
Correlation of Coding and Non-Coding RNAs on the Fat Deposition of Yaks Under Different Feeding Systems

 , ,
, ,
Abstract
1. Introduction
2. Results
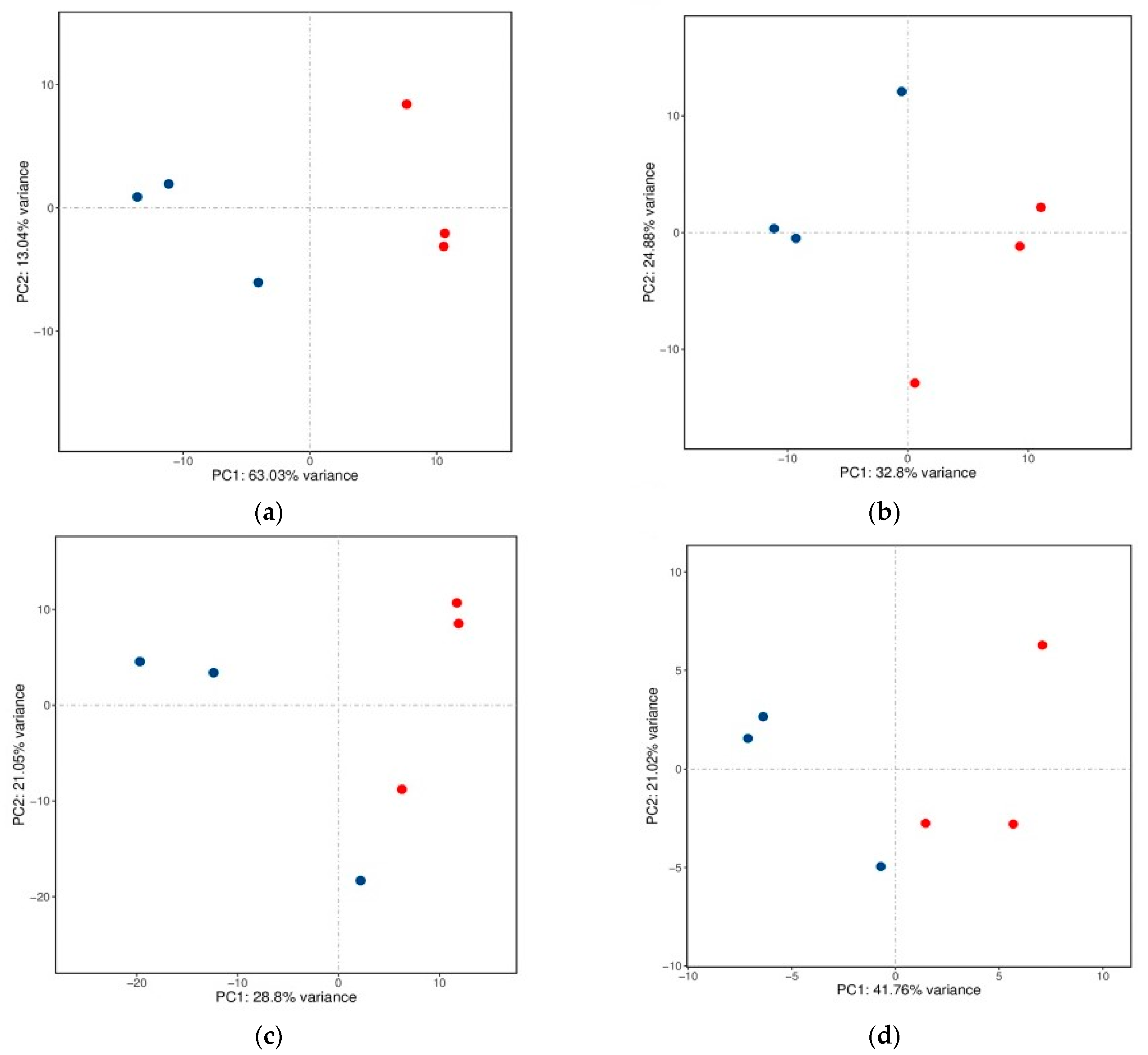
2.1. Overview of Sequencing Data from Yak Subcutaneous Fat
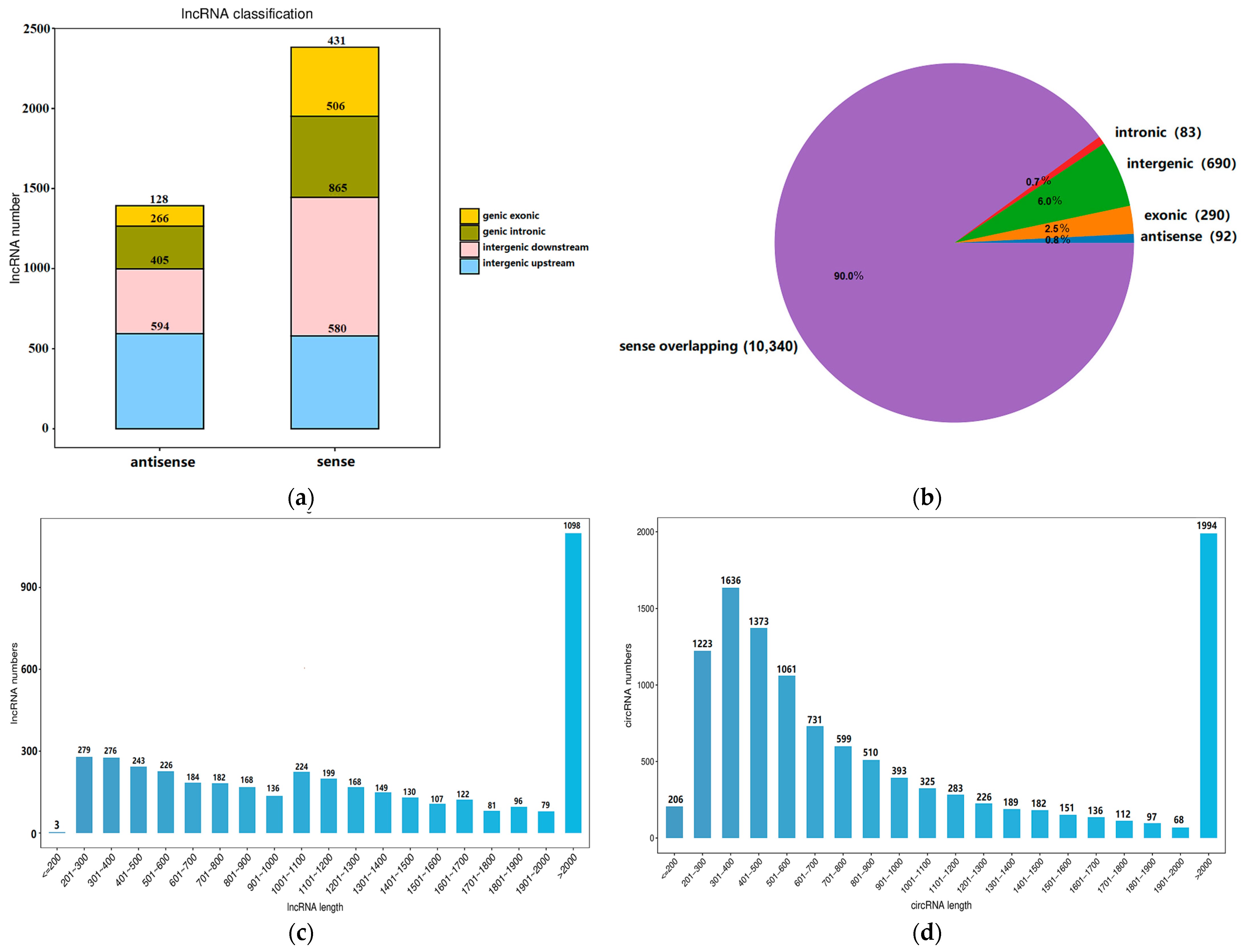
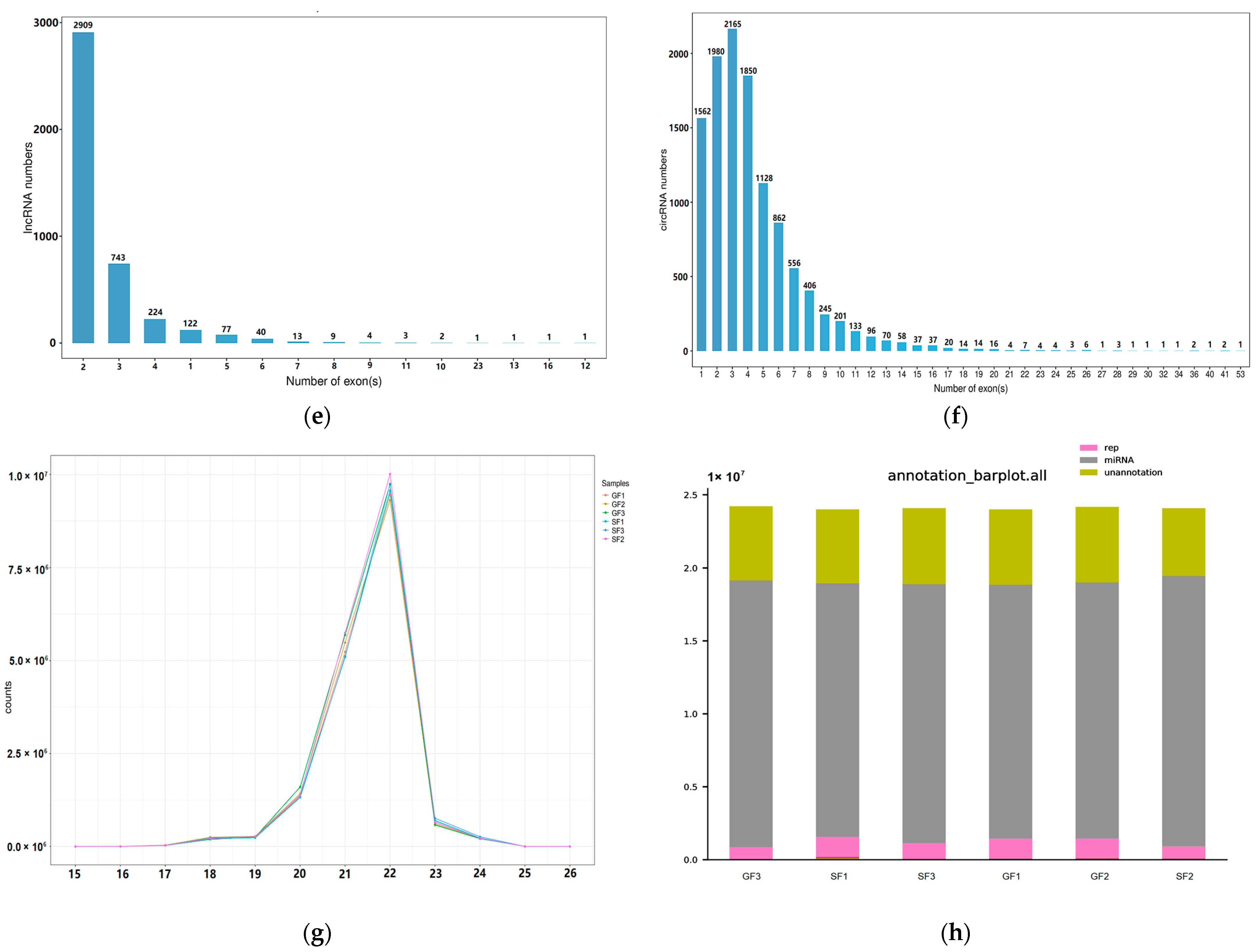
2.2. Properties of the RNAs in Yak Subcutaneous Fat
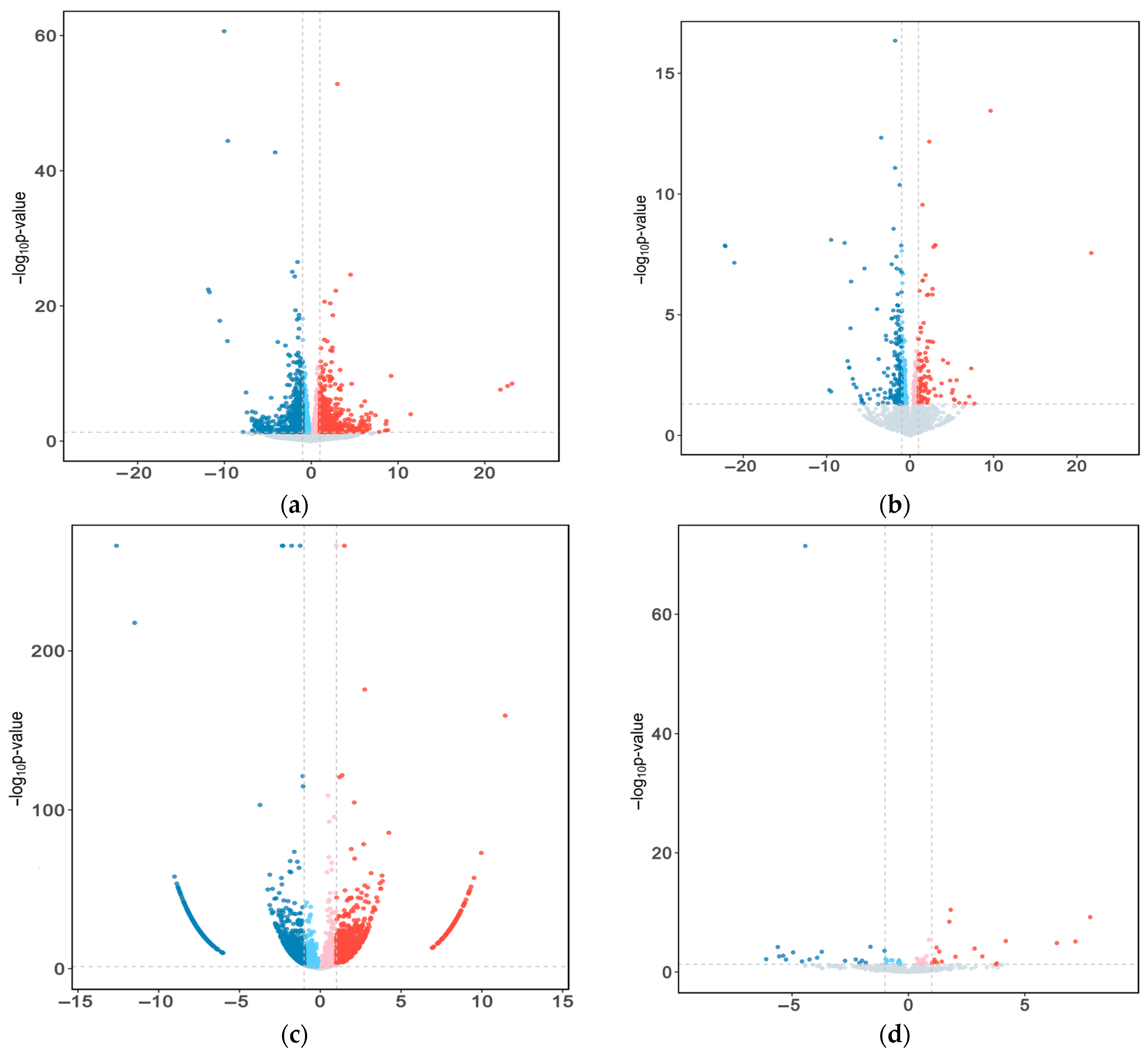
2.3. Differential Expression RNAs in Yak Subcutaneous Fat
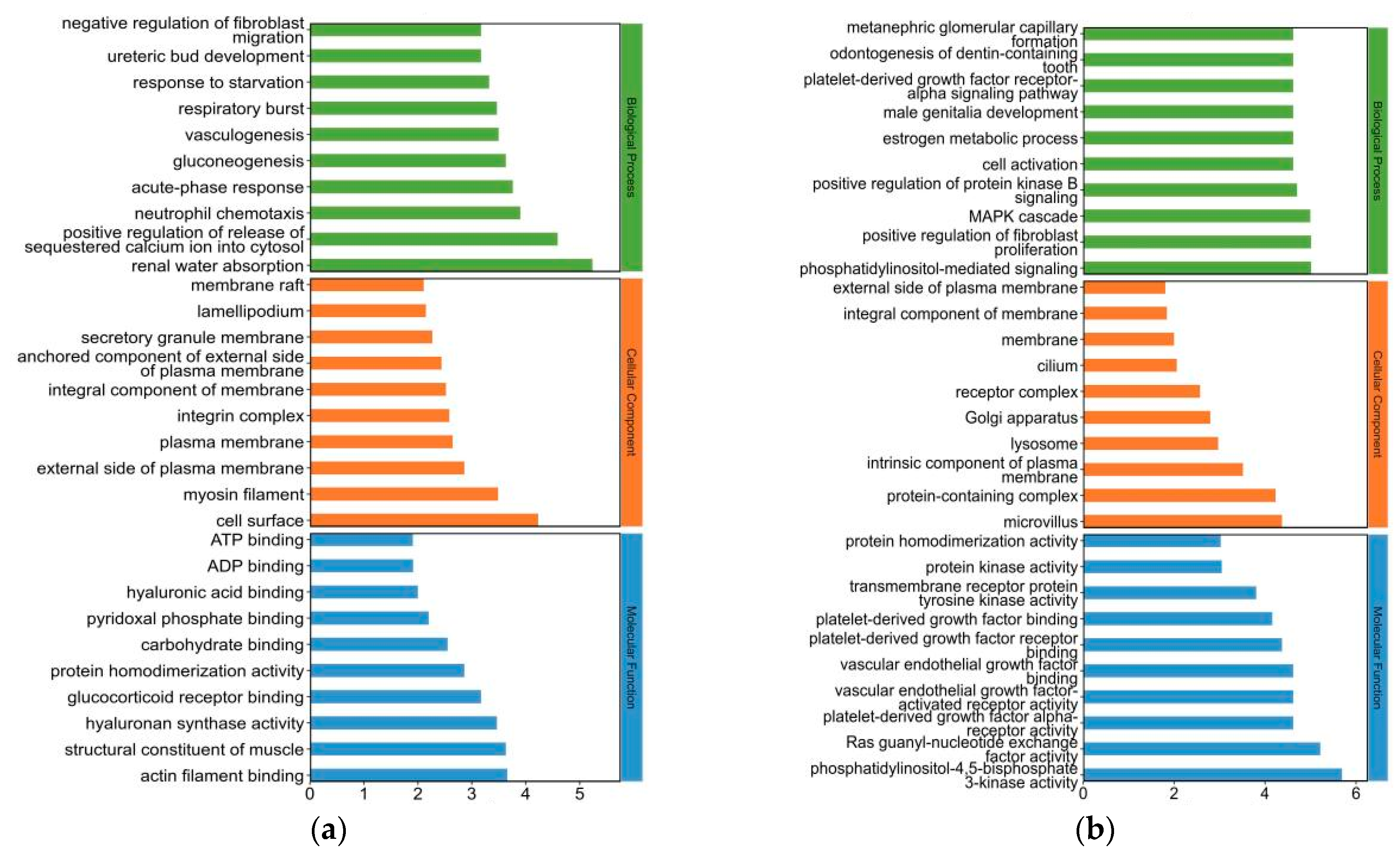
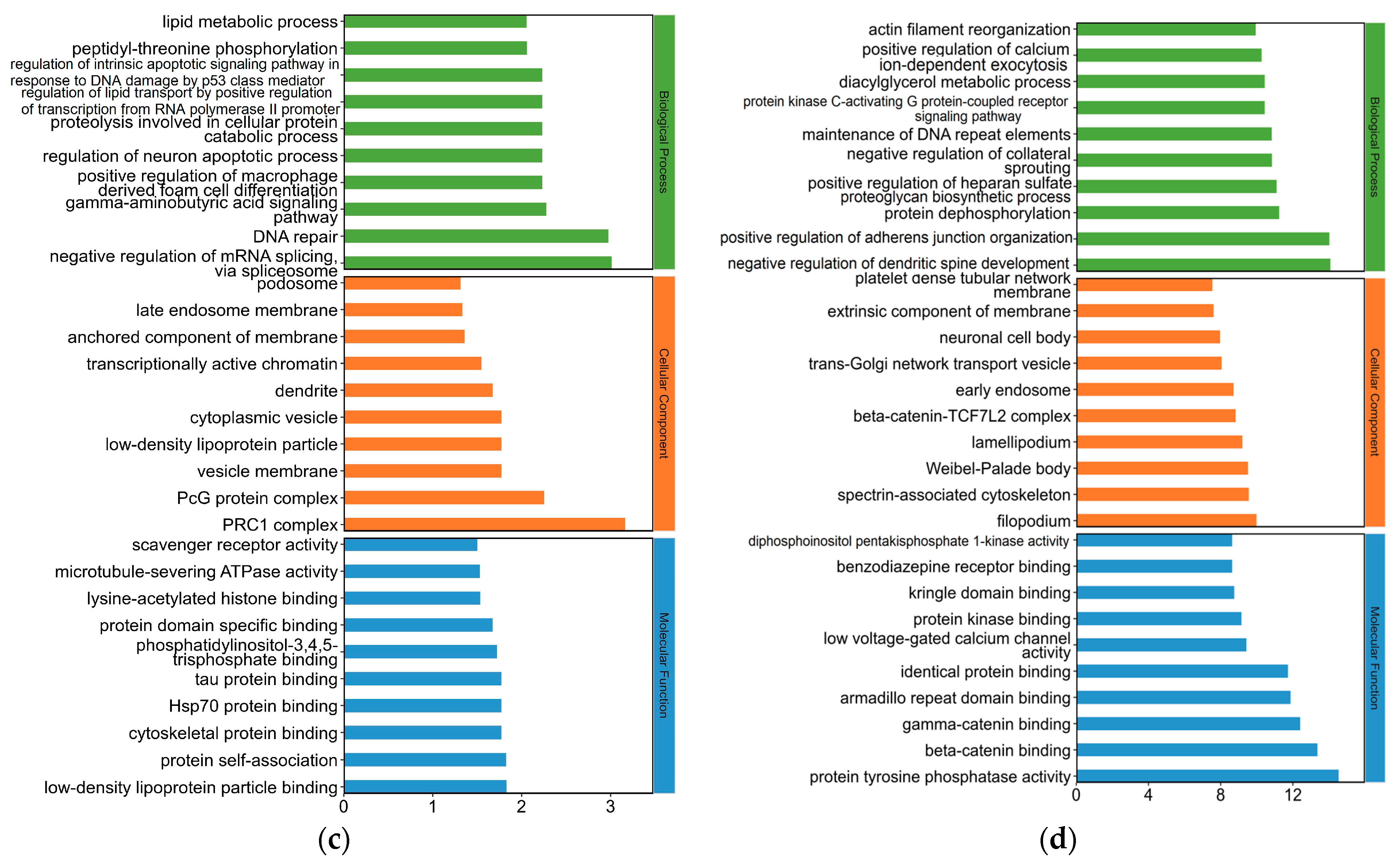
2.4. Gene Ontology (GO) Analysis for Differentially Expressed (DE) mRNAs in Yak Subcutaneous Fat
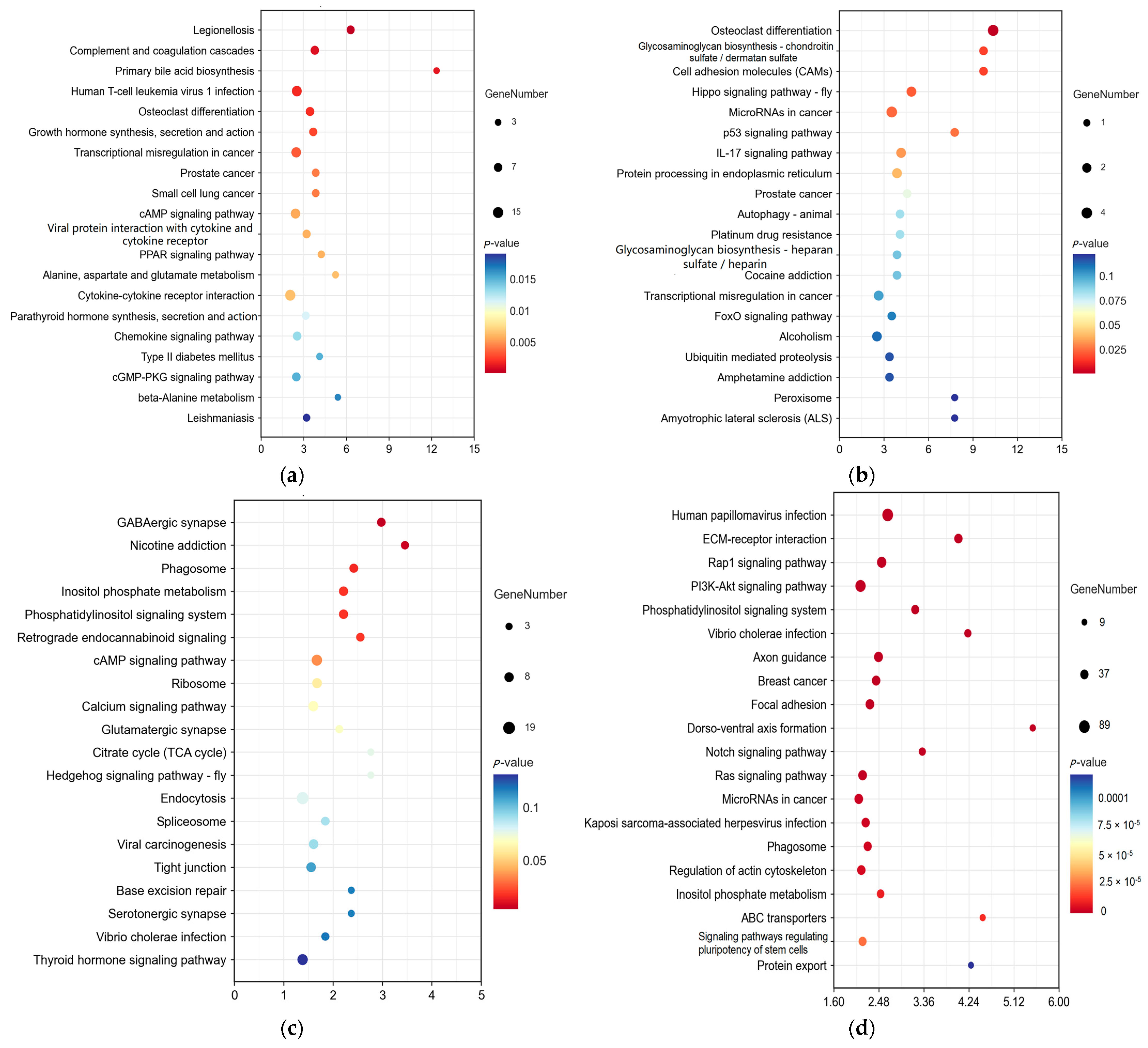
2.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis for DE RNAs in Yak Subcutaneous Fat
2.6. Co-Expression RNA (CeRNA) Regulatory Network for DE RNAs in Yak Subcutaneous Fat
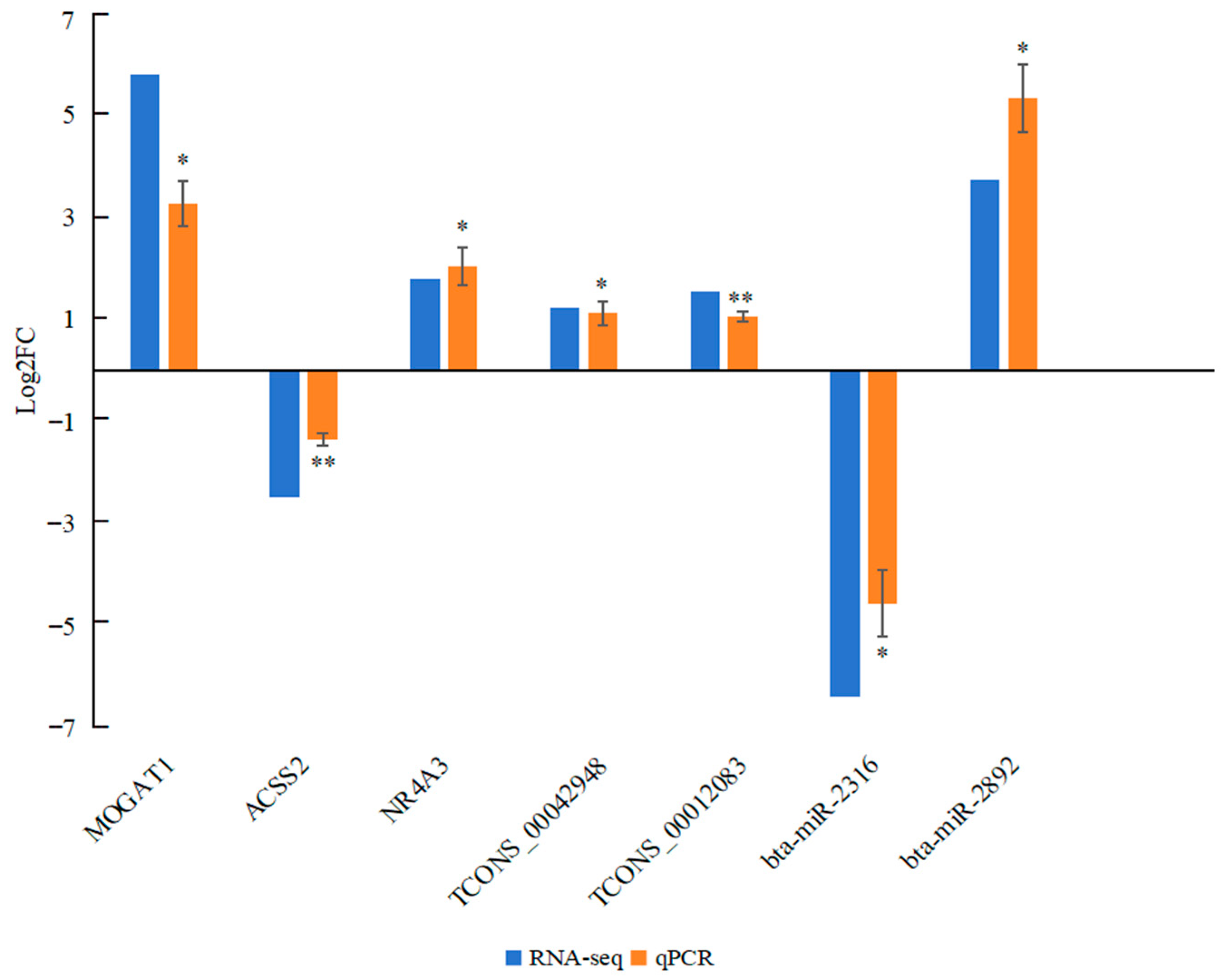
2.7. Comparative Results of Transcriptome and Quantitative Reverse-Transcription PCR (qPCR) for RNAs
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. Total RNA Extraction
4.3. Sequencing and Raw Data Analysis
4.3.1. Sequential Detection
4.3.2. Raw Data Quality Control, Comparison, and Stitching
4.3.3. Identification of lncRNAs and circRNAs
4.3.4. Identification and Prediction of miRNAs
4.3.5. Differentially Expressed RNA Analysis and Target Genes of DE miRNA Prediction
4.3.6. Functional Enrichment Analysis
4.3.7. Co-Expression RNA (CeRNA) Networks’ Construction
4.4. qPCR RNA Expression Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.L.; Han, L.; Ma, X.L.; Yu, Q.L.; Zhao, S.N. Effect of mitochondrial apoptotic activation through the mitochondrial membrane permeability transition pore on yak meat tenderness during postmortem aging. Food Chem. 2017, 234, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zou, H.W.; Wang, Z.S.; Cao, B.H.; Peng, Q.H.; Jing, X.P.; Wang, Y.X.; Shao, Y.Q.; Pei, Z.X.; Zhang, X.F.; et al. Nutritional interventions improved rumen functions and promoted compensatory growth of growth-retarded yaks as revealedby integrated transcripts and microbiome analyses. Front. Microbiol. 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.; Jeong, J.Y.; Vu, T.T.T.; Piao, M.Y.; Kang, H.J. Effects of castration on the adiposity and expression of lipid metabolism genes in various fat depots of Korean cattle. Livest. Sci. 2014, 168, 168–176. [Google Scholar] [CrossRef]
- Gui, L.S.; Hong, J.Y.; Raza, S.H.A.; Zan, L. Genetic variants in SIRT3 transcriptional regulatory region affect promoter activity and fat deposition in three cattle breeds. Mol. Cell. Probes 2007, 32, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Bucław, M.; Majewska, D.; Szczerbiňska, D.; Ligocki, M. The infuence of age and gender on emu (Dromaius novaehollandiae) fat. Sci. Rep. 2020, 10, 11082. [Google Scholar] [CrossRef]
- Xiong, L.; Pei, J.; Bao, P.; Wang, X.; Guo, S.; Cao, M.; Kang, Y.; Yan, P.; Guo, X. The Effect of the Feeding System on Fat Deposition in Yak Subcutaneous Fat. Int. J. Mol. Sci. 2023, 24, 7381. [Google Scholar] [CrossRef]
- Gui, L.; Raza, S.H.A.; Ma, B.; Easa, A.A.; Althobaiti, F.; Shukry, M.; Alotaibi, M.A.; Al Hazani, T.M.I.; Dawood, M.A.O.; Khan, R.; et al. CEBPbeta binding directly to the promoter region drives CEBP transcription and improves FABP4 transcriptional activity in adipose tissue of yak (Bos grunniens). Res. Veter Sci. 2021, 141, 174–179. [Google Scholar] [CrossRef]
- Sun, X.; Li, M.; Sun, Y.; Cai, H.; Lan, X.; Huang, Y.; Bai, Y.; Qi, X.; Chen, H. The developmental transcriptome sequencing of bovine skeletal muscle reveals a long noncoding RNA, lncMD, promotes muscle differentiation by sponging miR-125b. Biochim. Biophys. Acta 2016, 1863, 2835–2845. [Google Scholar] [CrossRef]
- Ding, Z.; Sun, D.; Han, J.; Shen, L.; Yang, F.; Sah, S.; Sui, X.Y.; Wu, G.H. Novel noncoding RNA circPTK2 regulates lipolysis and adipogenesis in cachexia. Mol. Metab. 2021, 53, 101310. [Google Scholar] [CrossRef]
- Wang, S.; Pan, C.; Sheng, H.; Yang, M.; Yang, C.; Feng, X.; Hu, C.; Ma, Y. Construction of a molecular regulatory network related to fat deposition by multi-tissue transcriptome sequencing of Jiaxian red cattle. iScience 2023, 26, 108346. [Google Scholar] [CrossRef]
- Tan, L.; Chen, Z.; Teng, M.; Chen, B.; Xu, H.Q. Genome-wide analysis of mRNAs, lncRNAs, and circRNAs during intramuscular adipogenesis in Chinese Guizhou Congjiang pigs. PLoS ONE 2022, 17, e0261293. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; Timoteo, G.D.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Han, K.; Zhang, G.X.; Wang, J.Y.; Xie, K.Z.; Xue, Q. Genome-wide analysis of lncRNA and mRNA expression during differentiation of abdominal preadipocytes in the chicken. G3 2017, 7, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhao, J.H.; Li, F.; Aloufi, B.H.; Alshammari, A.M.; Ma, Y. Weighted gene coexpression network analysis revealed that circMARK3 is a potential circRNA affects fat deposition in buffalo. Front. Vet. Sci. 2022, 9, 946447. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Wang, X.; Wan, Y.; Zhang, Z.; Zhang, W.; Zan, L.; Cheng, G. Bta-miR-493 Inhibits Bovine Preadipocytes Differentiation by Targeting BMPR1A via the TGFβ/BMP and p38MAPK Signaling Pathways. J. Agric. Food Chem. 2022, 70, 14641–14653. [Google Scholar] [CrossRef]
- Li, M.X.; Sun, X.M.; Cai, H.F.; Sun, Y.J.; Plath, M.; Li, C.J.; Lan, X.Y.; Lei, C.Z.; Lin, F.P.; Bai, Y.Y.; et al. Long non-coding RNA ADNCR suppresses adipogenic differentiation by targeting miR-204. Biochim. Biophys. Acta 2016, 1859, 871–882. [Google Scholar] [CrossRef]
- Jiang, R.; Li, H.; Yang, J.M.; Shen, X.M.; Song, C.C.; Yang, Z.X.; Wang, X.G.; Huang, Y.Z.; Lan, X.Y.; Lei, C.Z.; et al. circRNA Profiling Reveals an Abundant circFUT10 that Promotes Adipocyte Proliferation and Inhibits Adipocyte Differentiation via Sponging let-7. Mol. Ther. Nucleic Acids 2020, 20, 491–501. [Google Scholar] [CrossRef]
- Schäff, C.; Börner, S.; Hacke, S.; Kautzsch, U.; Albrecht, D.; Hammon, H.M. Increased anaplerosis, TCA cycling, and oxidative phosphorylation in the liver of dairy cows with intensive body fat mobilization during early lactation. J. Proteome Res. 2012, 11, 5503–5514. [Google Scholar] [CrossRef]
- Veshkini, A.M.; Hammon, H.; Sauerwein, H.; Tröscher, A.; Viala, D.; Delosière, M. Longitudinal liver proteome profiling in dairy cows during the transition from gestation to lactation: Investigating metabolic adaptations and their interactions with fatty acids supplementation via repeated measurements ANOVA-simultaneous component analysis. J. Proteom. 2022, 252, 104435. [Google Scholar] [CrossRef]
- Xu, M.; Liu, X.; Bao, P.; Wang, Y.; Zhu, X.; Liu, Y.; Ni, X.; Lu, J. Skeletal Muscle CSE Deficiency Leads to Insulin Resistance in Mice. Antioxidants 2022, 11, 2216. [Google Scholar] [CrossRef]
- Shi, L.; Liu, L.; Ma, Z.; Lv, X.; Li, C.; Xu, L.; Han, B.; Li, Y.; Zhao, F.; Yang, Y.; et al. Identification of genetic associations of ECHS1 gene with milk fatty acid traits in dairy cattle. Anim. Genet. 2019, 50, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Iqbal, A.; Wang, M.Y.; Li, X.H.; Fang, X.B.; Yu, H.B.; Zhao, Z.H. Transcriptomic Analysis of Short/Branched-Chain Acyl-Coenzyme a Dehydrogenase Knocked Out bMECs Revealed Its Regulatory Effect on Lipid Metabolism. Front. Veter Sci. 2021, 8, 744287. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Liu, L.; Lv, X.; Ma, Z.; Yang, Y.; Li, Y.; Zhao, F.; Sun, D.; Han, B. Polymorphisms and genetic effects of PRLR, MOGAT1, MINPP1 and CHUK genes on milk fatty acid traits in Chinese Holstein. BMC Genet. 2019, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Grala, T.M.; Roche, J.R.; Phyn, C.V.; Rius, A.G.; Boyle, R.H.; Snell, R.G.; Kay, J.K. Expression of key lipid metabolism genes in adipose tissue is not altered by once-daily milking during a feed restriction of grazing dairy cows. J. Dairy Sci. 2013, 96, 7753–7764. [Google Scholar] [CrossRef]
- Romao, J.M.; He, M.L.; McAllister, T.A.; Guan, L.L. Effect of age on bovine subcutaneous fat proteome: Molecular mechanisms of physiological variations during beef cattle growth. J. Anim. Sci. 2014, 92, 3316–3327. [Google Scholar] [CrossRef]
- Salcedo-Tacuma, D.; Parales-Giron, J.; Prom, C.; Chirivi, M.; Laguna, J.; Lock, A.L.; Contreras, G.A. Transcriptomic profiling of adipose tissue inflammation, remodeling, and lipid metabolism in periparturient dairy cows (Bos taurus). BMC Genom. 2020, 21, 824. [Google Scholar] [CrossRef]
- Lyu, C.C.; Meng, Y.; Che, H.Y.; Suo, J.L.; He, Y.T.; Zheng, Y.; Jiang, H.; Zhang, J.B.; Yuan, B. MSI2 Modulates Unsaturated Fatty Acid Metabolism by Binding FASN in Bovine Mammary Epithelial Cells. J. Agric. Food Chem. 2023, 71, 20359–20371. [Google Scholar] [CrossRef]
- Matosinho, C.G.R.; Fonseca, P.A.S.; Peixoto, M.G.C.D.; Rosse, I.C.; Lopes, F.C.F.; Zózimo, T.; Filho, A.E.V.; Bruneli, F.Â.T.; Carvalho, M.R.S.; Gama, M.A.S. Phenotypic variation in milk fatty acid composition and its association with stearoyl-CoA desaturase 1 (SCD1) gene polymorphisms in Gir cows. J. Anim. Breed Genet. 2023, 140, 532–548. [Google Scholar] [CrossRef]
- Weikard, R.; Hadlich, F.; Kuehn, C. Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genom. 2013, 14, 789. [Google Scholar] [CrossRef]
- Jiang, R.; Li, H.; Huang, Y.Z.; Lan, X.Y.; Lei, C.Z.; Chen, H. Transcriptome profiling of lncRNA related to fat tissues of Qinchuan cattle. Gene 2020, 742, 144587. [Google Scholar] [CrossRef]
- Mu, T.; Hu, H.H.; Ma, Y.F.; Yang, C.Y.; Feng, X.F.; Wang, Y.; Liu, J.M.; Yu, B.J.; Zhang, J.; Gu, Y.L. Identification of critical lncRNAs for milk fat metabolism in dairy cows using WGCNA and the construction of a ceRNAs network. Anim. Genet. 2022, 53, 740–760. [Google Scholar] [CrossRef] [PubMed]
- Nikołajuk, A.; Stefanowicz, M.; Strączkowski, M.; Karczewska-Kupczewska, M. Changes in Adipose Tissue Gene Expression of the Core Components of the Hippo Signaling Pathway in Young Adults with Uncomplicated Overweight or Obesity Following Weight Loss. J. Nutr. 2023, 153, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.Z.; Dong, Y.J. Different expression of lipid metabolism-related genes in shandong black cattle and Luxi cattle based on transcriptome analysis. Sci. Rep. 2020, 10, 21915. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, J.; Zheng, Q.; Wang, S.; Wei, X.; Li, F.; Shang, J.; Lei, C.; Ma, Y. Characterization of Circular RNAs in Chinese Buffalo (Bubalus bubalis) Adipose Tissue: A Focus on Circular RNAs Involved in Fat Deposition. Animals 2019, 9, 403. [Google Scholar] [CrossRef]
- Li, H.; Yang, J.M.; Wei, X.F.; Song, C.C.; Dong, D.; Huang, Y.Z.; Lan, X.Y.; Plath, M.; Lei, C.Z.; Ma, Y.; et al. CircFUT10 reduces proliferation and facilitates differentiation of myoblasts by sponging miR-133a. J. Cell Physiol. 2018, 233, 4643–4651. [Google Scholar] [CrossRef]
- Guo, H.; Khan, R.; Abbas Raza, S.H.; Suhail, S.M.; Khan, H.; Khan, S.B.; Abd El-Aziz, A.H.; Zan, L. RNA-Seq Reveals Function of Bta-miR-149-5p in the Regulation of Bovine Adipocyte Differentiation. Animals 2021, 11, 1207. [Google Scholar] [CrossRef]
- Jin, W.; Dodson, M.V.; Moore, S.S.; Basarab, J.A.; Guan, L.L. Characterization of microRNA expression in bovine adipose tissues: A potential regulatory mechanism of subcutaneous adipose tissue development. BMC Mol. Biol. 2010, 11, 29. [Google Scholar] [CrossRef]
- Pan, C.; Yang, C.; Wang, S.; Ma, Y. Identifying Key Genes and Functionally Enriched Pathways of Diverse Adipose Tissue Types in Cattle. Front. Genet. 2022, 13, 790690. [Google Scholar] [CrossRef]
- He, J.; Si, R.; Wang, Y.; Ji, R.; Ming, L. Lipidomic and proteomic profiling identifies the milk fat globule membrane composition of milk from cows and camels. Food Res. Int. 2024, 179, 113816. [Google Scholar] [CrossRef]
- He, Z.; Wang, X.; Qi, Y.; Zhu, C.; Zhao, Z.; Zhang, X.; Liu, X.; Li, S.; Zhao, F.; Wang, J.; et al. Long-stranded non-coding RNAs temporal-specific expression profiles reveal longissimus dorsi muscle development and intramuscular fat deposition in Tianzhu white yak. J. Anim. Sci. 2023, 101, skad394. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Y.; Wang, D.H.; Kothapalli, K.S.D.; Brenna, J.T. Branched chain fatty acid composition of yak milk and manure during full-lactation and half-lactation. Prostaglandins Leukot Essent Fat. Acids 2019, 150, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Xia, L.; Yu, H.; He, W.; Bai, Z.; Qin, L.; Jiang, P.; Zhao, Y.; Zhao, Z.; Yang, R. Comparative Genome-Wide Alternative Splicing Analysis of Longissimus Dorsi Muscles Between Japanese Black (Wagyu) and Chinese Red Steppes Cattle. Front. Vet. Sci. 2021, 8, 634577. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Kondo, J.; Ishikawa, M.; Yamamoto, T.T. Acetyl-Co A synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem. 2001, 276, 11420–11426. [Google Scholar] [CrossRef]
- Luong, A.; Hannah, V.C.; Brown, M.S.; Goldstein, J.L. Molecular characterization of human acetyl-Co A synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2000, 275, 26458–26466. [Google Scholar] [CrossRef]
- Hudson, N.J.; Reverter, A.; Griffiths, W.J.; Yutuc, E.; Wang, Y.Q.; Jeanes, A.; McWilliam, S.; Pethick, D.W.; Greenwood, P.L. Gene expression identifies metabolic and functional differences between intramuscular and subcutaneous adipocytes in cattle. BMC Genom. 2020, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.N.; Wang, Y.; Xu, Y.O.; Li, Y.C.; Tian, F.; Jiang, M.F. Characterisation of gene expression related to milk fat synthesis in the mammary tissue of lactating yaks. J. Dairy Res. 2017, 84, 283–288. [Google Scholar] [CrossRef]
- Silva, T.D.L.; Gondro, C.; Fonseca, P.A.D.S.; Silva, D.A.S.; Vargas, G.; Neves, H.H.D.R.; Filho, I.C.; Teixeira, C.D.S.; Albuquerque, L.G.D.; Carvalheiro, R. Feet and legs malformation in Nellore cattle: Genetic analysis and prioritization of GWAS results. Front. Genet. 2023, 14, 1118308. [Google Scholar] [CrossRef]
- He, W.; Fang, X.B.; Lu, X.; Liu, Y.; Li, G.H.; Zhao, Z.H.; Li, J.Y.; Yang, R.J. Function Identification of Bovine ACSF3 Gene and Its Association with Lipid Metabolism Traits in Beef Cattle. Front. Vet. Sci. 2022, 8, 766765. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Z.Y.; Zhang, T.; Zhang, L.; Hou, X.; Yan, H.; Wang, L. IRLnc: A novel functional noncoding RNA contributes to intramuscular fat deposition. BMC Genom. 2021, 22, 95. [Google Scholar] [CrossRef]
- Peng, S.Q.; Zhang, X.Y.; Yu, L.L.; Xu, Y.H.; Zhou, Y.; Qian, S.N.; Cao, X.Y.; Ye, X.T.; Yang, J.J.; Jia, W.P.; et al. NF-kB regulates brown adipocyte function through suppression of ANT2. Acta Pharm. Sin. B 2022, 12, 1186–1197. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Sun, K.; An, Y.A.; Gu, X.; Scherer, P.E. VEGF-AExpressing adipose tissue shows rapid beiging and enhanced survival after transplantation and confers IL-4-independent metabolic improvements. Diabetes 2017, 66, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Romao, J.M.; Jin, W.; He, M.; Mcallister, T.; Guan, L.L. MicroRNAs in bovine adipogenesis: Genomic context, expression and function. BMC Genom. 2014, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- MacPhillamy, C.; Ren, Y.; Chen, T.; Hiendleder, S.; Low, W.Y. MicroRNA breed and parent-of-origin effects provide insights into biological pathways differentiating cattle subspecies in fetal liver. Front. Genet. 2023, 14, 1329939. [Google Scholar] [CrossRef] [PubMed]
- GB/T17377-2008; Animal and Vegetable Fats and Oils—Analysis by Gas Chromatography of Methyl Ester of Fattys Acids. General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China; Standardization Administration of the People’s Republic of China: Beijing, China, 2009.
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; Cunto, F.D.; et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011, 147, 344–357. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Description | log2 Fold Change | q-Value |
|---|---|---|---|
| MOGAT1 | Monoacylglycerol O-acyltransferase 1 | −5.78 | 3.76 × 10−2 |
| ELOVL6 | ELOVL fatty acid elongase 6 | 2.44 | 8.33 × 10−12 |
| GPD1 | Glycerol-3-phosphate dehydrogenase 1 | 1.06 | 5.27 × 10−3 |
| APOC3 | Apolipoprotein C3 | 21.80 | 3.11 × 10−6 |
| FASN | Fatty acid synthase | 3.30 | 2.01 × 10−3 |
| LIPK | Lipase family member K | −1.98 | 1.04 × 10−2 |
| LIPA | Lysosomal acid lipase | −1.54 | 5.13 × 10−4 |
| SCD | Stearoyl-CoA desaturase | 2.77 | 1.96 × 10−4 |
| UNC119 | Unc-119 lipid binding chaperone | −1.11 | 1.26 × 10−5 |
| ACSS2 | Acyl-CoA synthetase short-chain family member 2 | 2.49 | 2.68 × 10−16 |
| TNFRSF21 | TNF receptor superfamily member 21 | 1.09 | 9.84 × 10−5 |
| MCAT | Malonyl-CoA-acyl carrier protein transacylase | −0.64 | 1.03 × 10−2 |
| NR4A3 | Nuclear receptor subfamily 4 group A member 3 | −1.79 | 5.18 × 10−5 |
| MAST3 | Microtubule-associated serine/threonine kinase 3 | −1.21 | 3.14 × 10−4 |
| SHOX | Short-stature homeobox | 6.11 | 1.73 × 10−2 |
| Pathway | List Hits | p-Value | Enrichment Score |
|---|---|---|---|
| PPAR signaling pathway | 5 | 5.88 × 10−3 | 4.24 |
| PI3K–Akt signaling pathway | 76 | 1.91 × 10−10 | 2.12 |
| cGMP–PKG signaling pathway | 8 | 1.50 × 10−2 | 2.48 |
| lncRNA ID | Direction | lncRNA Symbol | Target mRNA |
|---|---|---|---|
| TCONS00042948 | Sense | XLOC012001 | ENSBGRT00000031389 |
| TCONS00012083 | Sense | XLOC003393 | ENSBGRT00000038536 |
| circRNA | circRNA Length | Type | Transcript Position | Gene |
|---|---|---|---|---|
| circRNA03372 | 796 | Exonic | 16:60894938_60921423 | SH2B3 |
| circRNA06073 | 39,774 | Sense-overlapping | 24:7788337_7802802 | STK19 |
| circRNA06078 | 44,254 | Sense-overlapping | 24:7788337_7802802 | STK19 |
| circRNA00274 | 380 | Sense-overlapping | 1:77318523_77430765 | PAK2 |
| circRNA00679 | 548 | Sense-overlapping | 1:172082712_172194902 | KAT2B |
| circRNA01768 | 1345 | Sense-overlapping | 12:1412261_1546561 | USP6NL |
| circRNA05277 | 558 | Sense-overlapping | 20:65208274_65233932 | LIPE |
| circRNA06073 | 39,774 | Sense-overlapping | 24:7788337_7802802 | STK19 |
| circRNA06078 | 44,386 | Sense-overlapping | 24:7788337_7802802 | STK19 |
| circRNA06528 | 699 | Sense-overlapping | 26:7744912_7746357 | ZNHIT1 |
| circRNA07560 | 366 | Intronic | 3:74687137_74695810 | RABGGTB |
| circRNA07750 | 2067 | Sense-overlapping | 3:109743508_109751164 | TOE1 |
| circRNA08402 | 1018 | Sense-overlapping | 5:11880919_12172221 | PLEKHA5 |
| circRNA10133 | 432 | Exonic | 8:47906904_47933260 | CSNK1G2 |
| circRNA10476 | 28,908 | Sense-overlapping | 9:8107091_8173022 | SPOUT1 |
| circRNA11309 | 53,606 | Sense-overlapping | X:93853225_93854235 | UXT |
| miRNA ID | Sequence | miRNA Length | Target mRNA (Predict) |
|---|---|---|---|
| bta-miR-2892 | ACTCCGGCCTGGACTGCGGCGGG | 23 | ACSF3 |
| bta-miR-2316 | GGCGACGGAGGCGCGACCCCCC | 22 | UNC119, ACSS2, TNFRSF21, MCAT, NR4A3, MAST3, SHOX |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, L.; Pei, J.; Guo, S.; Cao, M.; Ding, Z.; Kang, Y.; Wu, X.; Guo, X. Correlation of Coding and Non-Coding RNAs on the Fat Deposition of Yaks Under Different Feeding Systems. Int. J. Mol. Sci. 2025, 26, 5359. https://doi.org/10.3390/ijms26115359
Xiong L, Pei J, Guo S, Cao M, Ding Z, Kang Y, Wu X, Guo X. Correlation of Coding and Non-Coding RNAs on the Fat Deposition of Yaks Under Different Feeding Systems. International Journal of Molecular Sciences. 2025; 26(11):5359. https://doi.org/10.3390/ijms26115359
Chicago/Turabian StyleXiong, Lin, Jie Pei, Shaoke Guo, Mengli Cao, Zhiqiang Ding, Yandong Kang, Xiaoyun Wu, and Xian Guo. 2025. "Correlation of Coding and Non-Coding RNAs on the Fat Deposition of Yaks Under Different Feeding Systems" International Journal of Molecular Sciences 26, no. 11: 5359. https://doi.org/10.3390/ijms26115359
APA StyleXiong, L., Pei, J., Guo, S., Cao, M., Ding, Z., Kang, Y., Wu, X., & Guo, X. (2025). Correlation of Coding and Non-Coding RNAs on the Fat Deposition of Yaks Under Different Feeding Systems. International Journal of Molecular Sciences, 26(11), 5359. https://doi.org/10.3390/ijms26115359