
Comparative Transcriptomic Analysis Reveals the Potential Molecular Mechanism Underlying Squalene Biosynthesis in Developing Seeds of Oil-Tea (Camellia oleifera)

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
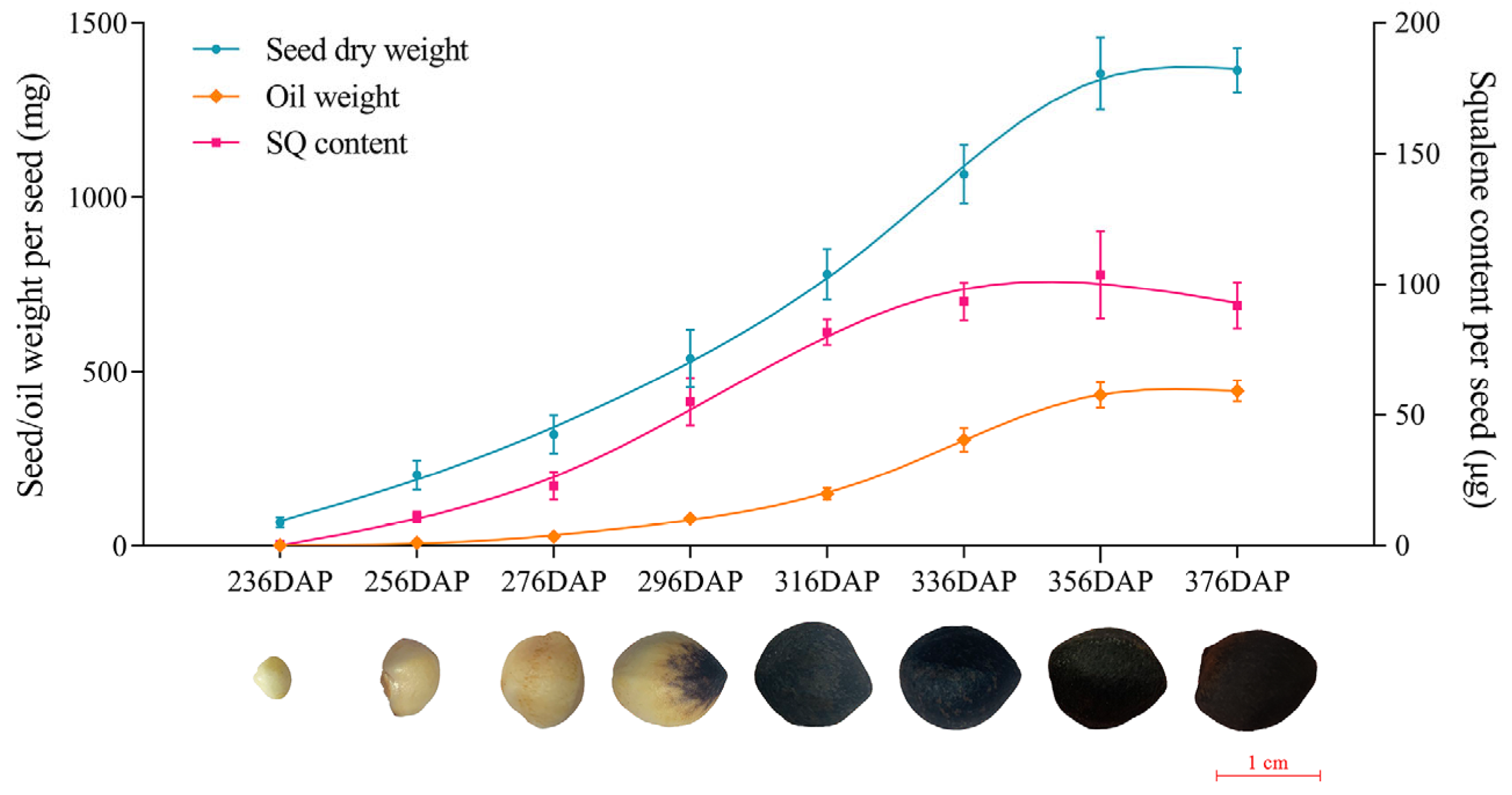
2.1. Determination of Squalene Accumulation with Seed Development
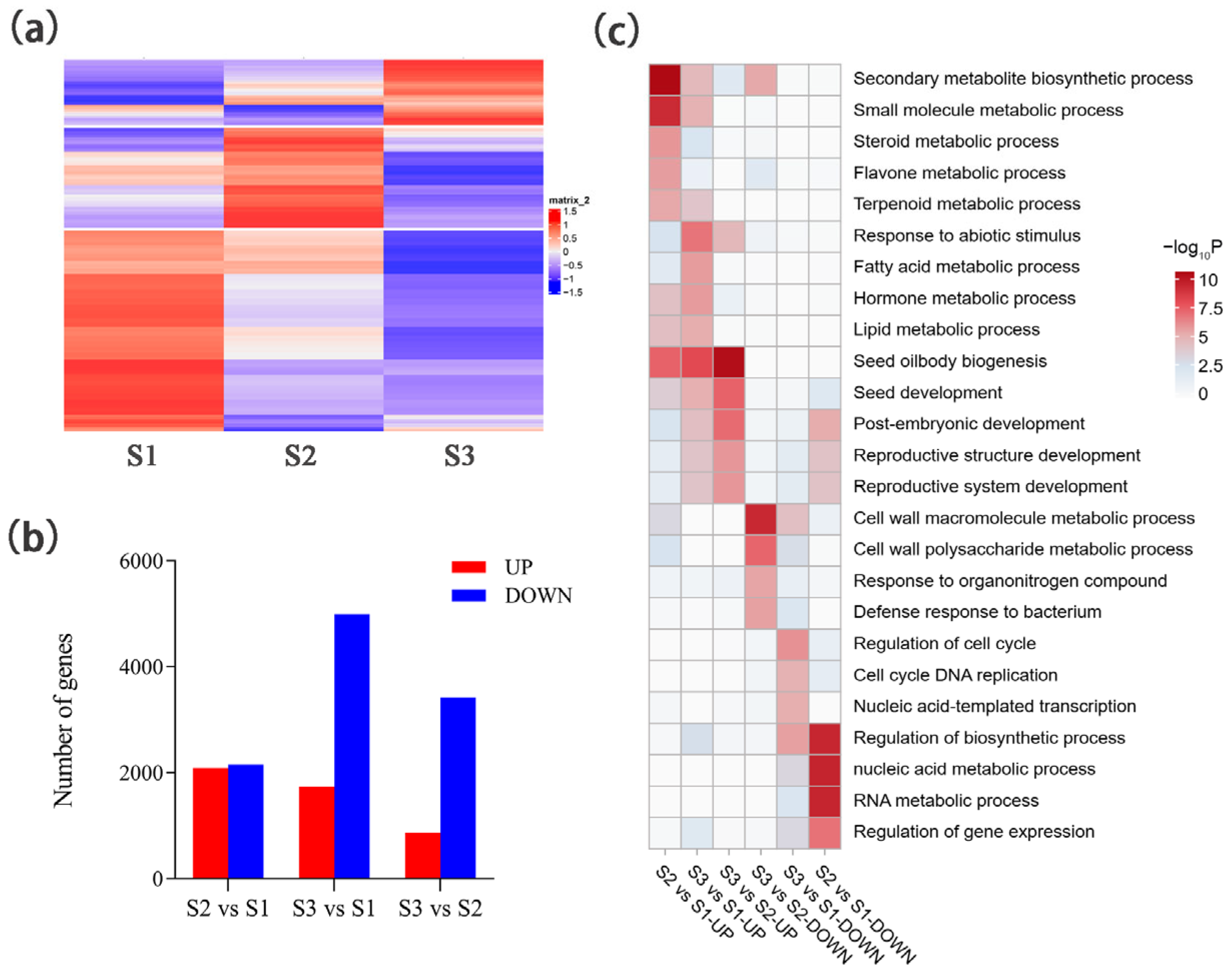
2.2. Comparative Transcriptomic Analysis of Developing Seeds
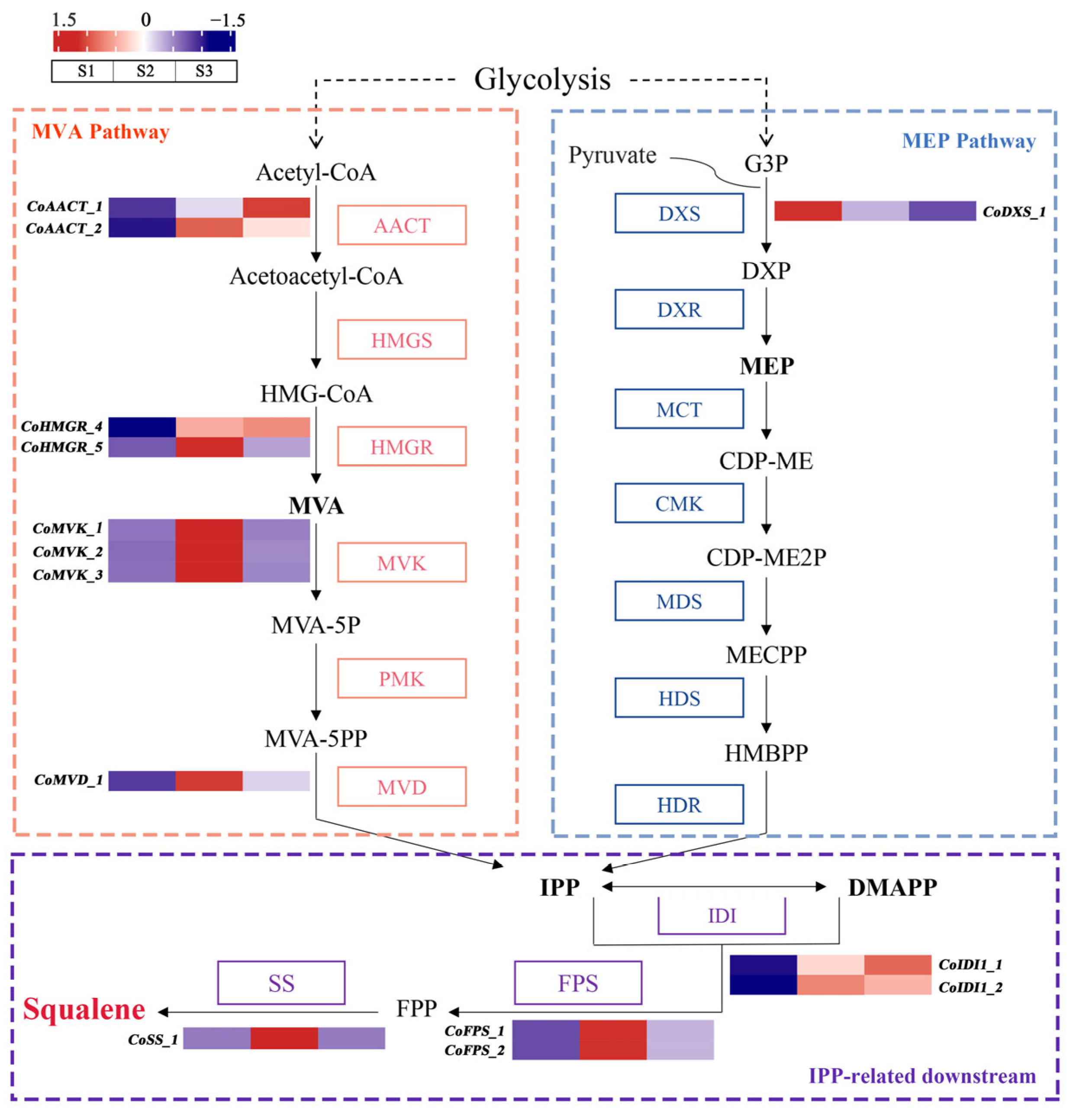
2.3. Identification of Critical Candidate Genes Involved in Squalene Biosynthesis
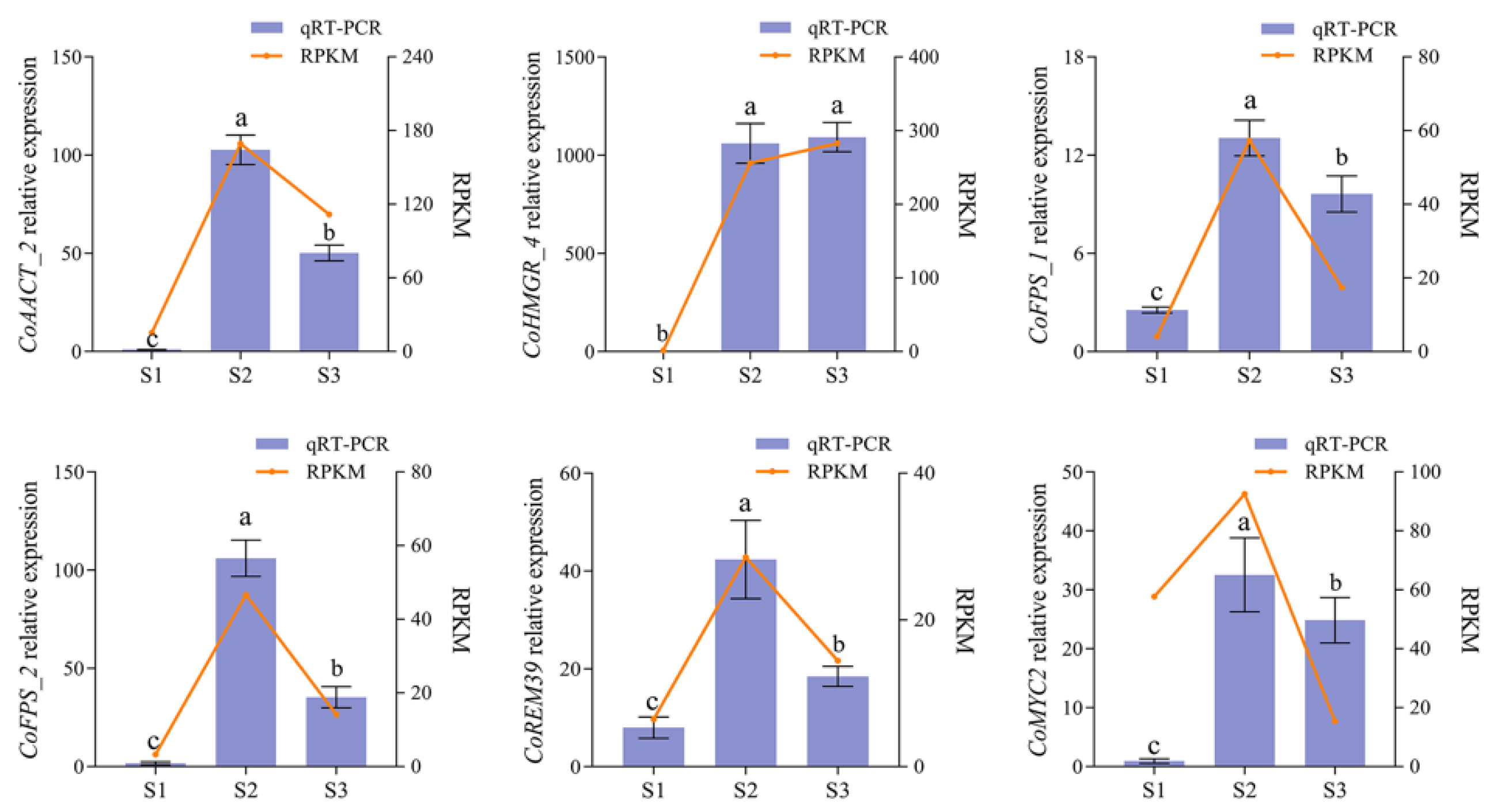
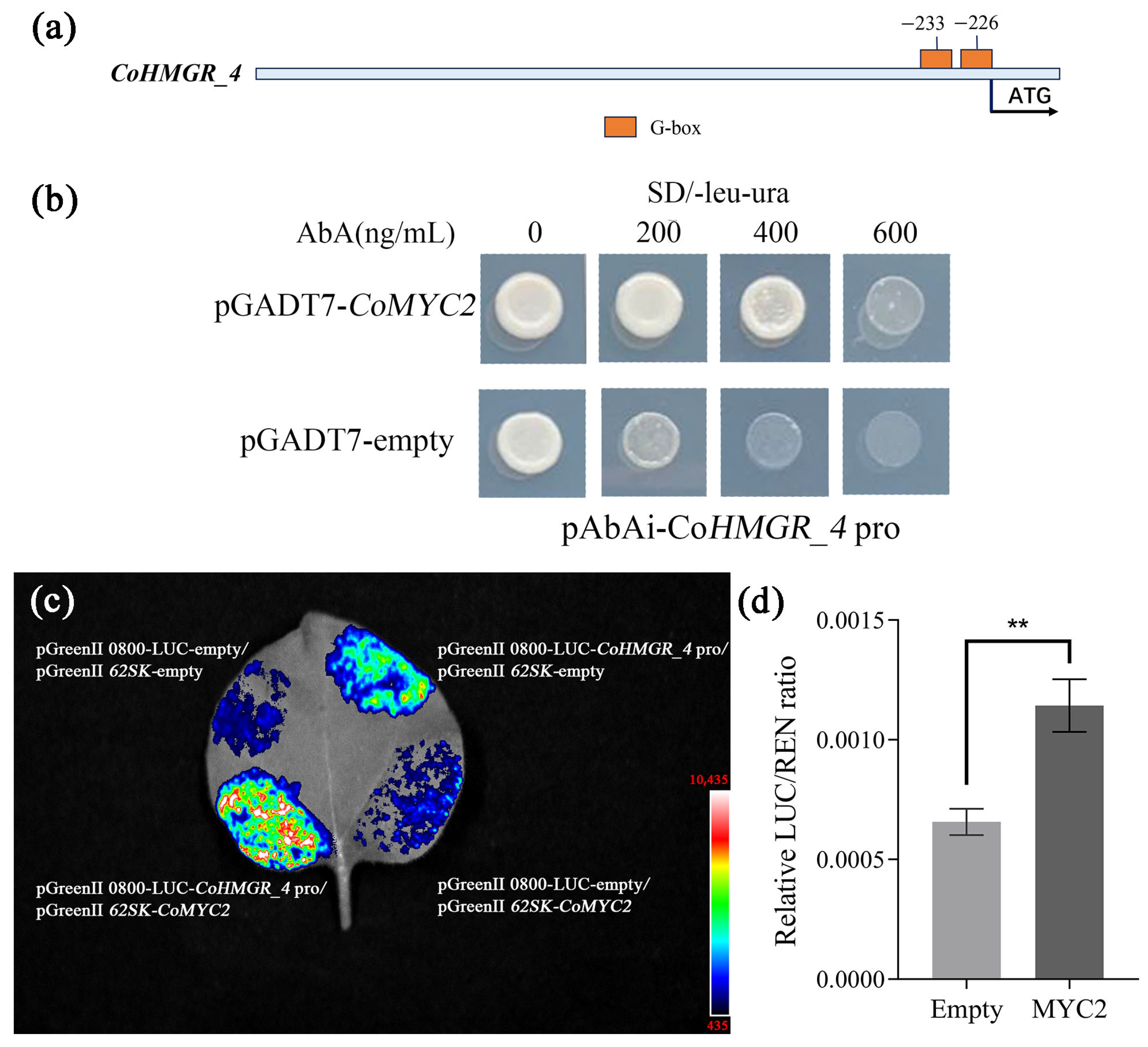
2.4. Validation of the Regulation of Squalene Biosynthesis Genes by Transcription Factor CoMYC2
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Measurement of Seed Dry Weight, Oil Weight, and Squalene Content
4.3. Construction of Illumina RNA-Seq Library for Sequencing and Functional Annotation
4.4. Identification of Squalene Biosynthesis Genes and Transcription Factors
4.5. Quantitative Real-Time PCR (qRT-PCR)
4.6. Construction of Co-Expression Network and Cis-Acting Element Analysis
4.7. Yeast One-Hybrid Assays
4.8. Dual-Luciferase Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Zeng, Q.; Verardo, V.; Contreras, M.d.M. Fatty acid and sterol composition of tea seed oils: Their comparison by the “FancyTiles” approach. Food Chem. 2017, 233, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ruan, C.; Han, P.; Ruan, D.; Xiong, C.; Ding, J.; Liu, S. Comparative transcriptomic analysis of high- and low-oil Camellia oleifera reveals a coordinated mechanism for the regulation of upstream and downstream multigenes for high oleic acid accumulation. 3 Biotech 2019, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, X.; Chen, Z.; Lin, Y.; Wang, S. Comparison of Oil Content and Fatty Acid Profile of Ten New Camellia oleifera Cultivars. J. Lipids 2016, 2016, 3982486. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed Transcriptomics Analysis in Camellia oleifera Uncovers Genes Associated with Oil Content and Fatty Acid Composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Z.; Zhou, J.; Gu, Y.; Tan, X. Comparative study on fruit development and oil synthesis in two cultivars of Camellia oleifera. BMC Plant Biol. 2021, 21, 348. [Google Scholar] [CrossRef]
- Qin, P.; Shen, J.; Wei, J.; Chen, Y. A critical review of the bioactive ingredients and biological functions of Camellia oleifera oil. Curr. Res. Food Sci. 2024, 8, 100753. [Google Scholar] [CrossRef]
- Chou, T.-Y.; Lu, Y.-F.; Inbaraj, B.S.; Chen, B.-H. Camelia oil and soybean-camelia oil blend enhance antioxidant activity and cardiovascular protection in hamsters. Nutrition 2018, 51–52, 86–94. [Google Scholar] [CrossRef]
- Guo, L.; Guo, Y.; Wu, P.; Lu, F.; Zhu, J.; Ma, H.; Chen, Y.; Zhang, T. Camellia oil lowering blood pressure in spontaneously hypertension rats. J. Funct. Foods 2020, 70, 103915. [Google Scholar] [CrossRef]
- Lozano-Grande, M.A.; Gorinstein, S.; Espitia-Rangel, E.; Dávila-Ortiz, G.; Martínez-Ayala, A.L. Plant Sources, Extraction Methods, and Uses of Squalene. Int. J. Agron. 2018, 2018, 1829160. [Google Scholar] [CrossRef]
- Ravi Kumar, S.; Yamauchi, I.; Narayan, B.; Katsuki, A.; Hosokawa, M.; Miyashita, K. Squalene modulates fatty acid metabolism: Enhanced EPA/DHA in obese/diabetic mice (KK-Ay) model. Eur. J. Lipid Sci. Technol. 2016, 118, 1935–1941. [Google Scholar] [CrossRef]
- Spanova, M.; Daum, G. Squalene—Biochemistry, molecular biology, process biotechnology, and applications. Eur. J. Lipid Sci. Technol. 2011, 113, 1299–1320. [Google Scholar] [CrossRef]
- He, H.-P.C.H. Oil and Squalene in Amaranthus Grain and Leaf. J. Agric. Food Chem. 2023, 51, 7913–7920. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.; Stanzione, V.; Mariotti, R.; Mastio, V.; Azariadis, A.; Passeri, V.; Valeri, M.C.; Baldoni, L.; Bufacchi, M. Bioactive Compound Profiling of Olive Fruit: The Contribution of Genotype. Antioxidants 2022, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Vranová, E.; Coman, D.; Gruissem, W. Network Analysis of the MVA and MEP Pathways for Isoprenoid Synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Soto, G.; Stritzler, M.; Lisi, C.; Alleva, K.; Pagano, M.E.; Ardila, F.; Mozzicafreddo, M.; Cuccioloni, M.; Angeletti, M.; Ayub, N.D. Acetoacetyl-CoA thiolase regulates the mevalonate pathway during abiotic stress adaptation. J. Exp. Bot. 2011, 62, 5699–5711. [Google Scholar] [CrossRef]
- Johnson, E.E.; Jetter, R.; Wasteneys, G. Rapid induction of the triterpenoid pathway in mesophyll protoplasts. Biotechnol. Lett. 2014, 36, 855–858. [Google Scholar] [CrossRef]
- Suzuki, M.; Kamide, Y.; Nagata, N.; Seki, H.; Ohyama, K.; Kato, H.; Masuda, K.; Sato, S.; Kato, T.; Tabata, S.; et al. Loss of function of 3-hydroxy-3-methylglutaryl coenzyme A reductase 1 (HMG1) in Arabidopsis leads to dwarfing, early senescence and male sterility, and reduced sterol levels. Plant J. 2004, 37, 750–761. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Kim, Y.B.; Uddin, M.R.; Lee, S.; Kim, S.-U.; Park, S.U. Enhanced Triterpene Accumulation in Panax ginseng Hairy Roots Overexpressing Mevalonate-5-pyrophosphate Decarboxylase and Farnesyl Pyrophosphate Synthase. ACS Synth. Biol. 2014, 3, 773–779. [Google Scholar] [CrossRef]
- Qu, L.; Li, H.-L.; Guo, D.; Wang, Y.; Zhu, J.-H.; Yin, L.-Y.; Peng, S.-Q. HbWRKY27, a group IIe WRKY transcription factor, positively regulates HbFPS1 expression in Hevea brasiliensis. Sci. Rep. 2020, 10, 20639. [Google Scholar] [CrossRef]
- Deng, B.; Huang, Z.; Ge, F.; Liu, D.; Lu, R.; Chen, C. An AP2/ERF Family Transcription Factor PnERF1 Raised the Biosynthesis of Saponins in Panax notoginseng. J. Plant Growth Regul. 2017, 36, 691–701. [Google Scholar] [CrossRef]
- Yin, J.; Sun, L.; Li, Y.; Xiao, J.; Wang, S.; Yang, J.; Qu, Z.; Zhan, Y. Functional identification of BpMYB21 and BpMYB61 transcription factors responding to MeJA and SA in birch triterpenoid synthesis. BMC Plant Biol. 2020, 20, 374. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Pollier, J.; Vanden Bossche, R.; Lopez-Vidriero, I.; Franco-Zorrilla, J.M.; Goossens, A. The bHLH Transcription Factors TSAR1 and TSAR2 Regulate Triterpene Saponin Biosynthesis in Medicago truncatula. Plant Physiol. 2016, 170, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liao, J.; Shi, M.; Li, L.; Liu, Q.; Cui, X.; Ning, W.; Kai, G. A jasmonate-responsive bHLH transcription factor TaMYC2 positively regulates triterpenes biosynthesis in Taraxacum antungense Kitag. Plant Sci. 2023, 326, 111506. [Google Scholar] [CrossRef]
- Sharma, A.; Rather, G.A.; Misra, P.; Dhar, M.K.; Lattoo, S.K. Jasmonate responsive transcription factor WsMYC2 regulates the biosynthesis of triterpenoid withanolides and phytosterol via key pathway genes in Withania somnifera (L.) Dunal. Plant Mol. Biol. 2019, 100, 543–560. [Google Scholar] [CrossRef]
- Wei, T.; Dong, L.; Zhong, S.; Jing, H.; Deng, Z.; Wen, Q.; Li, J. Chemical composition of Camellia chekiangoleosa Hu. seeds during ripening and evaluations of seed oils quality. Ind. Crops Prod. 2022, 177, 114499. [Google Scholar] [CrossRef]
- Mišina, I.; Sipeniece, E.; Rudzińska, M.; Grygier, A.; Radzimirska-Graczyk, M.; Kaufmane, E.; Segliņa, D.; Lācis, G.; Górnaś, P. Associations between Oil Yield and Profile of Fatty Acids, Sterols, Squalene, Carotenoids, and Tocopherols in Seed Oil of Selected Japanese Quince Genotypes during Fruit Development. Eur. J. Lipid Sci. Technol. 2020, 122, 1900386. [Google Scholar] [CrossRef]
- Herchi, W.; Harrabi, S.; Rochut, S.; Boukhchina, S.; Kallel, H.; Pepe, C. Characterization and Quantification of the Aliphatic Hydrocarbon Fraction during Linseed Development (Linum usitatissimum L.). J. Agric. Food Chem. 2009, 57, 5832–5836. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, H.; Kang, J.; Warusawitharana, H.K.; Chen, S.; Wang, K.; Yu, F.; Wu, Y.; He, P.; Tu, Y.; et al. Comparative Transcriptome and Phytochemical Analysis Provides Insight into Triterpene Saponin Biosynthesis in Seeds and Flowers of the Tea Plant (Camellia sinensis). Metabolites 2022, 12, 204. [Google Scholar] [CrossRef]
- Sando, T.; Takaoka, C.; Mukai, Y.; Yamashita, A.; Hattori, M.; Ogasawara, N.; Fukusaki, E.; Kobayashi, A. Cloning and Characterization of Mevalonate Pathway Genes in a Natural Rubber Producing Plant, Hevea brasiliensis. Biosci. Biotechnol. Biochem. 2008, 72, 2049–2060. [Google Scholar] [CrossRef]
- Pütter, K.M.; van Deenen, N.; Unland, K.; Prüfer, D.; Gronover, C.S. Isoprenoid biosynthesis in dandelion latex is enhanced by the overexpression of three key enzymes involved in the mevalonate pathway. BMC Plant Biol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Z.; Zheng, Z.; Tian, Z.; Zhang, H.; Zhu, C.; Yao, X.; Yang, Y.; Cai, X. Molecular Cloning and Analysis of an Acetyl-CoA C-acetyltransferase Gene (EkAACT) from Euphorbia kansui Liou. Plants 2022, 11, 1539. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Zhang, Z.; Shi, Q.; Niu, R.; Duan, X.; Shen, B.; Li, X. Transcription Factors ZjMYB39 and ZjMYB4 Regulate Farnesyl Diphosphate Synthase- and Squalene Synthase-Mediated Triterpenoid Biosynthesis in Jujube. J. Agric. Food Chem. 2023, 71, 4599–4614. [Google Scholar] [CrossRef] [PubMed]
- Closa, M.; Vranová, E.; Bortolotti, C.; Bigler, L.; Arró, M.; Ferrer, A.; Gruissem, W. The Arabidopsis thaliana FPP synthase isozymes have overlapping and specific functions in isoprenoid biosynthesis, and complete loss of FPP synthase activity causes early developmental arrest. Plant J. 2010, 63, 512–525. [Google Scholar] [CrossRef]
- Wang, S.; Feng, Y.; Lou, Y.; Niu, J.; Yin, C.; Zhao, J.; Du, W.; Yue, A. 3-Hydroxy-3-methylglutaryl coenzyme A reductase genes from Glycine max regulate plant growth and isoprenoid biosynthesis. Sci. Rep. 2023, 13, 3902. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Lee, O.R.; Oh, J.Y.; Jang, M.-G.; Yang, D.-C. Functional Analysis of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Encoding Genes in Triterpene Saponin-Producing Ginseng. Plant Physiol. 2014, 165, 373–387. [Google Scholar] [CrossRef]
- Harker, M.H.N.; Clayton, J.C.; Gibbard, C.L.; Wallace, A.D.; Rawlins, S.S.; Hellyer, A.; Lanot, A.; Safford, R. Enhancement of seed phytosterol levels by expression of an N-terminal truncated Hevea brasiliensis (rubber tree) 3-hydroxy-3-methylglutaryl-CoA reductase. Plant Biotechnol. J. 2003, 1, 113–121. [Google Scholar] [CrossRef]
- Jayashree, R.; Nazeem, P.A.; Rekha, K.; Sreelatha, S.; Thulaseedharan, A.; Krishnakumar, R.; Kala, R.G.; Vineetha, M.; Leda, P.; Jinu, U.; et al. Over-expression of 3-hydroxy-3- methylglutaryl-coenzyme A reductase 1 (hmgr1) gene under super-promoter for enhanced latex biosynthesis in rubber tree (Hevea brasiliensis Muell. Arg.). Plant Physiol. Biochem. 2018, 127, 414–424. [Google Scholar] [CrossRef]
- Ribeiro, B.; Erffelinck, M.-L.; Lacchini, E.; Ceulemans, E.; Colinas, M.; Williams, C.; Van Hamme, E.; De Clercq, R.; Perassolo, M.; Goossens, A. Interference between ER stress-related bZIP-type and jasmonate-inducible bHLH-type transcription factors in the regulation of triterpene saponin biosynthesis in Medicago truncatula. Front. Plant Sci. 2022, 13, 903793. [Google Scholar] [CrossRef]
- Li, C.; Jiao, M.; Zhao, X.; Ma, J.; Cui, Y.; Kou, X.; Long, Y.; Xing, Z. bZIP transcription factor responds to changes in light quality and affects saponins synthesis in Eleutherococcus senticosus. Int. J. Biol. Macromol. 2024, 279, 135273. [Google Scholar] [CrossRef]
- Cárdenas, P.D.; Sonawane, P.D.; Pollier, J.; Vanden Bossche, R.; Dewangan, V.; Weithorn, E.; Tal, L.; Meir, S.; Rogachev, I.; Malitsky, S.; et al. GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 2016, 7, 10654. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Sugawara, S.; Mori, T.; Kobayashi, M.; Kusano, M.; Saito, K.; Bayer, E. Induced production of specialized steroids by transcriptional reprogramming in Petunia hybrida. PNAS Nexus 2023, 2, pgad326. [Google Scholar] [CrossRef] [PubMed]
- Thagun, C.; Imanishi, S.; Kudo, T.; Nakabayashi, R.; Ohyama, K.; Mori, T.; Kawamoto, K.; Nakamura, Y.; Katayama, M.; Nonaka, S.; et al. Jasmonate-Responsive ERF Transcription Factors Regulate Steroidal Glycoalkaloid Biosynthesis in Tomato. Plant Cell Physiol. 2016, 57, 961–975. [Google Scholar] [CrossRef]
- Li, A.; Du, Q.; Zeng, Y.; Yang, R.; Ge, L.; Zhu, Z.; Li, C.; Tan, X. Light Regulated CoWRKY15 Acts on CoSQS Promoter to Promote Squalene Synthesis in Camellia oleifera Seeds. Int. J. Mol. Sci. 2024, 25, 1134. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, S.R.; Dwivedi, V.; Rai, A.; Pal, S.; Shasany, A.K.; Nagegowda, D.A. A WRKY transcription factor from Withania somnifera regulates triterpenoid withanolide accumulation and biotic stress tolerance through modulation of phytosterol and defense pathways. New Phytol. 2017, 215, 1115–1131. [Google Scholar] [CrossRef]
- Zhang, F.; Kong, C.; Ma, Z.; Chen, W.; Li, Y.; Lou, H.; Wu, J. Molecular characterization and transcriptional regulation analysis of the Torreya grandis squalene synthase gene involved in sitosterol biosynthesis and drought response. Front. Plant Sci. 2023, 14, 1136643. [Google Scholar] [CrossRef]
- Huang, D.; Li, J.; Chen, J.; Yao, S.; Li, L.; Huang, R.; Tan, Y.; Ming, R.; Huang, Y. Genome-wide identification and characterization of the JAZ gene family in Gynostemma pentaphyllum reveals the COI1/JAZ/MYC2 complex potential involved in the regulation of the MeJA-induced gypenoside biosynthesis. Plant Physiol. Biochem. 2024, 214, 108952. [Google Scholar] [CrossRef]
- Gong, W.S.Q.; Ji, K.; Gong, S.; Wang, L.; Chen, L.; Zhang, J.; Yuan, D. Full-Length Transcriptome from Camellia oleifera Seed Provides Insight into the Transcript Variants Involved in Oil Biosynthesis. J. Agric. Food Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.F.M. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.I.A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hellens, R.P.; Allan, A.C.; Friel, E.N.; Bolitho, K.; Grafton, K.; Templeton, M.D.; Karunairetnam, S.; Gleave, A.P.; Laing, W.A. Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 2005, 1, 13. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, X.; Yu, A.; Li, P.; Zhang, M.; Lv, Y.; Xu, D.; Liu, A. Comparative Transcriptomic Analysis Reveals the Potential Molecular Mechanism Underlying Squalene Biosynthesis in Developing Seeds of Oil-Tea (Camellia oleifera). Int. J. Mol. Sci. 2025, 26, 5465. https://doi.org/10.3390/ijms26125465
Gu X, Yu A, Li P, Zhang M, Lv Y, Xu D, Liu A. Comparative Transcriptomic Analysis Reveals the Potential Molecular Mechanism Underlying Squalene Biosynthesis in Developing Seeds of Oil-Tea (Camellia oleifera). International Journal of Molecular Sciences. 2025; 26(12):5465. https://doi.org/10.3390/ijms26125465
Chicago/Turabian StyleGu, Xu, Anmin Yu, Ping Li, Meihong Zhang, Ya Lv, Debing Xu, and Aizhong Liu. 2025. "Comparative Transcriptomic Analysis Reveals the Potential Molecular Mechanism Underlying Squalene Biosynthesis in Developing Seeds of Oil-Tea (Camellia oleifera)" International Journal of Molecular Sciences 26, no. 12: 5465. https://doi.org/10.3390/ijms26125465
APA StyleGu, X., Yu, A., Li, P., Zhang, M., Lv, Y., Xu, D., & Liu, A. (2025). Comparative Transcriptomic Analysis Reveals the Potential Molecular Mechanism Underlying Squalene Biosynthesis in Developing Seeds of Oil-Tea (Camellia oleifera). International Journal of Molecular Sciences, 26(12), 5465. https://doi.org/10.3390/ijms26125465