
scDown: A Pipeline for Single-Cell RNA-Seq Downstream Analysis

 , ,
, , 


Abstract
1. Introduction
2. Results
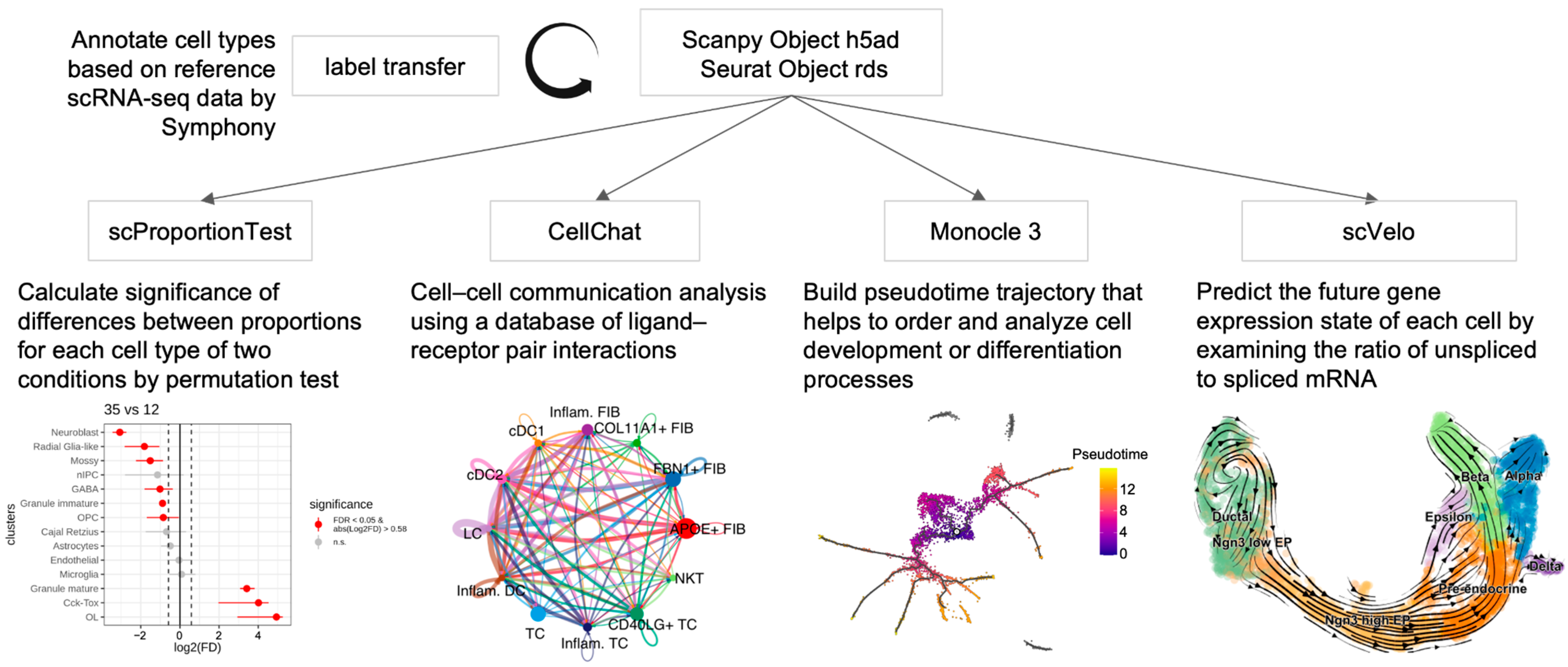
2.1. Functional Features of scDown
2.2. Case Study—Application to the Published Dataset
3. Discussion
4. Materials and Methods
4.1. Case Study Datasets
4.2. Cell Proportion Differentiation Analysis
4.3. Cell–Cell Communication Analysis
4.4. Pseudotime Analysis
4.5. RNA Velocity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valenzi, E.; Tabib, T.; Papazoglou, A.; Sembrat, J.; Bittar, H.E.T.; Rojas, M.; Lafyatis, R. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Immunol. 2021, 12, 595811. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma Heterogeneity at Single Cell Resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Sánchez, E.; Carbayo, Á.; Valle-Tamayo, N.; Muñoz, L.; Aumatell, J.; Torres, S.; Rubio-Guerra, S.; García-Castro, J.; Selma-González, J.; Alcolea, D.; et al. Single-Cell RNA Sequencing Highlights the Role of Distinct Natural Killer Subsets in Sporadic Amyotrophic Lateral Sclerosis. J. Neuroinflamm. 2025, 22, 15. [Google Scholar] [CrossRef]
- Miller, S.A.; Policastro, R.A.; Sriramkumar, S.; Lai, T.; Huntington, T.D.; Ladaika, C.A.; Kim, D.; Hao, C.; Zentner, G.E.; O’Hagan, H.M. LSD1 and Aberrant DNA Methylation Mediate Persistence of Enteroendocrine Progenitors That Support BRAF-Mutant Colorectal Cancer. Cancer Res. 2021, 81, 3791–3805. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for Systematic Analysis of Cell–Cell Communication from Single-Cell Transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed Graph Embedding Resolves Complex Single-Cell Trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Enver, T. Single-Cell Entropy for Accurate Estimation of Differentiation Potency from a Cell’s Transcriptome. Nat. Commun. 2017, 8, 15599. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The Single-Cell Transcriptional Landscape of Mammalian Organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Manno, G.L.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA Velocity of Single Cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph Abstraction Reconciles Clustering with Trajectory Inference through a Topology Preserving Map of Single Cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA Velocity to Transient Cell States through Dynamical Modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Polański, K.; Young, M.D.; Miao, Z.; Meyer, K.B.; Teichmann, S.A.; Park, J.-E. BBKNN: Fast Batch Alignment of Single Cell Transcriptomes. Bioinformatics 2019, 36, 964–965. [Google Scholar] [CrossRef]
- Kang, J.B.; Nathan, A.; Weinand, K.; Zhang, F.; Millard, N.; Rumker, L.; Moody, D.B.; Korsunsky, I.; Raychaudhuri, S. Efficient and Precise Single-Cell Reference Atlas Mapping with Symphony. Nat. Commun. 2021, 12, 5890. [Google Scholar] [CrossRef]
- Dias, C.; Mo, A.; Cai, C.; Sun, L.; Cabral, K.; Brownstein, C.A.; Rockowitz, S.; Walsh, C.A. Cell-Type-Specific Effects of Autism-Associated 15q Duplication Syndrome in the Human Brain. Am. J. Hum. Genet. 2024, 111, 1544–1558. [Google Scholar] [CrossRef]
- Crowell, H.L.; Soneson, C.; Germain, P.-L.; Calini, D.; Collin, L.; Raposo, C.; Malhotra, D.; Robinson, M.D. Muscat Detects Subpopulation-Specific State Transitions from Multi-Sample Multi-Condition Single-Cell Transcriptomics Data. Nat. Commun. 2020, 11, 6077. [Google Scholar] [CrossRef]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of Autism: Increased Ratio of Excitation/Inhibition in Key Neural Systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Culotta, L.; Penzes, P. Exploring the Mechanisms Underlying Excitation/Inhibition Imbalance in Human IPSC-Derived Models of ASD. Mol. Autism 2020, 11, 32. [Google Scholar] [CrossRef]
- Wei, W.; Xia, X.; Li, T.; Chen, Q.; Feng, X. Shaoxia: A Web-Based Interactive Analysis Platform for Single Cell RNA Sequencing Data. BMC Genom. 2024, 25, 402. [Google Scholar] [CrossRef]
- Büttner, M.; Ostner, J.; Müller, C.L.; Theis, F.J.; Schubert, B. ScCODA Is a Bayesian Model for Compositional Single-Cell Data Analysis. Nat. Commun. 2021, 12, 6876. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Aguilar, S.; Alame, M.; Kon-Sun-Tack, F.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of Intercellular Networks from Single-Cell Transcriptomics. Nucleic Acids Res. 2020, 48, e55. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling Intercellular Communication by Linking Ligands to Target Genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Denisenko, E.; Ong, H.T.; Ramilowski, J.A.; Forrest, A.R.R. Predicting Cell-to-Cell Communication Networks Using NATMI. Nat. Commun. 2020, 11, 5011. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring Cell–Cell Communication from Combined Expression of Multi-Subunit Ligand–Receptor Complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell Lineage and Pseudotime Inference for Single-Cell Transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Setty, M.; Kiseliovas, V.; Levine, J.; Gayoso, A.; Mazutis, L.; Pe’er, D. Characterization of Cell Fate Probabilities in Single-Cell Data with Palantir. Nat. Biotechnol. 2019, 37, 451–460. [Google Scholar] [CrossRef]
- Cannoodt, R.; Saelens, W.; Sichien, D.; Tavernier, S.; Janssens, S.; Guilliams, M.; Lambrecht, B.; Preter, K.D.; Saeys, Y. SCORPIUS Improves Trajectory Inference and Identifies Novel Modules in Dendritic Cell Development. bioRxiv 2016, 079509. [Google Scholar] [CrossRef]
- Bergen, V.; Soldatov, R.A.; Kharchenko, P.V.; Theis, F.J. RNA Velocity—Current Challenges and Future Perspectives. Mol. Syst. Biol. 2021, 17, MSB202110282. [Google Scholar] [CrossRef]
- Cui, H.; Maan, H.; Vladoiu, M.C.; Zhang, J.; Taylor, M.D.; Wang, B. DeepVelo: Deep Learning Extends RNA Velocity to Multi-Lineage Systems with Cell-Specific Kinetics. Genome Biol. 2024, 25, 27. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhang, Y.; Martin-Rufino, J.D.; Weng, C.; Hosseinzadeh, S.; Yang, D.; Pogson, A.N.; Hein, M.Y.; Hoi Joseph Min, K.; Wang, L.; et al. Mapping Transcriptomic Vector Fields of Single Cells. Cell 2022, 185, 690–711. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Dou, J.; Tan, Y.; Kock, K.H.; Wang, J.; Cheng, X.; Tan, L.M.; Han, K.Y.; Hon, C.-C.; Park, W.-Y.; Shin, J.W.; et al. Single-Nucleotide Variant Calling in Single-Cell Sequencing Data with Monopogen. Nat. Biotechnol. 2024, 42, 803–812. [Google Scholar] [CrossRef]
- Muyas, F.; Sauer, C.M.; Valle-Inclán, J.E.; Li, R.; Rahbari, R.; Mitchell, T.J.; Hormoz, S.; Cortés-Ciriano, I. De Novo Detection of Somatic Mutations in High-Throughput Single-Cell Profiling Data Sets. Nat. Biotechnol. 2024, 42, 758–767. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Module | Function | Function Description | Required scRNA-Seq Data | ||
|---|---|---|---|---|---|
| Unannotated Data | Annotated Data | ||||
| One Condition | Two or More Conditions | ||||
| 1 | doTransferLabel | Automated cell type annotation by transferring cell type annotation from a reference Seurat object to a query unannotated Seurat object | ✓ | ||
| 2 | run_scproportion | Statistically assess the significance of differences in cell type proportions for different condition comparisons | ✓ | ||
| 3 | run_cellchatV2 | Perform comprehensive intercellular communications analysis based on ligand–receptor pair interactions across cell types using CellChat. | ✓ | ✓ | |
| 4 | run_monocle3 | Construct pseudotime trajectories to model the progression of cellular differentiation utilizing monocle3 | ✓ | ✓ | |
| 5 | run_scvelo | Incorporate spliced and unspliced counts using velocyto.R and estimate RNA velocity utilizing velociraptor | ✓ | ✓ | |
| run_scvelo_full | Conduct RNA velocity analysis with enhanced visualizations and PAGA trajectory inference using scVelo | ✓ | ✓ | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Ma, Q.; Cai, C.; Labaf, M.; Jain, A.; Dias, C.; Rockowitz, S.; Sliz, P. scDown: A Pipeline for Single-Cell RNA-Seq Downstream Analysis. Int. J. Mol. Sci. 2025, 26, 5297. https://doi.org/10.3390/ijms26115297
Sun L, Ma Q, Cai C, Labaf M, Jain A, Dias C, Rockowitz S, Sliz P. scDown: A Pipeline for Single-Cell RNA-Seq Downstream Analysis. International Journal of Molecular Sciences. 2025; 26(11):5297. https://doi.org/10.3390/ijms26115297
Chicago/Turabian StyleSun, Liang, Qianyi Ma, Chunhui Cai, Maryam Labaf, Ashish Jain, Caroline Dias, Shira Rockowitz, and Piotr Sliz. 2025. "scDown: A Pipeline for Single-Cell RNA-Seq Downstream Analysis" International Journal of Molecular Sciences 26, no. 11: 5297. https://doi.org/10.3390/ijms26115297
APA StyleSun, L., Ma, Q., Cai, C., Labaf, M., Jain, A., Dias, C., Rockowitz, S., & Sliz, P. (2025). scDown: A Pipeline for Single-Cell RNA-Seq Downstream Analysis. International Journal of Molecular Sciences, 26(11), 5297. https://doi.org/10.3390/ijms26115297