
Assessment of 1863 GRIN2A Variants Contradicts a Role in Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
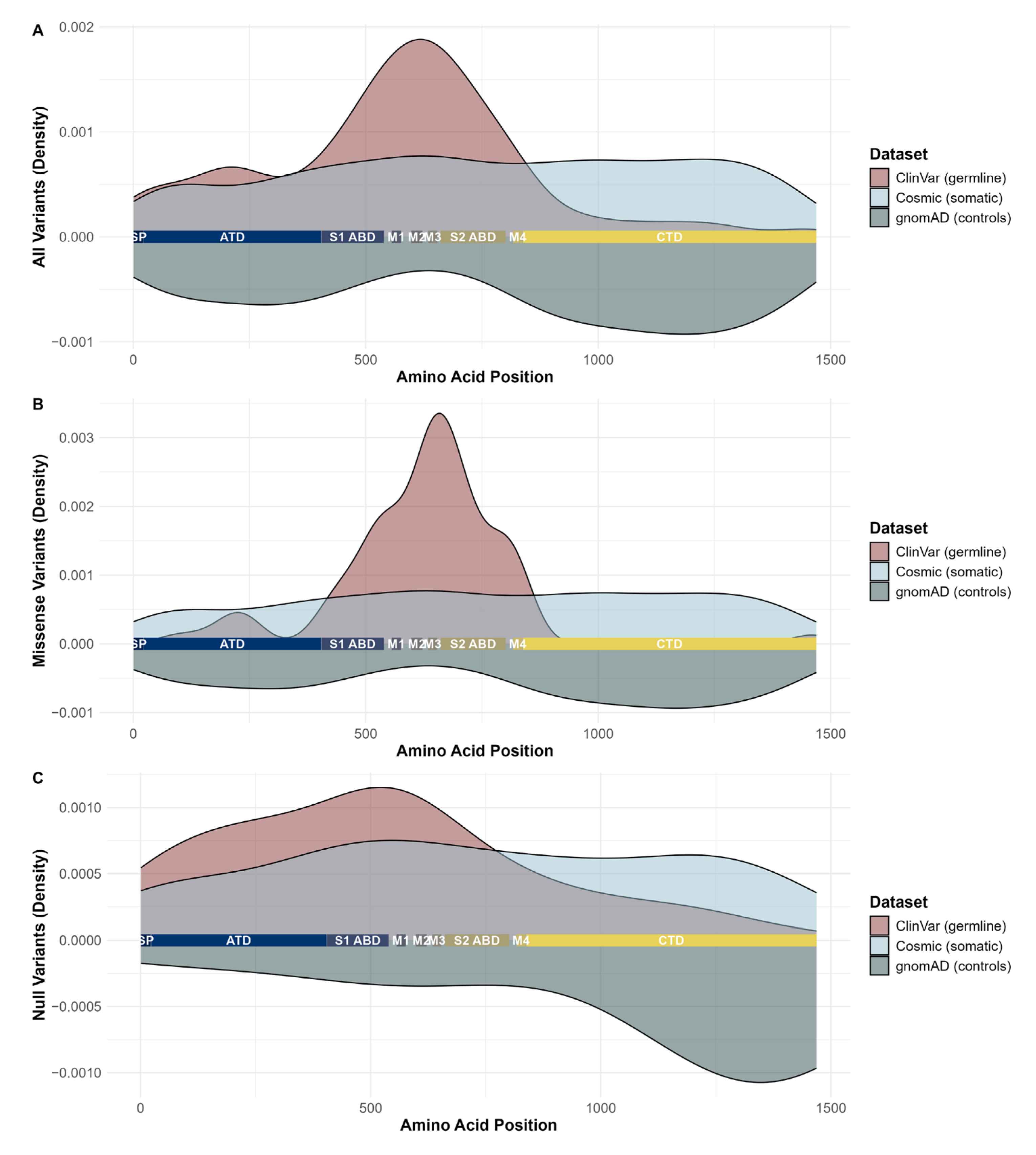
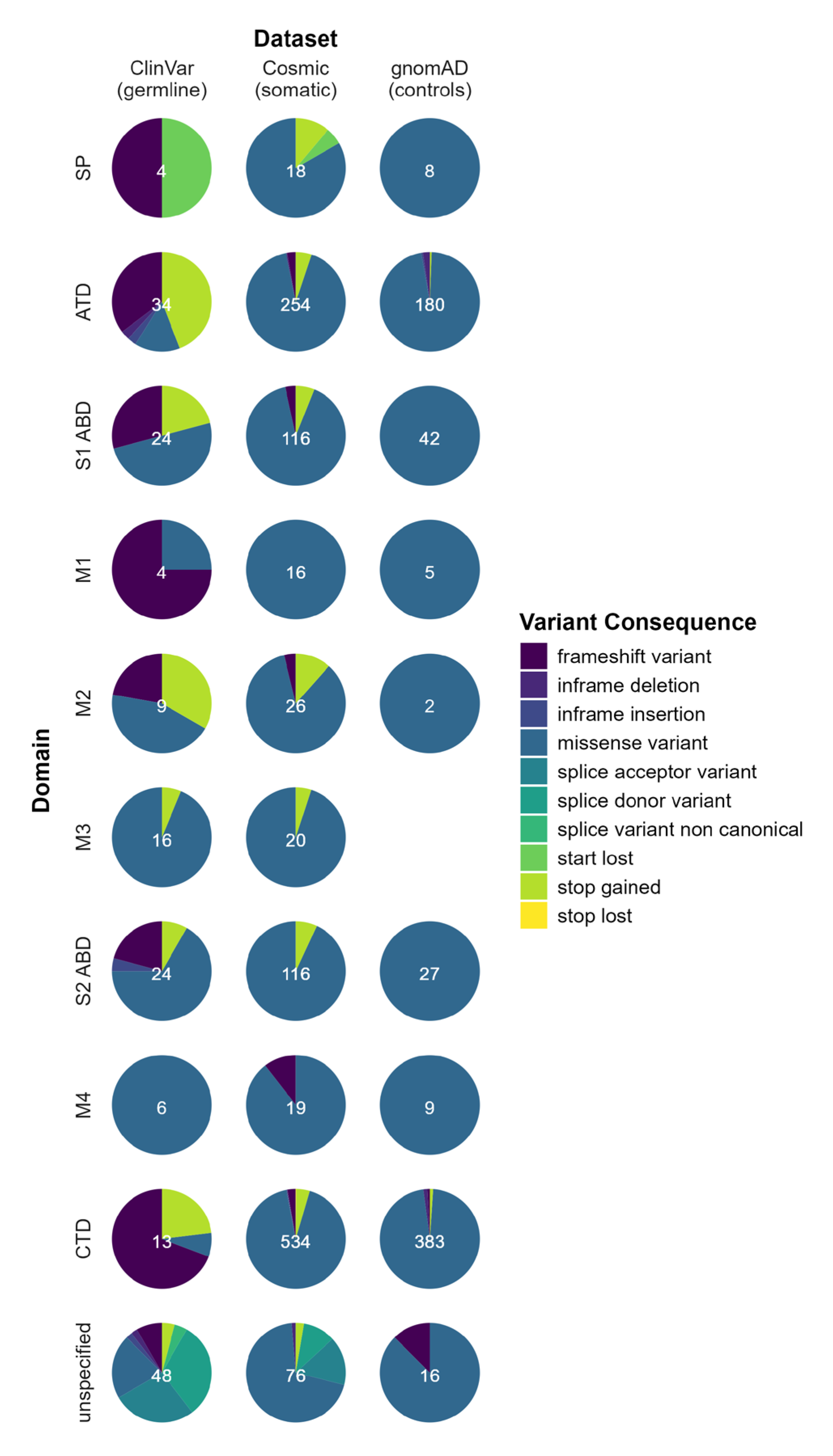
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T.R. MGluR5 and NMDA Receptors Drive the Experience- and Activity-Dependent NMDA Receptor NR2B to NR2A Subunit Switch. Neuron 2011, 70, 339. [Google Scholar] [CrossRef] [PubMed]
- Strehlow, V.; Heyne, H.O.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; De Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A-Related Disorders: Genotype and Functional Consequence Predict Phenotype. Brain 2019, 142, 80. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A Cause Idiopathic Focal Epilepsy with Rolandic Spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Wangari-Talbot, J.; Chen, S. Genetics of Melanoma. Front. Genet. 2013, 3, 38084. [Google Scholar] [CrossRef] [PubMed]
- D’mello, S.A.N.; Flanagan, J.U.; Green, T.N.; Leung, E.Y.; Askarian-Amiri, M.E.; Joseph, W.R.; McCrystal, M.R.; Isaacs, R.J.; Shaw, J.H.F.; Furneaux, C.E.; et al. Evidence That GRIN2A Mutations in Melanoma Correlate with Decreased Survival. Front. Oncol. 2014, 3, 333. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Zerlanko, B.J.; Hill, V.K.; Gartner, J.J.; Qutob, N.; Jiang, J.; Simaan, M.; Wunderlich, J.; Gutkind, J.S.; Rosenberg, S.A.; et al. Somatic Mutation of GRIN2A in Malignant Melanoma Results in Loss of Tumor Suppressor Activity via Aberrant NMDAR Complex Formation. J. Investig. Dermatol. 2014, 134, 2390. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; Stemke-Hale, K.; Davies, M.A.; Gershenwald, J.E.; et al. Exome Sequencing Identifies GRIN2A as Frequently Mutated in Melanoma. Nat. Genet. 2011, 43, 442. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Gartner, J.J.; Samuels, Y. Genetic and Functional Analysis of GRIN2A in Tumor Samples. Methods Mol. Biol. 2017, 1677, 93–116. [Google Scholar] [CrossRef] [PubMed]
- Aguissa-Touré, A.H.; Li, G. Genetic Alterations of PTEN in Human Melanoma. Cell. Mol. Life Sci. 2012, 69, 1475–1491. [Google Scholar] [CrossRef] [PubMed]
- Münger, K. Disruption of Oncogene/Tumor Suppressor Networks during Human Carcinogenesis. Cancer Investig. 2002, 20, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ardlie, K.G.; DeLuca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jauss, R.-T.; Lemke, J.R.; Strehlow, V. Assessment of 1863 GRIN2A Variants Contradicts a Role in Tumorigenesis. Int. J. Mol. Sci. 2025, 26, 5558. https://doi.org/10.3390/ijms26125558
Jauss R-T, Lemke JR, Strehlow V. Assessment of 1863 GRIN2A Variants Contradicts a Role in Tumorigenesis. International Journal of Molecular Sciences. 2025; 26(12):5558. https://doi.org/10.3390/ijms26125558
Chicago/Turabian StyleJauss, Robin-Tobias, Johannes R. Lemke, and Vincent Strehlow. 2025. "Assessment of 1863 GRIN2A Variants Contradicts a Role in Tumorigenesis" International Journal of Molecular Sciences 26, no. 12: 5558. https://doi.org/10.3390/ijms26125558
APA StyleJauss, R.-T., Lemke, J. R., & Strehlow, V. (2025). Assessment of 1863 GRIN2A Variants Contradicts a Role in Tumorigenesis. International Journal of Molecular Sciences, 26(12), 5558. https://doi.org/10.3390/ijms26125558