; Llancalahuen, F.M.; Aravena, C.; Eltit, F.; Echeverria, C.; Gatica, S.; Riedel, C.A.; Simon, F. Statins and Hemostasis: Therapeutic Potential Based on Clinical Evidence. Adv. Exp. Med. Biol. 2023, 1408, 25–47. [Google Scholar]
- Welch, K.M. Review of the SPARCL trial and its subanalyses. Curr. Atheroscler. Rep. 2009, 11, 315–321. [Google Scholar] [CrossRef]
- Goldstein, L.B.; Toth, P.P.; Dearborn-Tomazos, J.L.; Giugliano, R.P.; Hirsh, B.J.; Pena, J.M.; Selim, M.H.; Woo, D.; American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; et al. Aggressive LDL-C Lowering and the Brain: Impact on Risk for Dementia and Hemorrhagic Stroke: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e404–e442. [Google Scholar] [CrossRef]
- Sanz-Cuesta, B.E.; Saver, J.L. Lipid-Lowering Therapy and Hemorrhagic Stroke Risk: Comparative Meta-Analysis of Statins and PCSK9 Inhibitors. Stroke 2021, 52, 3142–3150. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, K.; Minematsu, K.; Yasaka, M.; Nagai, Y.; Hosomi, N.; Origasa, H.; Kitagawa, K.; Uchiyama, S.; Koga, M.; Matsumoto, M.; et al. The Japan Statin Treatment Against Recurrent Stroke (J-STARS) Echo Study: Rationale and Trial Protocol. J. Stroke Cerebrovasc. Dis. 2017, 26, 595–599. [Google Scholar] [CrossRef]
- Siniscalchi, C.; Di Micco, P.; Tufano, A.; Peris, M.L.; Lopez-Miguel, P.; Alda-Lozano, A.; Llamas, P.; Barata, D.D.; Jenab, Y.; Monreal, M.; et al. Baseline Hemoglobin Values and Clinical Outcomes in Acute Venous Thromboembolism: Insights From the RIETE Registry. Am. J. Hematol. 2025; early view. [Google Scholar]
- Zhai, F.F.; Yan, S.; Li, M.L.; Han, F.; Wang, Q.; Zhou, L.X.; Ni, J.; Yao, M.; Zhang, S.Y.; Cui, L.Y.; et al. Intracranial Arterial Dolichoectasia and Stenosis: Risk Factors and Relation to Cerebral Small Vessel Disease. Stroke 2018, 49, 1135–1140. [Google Scholar] [CrossRef]
- Del Brutto, O.H.; Mera, R.M.; Del Brutto, V.J.; Costa, A.F.; Zambrano, M.; Brorson, J. Basilar Artery Dolichoectasia: Prevalence and Correlates With Markers of Cerebral Small Vessel Disease in Community-Dwelling Older Adults. J. Stroke Cerebrovasc. Dis. 2017, 26, 2909–2914. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Pradhan, A.; MacFadyen, J.G.; Libby, P.; Glynn, R.J. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: An analysis from the JUPITER trial. Lancet 2012, 380, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Bhagavathula, A.S.; Vidyasaga, K.; Gebreyohannes, E.A.; Tesfaye, W. Risk of Gastrointestinal Bleeding on Treatment With Statin Alone or With Concomitant Administration of Warfarin: A Systematic Review and Meta-analysis of 5.3 Million Participants. Ann. Pharmacother. 2022, 56, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.I.; Freeman, P.R.; Moga, D.C. Statin Use and Gastrointestinal Hemorrhage: A Large Retrospective Cohort Study. Am. J. Cardiovasc. Drugs 2019, 19, 65–74. [Google Scholar] [CrossRef]
- Del Giorno, R.; Mazzolai, L.; Keller, S.; Siniscalchi, C.; Lopez-Jimenez, L.; Ballaz, A.; Montenegro, A.C.; Otero, R.; Rashidi, F.; Monreal, M.; et al. Assessment of bleeding events in patients receiving DOACs with or without statins to treat venous thromboembolism: Insights from the RIETE registry. BMJ Open 2024, 14, e085401. [Google Scholar] [CrossRef]
- Siniscalchi, C. Protective role of statins during anticoagulation for venous thromboembolism: Beyond their lipid lowering effect? Eur. J. Intern. Med. 2020, 79, 127–129. [Google Scholar] [CrossRef]
- Pisters, R.; Lane, D.A.; Nieuwlaat, R.; de Vos, C.B.; Crijns, H.J.; Lip, G.Y. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: The Euro Heart Survey. Chest 2010, 138, 1093–1100. [Google Scholar] [CrossRef]
- Costa, F.; van Klaveren, D.; James, S.; Heg, D.; Raber, L.; Feres, F.; Pilgrim, T.; Hong, M.K.; Kim, H.S.; Colombo, A.; et al. Derivation and validation of the predicting bleeding complications in patients undergoing stent implantation and subsequent dual antiplatelet therapy (PRECISE-DAPT) score: A pooled analysis of individual-patient datasets from clinical trials. Lancet 2017, 389, 1025–1034. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Clinical Implications |
|---|
|
| Translational perspectives |
|
| Study/Analysis | Population | LDL-C Target/Achieved | Type of Bleeding Observed | Key Findings | Reference |
|---|---|---|---|---|---|
| SPARCL | 4731 patients with stroke or TIA | Median LDL-C: ~73 mg/dL with atorvastatin 80 mg | Intracerebral hemorrhage | 66% increased risk of ICH (HR: 1.66; 95% CI: 1.08–2.55) | [47] |
| Meta-analysis 2021 | >300,000 patients (16 RCTs + 14 observational studies) | LDL-C < 50 mg/dL | Major bleeding (various sites) | 1.4–1.6-fold increased risk, especially with antithrombotic therapy | [58] |
| FOURIER + ODYSSEY OUTCOMES (pooled) | High CV risk patients treated with PCSK9 inhibitors | LDL-C < 30 mg/dL (achieved) | Epistaxis, GIB, retinal bleeding | Numerical increase in bleeding events, but low absolute incidence | [59] |
| J-STARS (Japan) | Patients with low baseline LDL-C at stroke risk | Already low baseline LDL-C | Intracranial bleeding | Increased hemorrhagic events, particularly in those with low initial LDL-C | [50] |
| JUPITER | 17,802 healthy individuals with elevated hsCRP | Median LDL-C: ~55 mg/dL with rosuvastatin 20 mg | GIB | Increased GIB (1.2% vs. 0.9%, p = 0.03) | [54] |
| NHIRD (Taiwan) | >30,000 patients on statin therapy | LDL-C < 70 mg/dL | GIB | 25% increased risk of GIB, particularly with concomitant antiplatelet agents | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siniscalchi, C.; Basaglia, M.; Meschi, T.; Imbalzano, E.; Futura Bernardi, F.; Perrella, A.; Trama, U.; Passannanti, A.; Di Micco, P.; Schiano, C. Low LDL-Cholesterol and Hemorrhagic Risk: Mechanistic Insights and Clinical Perspectives. Int. J. Mol. Sci. 2025, 26, 5612. https://doi.org/10.3390/ijms26125612
Siniscalchi C, Basaglia M, Meschi T, Imbalzano E, Futura Bernardi F, Perrella A, Trama U, Passannanti A, Di Micco P, Schiano C. Low LDL-Cholesterol and Hemorrhagic Risk: Mechanistic Insights and Clinical Perspectives. International Journal of Molecular Sciences. 2025; 26(12):5612. https://doi.org/10.3390/ijms26125612
Chicago/Turabian StyleSiniscalchi, Carmine, Manuela Basaglia, Tiziana Meschi, Egidio Imbalzano, Francesca Futura Bernardi, Alessandro Perrella, Ugo Trama, Angelica Passannanti, Pierpaolo Di Micco, and Concetta Schiano. 2025. "Low LDL-Cholesterol and Hemorrhagic Risk: Mechanistic Insights and Clinical Perspectives" International Journal of Molecular Sciences 26, no. 12: 5612. https://doi.org/10.3390/ijms26125612
APA StyleSiniscalchi, C., Basaglia, M., Meschi, T., Imbalzano, E., Futura Bernardi, F., Perrella, A., Trama, U., Passannanti, A., Di Micco, P., & Schiano, C. (2025). Low LDL-Cholesterol and Hemorrhagic Risk: Mechanistic Insights and Clinical Perspectives. International Journal of Molecular Sciences, 26(12), 5612. https://doi.org/10.3390/ijms26125612