

Iron-Mediated Overexpression of Amyloid Precursor Protein via Iron Responsive mRNA in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure and Function of IREs
2.1. IRE Structure
2.2. IRE Conservation
3. IRE/IRP Functional Interaction
4. Fe2+ Sensing IRE mRNA to Influence Protein Binding
5. Fe2+-Induced APP mRNA Translation
6. Brain Iron Transport and Regulation
7. Role of Iron in the Occurrence of Alzheimer’s Disease
8. Small Molecule Therapeutic Targeting mRNA for AD
9. Metal Chelator Therapeutics in AD
10. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Katsumata, Y.; Wu, X.; Aung, K.Z.; Fardo, D.W.; Forrest, S.L.; Nelson, P.T. Amyloid-beta predominant Alzheimer’s disease neuropathologic change. Brain 2025, 148, 401–407. [Google Scholar] [CrossRef]
- Lee, V.M.-Y.; Giasson, B.I.; Trojanowski, J.Q. More than just two peas in a pod: Common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004, 27, 129–134. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Lloret, A.; Fuchsberger, T.; Giraldo, E.; Viña, J. Molecular mechanisms linking amyloid β toxicity and Tau hyperphosphorylation in Alzheimer’s disease. Free Radic. Biol. Med. 2015, 83, 186–191. [Google Scholar] [CrossRef]
- Fułek, M.; Hachiya, N.; Gachowska, M.; Beszłej, J.A.; Bartoszewska, E.; Kurpas, D.; Kurpiński, T.; Adamska, H.; Poręba, R.; Urban, S.; et al. Cellular Prion Protein and Amyloid-beta Oligomers in Alzheimer’s Disease-Are There Connections? Int. J. Mol. Sci. 2025, 26, 2097. [Google Scholar] [CrossRef]
- Wisse, L.E.M.; Wuestefeld, A.; Murray, M.E.; Jagust, W.; La Joie, R. Role of tau versus TDP-43 pathology on medial temporal lobe atrophy in aging and Alzheimer’s disease. Alzheimers Dement. 2025, 21, e14582. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, X. Alzheimer’s disease: Insights into pathology, molecular mechanisms, and therapy. Protein Cell 2025, 16, 83–120. [Google Scholar] [CrossRef]
- Orobets, K.S.; Karamyshev, A.L. Amyloid Precursor Protein and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14794. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef]
- Valappil, D.K.; Mini, N.J.; Dilna, A.; Nath, S. Membrane interaction to intercellular spread of pathology in Alzheimer’s disease. Front. Neurosci. 2022, 16, 936897. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Cabronero, J.; Betts, M.J.; Cardenas-Blanco, A.; Yang, S.; Nestor, P.J. In Vivo MRI Mapping of Brain Iron Deposition across the Adult Lifespan. J. Neurosci. 2016, 36, 364–374. [Google Scholar] [CrossRef]
- Li, G.; Tong, R.; Zhang, M.; Gillen, K.M.; Jiang, W.; Du, Y.; Wang, Y.; Li, J. Age-dependent changes in brain iron deposition and volume in deep gray matter nuclei using quantitative susceptibility mapping. Neuroimage 2023, 269, 119923. [Google Scholar] [CrossRef]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1–42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef]
- Zecca, L.; Gallorini, M.; Schünemann, V.; Trautwein, A.X.; Gerlach, M.; Riederer, P.; Vezzoni, P.; Tampellini, D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: Consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001, 76, 1766–1773. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1460. [Google Scholar] [CrossRef]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef]
- Chaudhari, V.; Bagwe-Parab, S.; Buttar, H.S.; Gupta, S.; Vora, A.; Kaur, G. Challenges and Opportunities of Metal Chelation Therapy in Trace Metals Overload-Induced Alzheimer’s Disease. Neurotox. Res. 2023, 41, 270–287. [Google Scholar] [CrossRef]
- Paul, J.S.; Raj, A.; Raghavan, S.; Kesavadas, C. Comparative analysis of quantitative susceptibility mapping in preclinical dementia detection. Eur. J. Radiol. 2024, 178, 111598. [Google Scholar] [CrossRef]
- Loughnan, R.; Ahern, J.; Tompkins, C.; Palmer, C.E.; Iversen, J.; Thompson, W.K.; Andreassen, O.; Jernigan, T.; Sugrue, L.; Dale, A.; et al. Association of Genetic Variant Linked to Hemochromatosis With Brain Magnetic Resonance Imaging Measures of Iron and Movement Disorders. JAMA Neurol. 2022, 79, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Marchi, G.; Busti, F.; Lira Zidanes, A.; Castagna, A.; Girelli, D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.L.; Pilling, L.C.; Heales, C.J.; Savage, S.; Kuo, C.L.; Kuchel, G.A.; Steffens, D.C.; Melzer, D. Hemochromatosis Mutations, Brain Iron Imaging, and Dementia in the UK Biobank Cohort. J. Alzheimers Dis. 2021, 79, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Vance, E.; Gonzalez Murcia, J.D.; Miller, J.B.; Staley, L.; Crane, P.K.; Mukherjee, S.; Kauwe, J.S.K. Failure to detect synergy between variants in transferrin and hemochromatosis and Alzheimer’s disease in large cohort. Neurobiol. Aging 2020, 89, 142.e9–142.e12. [Google Scholar] [CrossRef]
- Casanova, F.; Tian, Q.; Atkins, J.L.; Wood, A.R.; Williamson, D.; Qian, Y.; Zweibaum, D.; Ding, J.; Melzer, D.; Ferrucci, L.; et al. Iron and risk of dementia: Mendelian randomisation analysis in UK Biobank. J. Med. Genet. 2024, 61, 435–442. [Google Scholar] [CrossRef]
- Lupton, M.K.; Benyamin, B.; Proitsi, P.; Nyholt, D.R.; Ferreira, M.A.; Montgomery, G.W.; Madden, P.A.; Medland, S.E.; Gordon, S.D.; Lovestone, S.; et al. No genetic overlap between circulating iron levels and alzheimer’s disease. J. Alzheimers Dis. 2017, 59, 85–99. [Google Scholar] [CrossRef]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef]
- Jiang, H.; Song, N.; Jiao, Q.; Shi, L.; Du, X. Iron Pathophysiology in Parkinson Diseases. Adv. Exp. Med. Biol. 2019, 1173, 45–66. [Google Scholar]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2012, 434, 365–381. [Google Scholar] [CrossRef]
- James, S.A.; Churches, Q.I.; de Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S277–S281. [Google Scholar] [CrossRef]
- Hofer, T.; Perry, G. Nucleic acid oxidative damage in Alzheimer’s disease—Explained by the hepcidin-ferroportin neuronal iron overload hypothesis. J. Trace Elem. Med. Biol. 2016, 38, 1–9. [Google Scholar] [CrossRef]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s Disease Based on the Metal Hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avramovich-Tirosh, Y.; Youdim, M.B.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem. 2006, 97, 527–536. [Google Scholar] [CrossRef]
- Zhu, M.J.; Zhang, L.; Wang, C.P. Copper Overload Promotes beta-amyloid Induced NLRP3/Caspase-1/GSDMD-Mediated Pyroptosis in Alzheimer’s Disease. J. Integr. Neurosci. 2024, 23, 194. [Google Scholar] [CrossRef]
- Svoboda, P.; Di Cara, A. Hairpin RNA: A secondary structure of primary importance. Cell. Mol. Life Sci. 2006, 63, 901–908. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Blose, J.M. Structures, kinetics, thermodynamics, and biological functions of RNA hairpins. Annu. Rev. Phys. Chem. 2008, 59, 79–103. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Lisi, V.; Major, F. A comparative analysis of the triloops in all high-resolution RNA structures reveals sequence structure relationships. RNA 2007, 13, 1537–1545. [Google Scholar] [CrossRef]
- Cho, H.H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef]
- dos Santos, C.O.; Dore, L.C.; Valentine, E.; Shelat, S.G.; Hardison, R.C.; Ghosh, M.; Wang, W.; Eisenstein, R.S.; Costa, F.F.; Weiss, M.J. An iron responsive element-like stem-loop regulates alpha-hemoglobin-stabilizing protein mRNA. J. Biol. Chem. 2008, 283, 26956–26964. [Google Scholar] [CrossRef]
- Khan, M.A.; Walden, W.E.; Goss, D.J.; Theil, E.C. Direct Fe2+ sensing by iron-responsive messenger RNA:repressor complexes weakens binding. J. Biol. Chem. 2009, 284, 30122–30128. [Google Scholar] [CrossRef]
- Khan, M.A.; Mohammad, T.; Malik, A.; Hassan, M.I.; Domashevskiy, A.V. Iron response elements (IREs)-mRNA of Alzheimer’s amyloid precursor protein binding to iron regulatory protein (IRP1): A combined molecular docking and spectroscopic approach. Sci. Rep. 2023, 13, 5073. [Google Scholar] [CrossRef]
- Volz, K. Conservation in the Iron Responsive Element Family. Genes 2021, 12, 1365. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hamada, M. Recent trends in RNA informatics: A review of machine learning and deep learning for RNA secondary structure prediction and RNA drug discovery. Brief. Bioinform. 2023, 24, bbad186. [Google Scholar]
- Leipuviene, R.; Theil, E.C. The family of iron responsive RNA structures regulated by changes in cellular iron and oxygen. Cell. Mol. Life Sci. 2007, 64, 2945–2955. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 2008, 18, 106–111. [Google Scholar] [CrossRef]
- Shen, M.; Goforth, J.B.; Eisenstein, R.S. Iron-dependent post transcriptional control of mitochondrial aconitase expression. Met. Integr. Biometal Sci. 2023, 15, mfac099. [Google Scholar] [CrossRef]
- Rogers, J.T.; Mikkilineni, S.; Cantuti-Castelvetri, I.; Smith, D.H.; Huang, X.; Bandyopadhyay, S.; Cahill, C.M.; Maccecchini, M.L.; Lahiri, D.K.; Greig, N.H. The alpha-synuclein 5′untranslated region targeted translation blockers: Anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J. Neural Transmmission 2011, 118, 493–507. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Mikkilineni, S.; Cantuti-Castelvetri, I.; Cahill, C.M.; Balliedier, A.; Greig, N.H.; Rogers, J.T. The anticholinesterase phenserine and its enantiomer posiphen as 5′untranslated-region-directed translation blockers of the Parkinson’s alpha synuclein expression. Park. Dis. 2012, 2012, 142372. [Google Scholar] [CrossRef]
- Goforth, J.B.; Anderson, S.A.; Nizzi, C.P.; Eisenstein, R.S. Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA 2010, 16, 154–169. [Google Scholar] [CrossRef]
- Theil, E.C.; Goss, D.J. Living with iron (and oxygen)): Questions and answers about iron homeostasis. Chem. Rev. 2009, 109, 4568–4579. [Google Scholar] [CrossRef]
- Khan, M.A.; Walden, W.E.; Theil, E.C.; Goss, D.J. Thermodynamic and kinetic analyses of iron response element (IRE)-mRNA binding to iron regulatory protein, IRP1. Sci. Rep. 2017, 7, 8532. [Google Scholar] [CrossRef]
- Piccinelli, P.; Samuelsson, T. Evolution of the iron-responsive element. RNA 2007, 13, 952–966. [Google Scholar] [CrossRef]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef]
- Bellingham, S.A.; Lahiri, D.K.; Maloney, B.; La Fontaine, S.; Multhaup, G.; Camakaris, J. Copper depletion down-regulates expression of the Alzheimer’s disease Amyloid-beta precursor protein gene. J. Biol. Chem. 2004, 279, 20378–20386. [Google Scholar] [CrossRef]
- Walden, W.E.; Selezneva, A.; Volz, K. Accommodating variety in iron-responsive elements: Crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. FEBS Lett. 2012, 586, 32–35. [Google Scholar] [CrossRef]
- Kim, Y.S.; Gu, M.B. Advances in aptamer screening and small molecule aptasensors. Adv. Biochem. Eng. Biotechnol. 2014, 140, 29–67. [Google Scholar]
- Khan, M.A.; Ma, J.; Walden, W.E.; Merrick, W.C.; Theil, E.C.; Goss, D.J. Rapid kinetics of iron responsive element (IRE) RNA/iron regulatory protein 1 and IRE-RNA/eIF4F complexes respond differently to metal ions. Nucleic Acids Res. 2014, 42, 6567–6577. [Google Scholar] [CrossRef]
- Selezneva, A.I.; Cavigiolio, G.; Theil, E.C.; Walden, W.E.; Volz, K. Crystallization and preliminary X-ray diffraction analysis of iron regulatory protein 1 in complex with ferritin IRE RNA. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 249–252. [Google Scholar] [CrossRef]
- Khan, M.A. α-Synuclein Iron-Responsive-Element RNA and Iron Regulatory Protein Affinity Is Specifically Reduced by Iron in Parkinson’s Disease. Biomolecules 2025, 15, 214. [Google Scholar] [CrossRef]
- Garza, K.R.; Clarke, S.L.; Ho, Y.H.; Bruss, M.D.; Vasanthakumar, A.; Anderson, S.A.; Eisenstein, R.S. Differential translational control of 5′ IRE-containing mRNA in response to dietary iron deficiency and acute iron overload. Met. Integr. Biometal Sci. 2020, 12, 2186–2198. [Google Scholar] [CrossRef]
- Goss, D.J.; Theil, E.C. Iron responsive mRNAs: A family of Fe2+ sensitive riboregulators. Acc. Chem. Res. 2011, 44, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. IRE mRNA riboregulators use metabolic iron (Fe(2+)) to control mRNA activity and iron chemistry in animals. Met. Integr. Biometal Sci. 2015, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Martick, M.; Horan, L.H.; Noller, H.F.; Scott, W.G. A discontinuous hammerhead ribozyme embedded in a mammalian messenger RNA. Nature 2008, 454, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Haldar, S.; Khan, M.A.; Sharma, S.D.; Merrick, W.C.; Theil, E.C.; Goss, D.J. Fe2+ binds iron responsive element-RNA, selectively changing protein-binding affinities and regulating mRNA repression and activation. Proc. Natl. Acad. Sci. USA 2012, 109, 8417–8422. [Google Scholar] [CrossRef]
- Rogers, J.T.; Bush, A.I.; Cho, H.H.; Smith, D.H.; Thomson, A.M.; Friedlich, A.L.; Lahiri, D.K.; Leedman, P.J.; Huang, X.; Cahill, C.M. Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: Riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem. Soc. Trans. 2008, 36 Pt 6, 1282–1287. [Google Scholar] [CrossRef]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: An update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. (Rev.) 2019, 13, 180. [Google Scholar] [CrossRef]
- Tucker, S.; Ahl, M.; Cho, H.H.; Bandyopadhyay, S.; Cuny, G.D.; Bush, A.I.; Goldstein, L.E.; Westaway, D.; Huang, X.; Rogers, J.T. RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr. Alzheimer Res. 2006, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Cooperman, S. Brain iron metabolism. Semin. Pediatr. Neurol. 2006, 13, 142–148. [Google Scholar] [CrossRef]
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads Between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients 2023, 15, 2067. [Google Scholar] [CrossRef]
- Li, X.; Lei, P.; Tuo, Q.; Ayton, S.; Li, Q.; Moon, S.; Volitakis, I.; Liu, R.; Masters, C.L.; Finkelstein, D.I.; et al. Enduring Elevations of Hippocampal Amyloid Precursor Protein and Iron Are Features of β-Amyloid Toxicity and Are Mediated by Tau. Neurotherapeutics 2015, 12, 862–873. [Google Scholar] [CrossRef]
- Uddin, M.S.; Ashraf, G.M. Dysregulation of Neuronal Iron in Alzheimer’s Disease. Curr. Neuropharmacol. 2023, 21, 2247–2250. [Google Scholar] [CrossRef]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- Liu, B.; Moloney, A.; Meehan, S.; Morris, K.; Thomas, S.E.; Serpell, L.C.; Hider, R.; Marciniak, S.J.; Lomas, D.A.; Crowther, D.C. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J. Biol. Chem. 2011, 286, 4248–4256. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. (Rev.) 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Kalionis, B.; Murthi, P.; Xia, S.; Guan, Y. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front. Neurol. 2019, 10, 607. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- LeVine, S.M. Exploring Potential Mechanisms Accounting for Iron Accumulation in the Central Nervous System of Patients with Alzheimer’s Disease. Cells 2024, 13, 689. [Google Scholar] [CrossRef]
- LeVine, S.M.; Tsau, S.; Gunewardena, S. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer’s Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sci. 2023, 13, 511. [Google Scholar] [CrossRef]
- Khan, M.A. Targeting iron responsive elements (IREs) of APP mRNA tnto novel therapeutics to control the translation of amyloid-beta precursor protein in Alzheimer’s disease. Pharmaceuticals 2024, 17, 1669. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.-W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Amit, T.; Avramovich-Tirosh, Y.; Youdim, M.B.; Mandel, S. Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008, 22, 1296–1305. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, P.; Zhong, M.L.; Wang, T.; Huang, X.S.; Li, J.Y.; Wang, Z.-Y. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Sahoo, A.; Anand, S.; Ganguly, A.; Righi, G.; Bovicelli, P.; Saso, L.; Chakrabarti, S. Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: Protective action of negletein. Neuromolecular Med. 2014, 16, 787–798. [Google Scholar] [CrossRef]
- Guillemot, J.; Canuel, M.; Essalmani, R.; Prat, A.; Seidah, N.G. Implication of the proprotein convertases in iron homeostasis: Proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology 2013, 57, 2514–2524. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Park, Y.H.; Kosman, D.J. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. 2014, 15, 809–815. [Google Scholar] [CrossRef]
- Johannesson, T.; Kristinsson, J.; Torsdottir, G.; Snaedal, J. Ceruloplasmin (Cp) and iron in connection with Parkinson’s disease (PD) and Alzheimer’s disease (AD). Laeknabladid 2012, 98, 531–537. [Google Scholar]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef]
- Jabeen, F.; Fu, M. Brain iron and neurodegenerative disorders. Glob. Drugs Ther. 2017, 2, 1–2. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Kagerer, S.M.; van Bergen, J.M.G.; Li, X.; Quevenco, F.C.; Gietl, A.F.; Studer, S.; Treyer, V.; Meyer, R.; Kaufmann, P.A.; Nitsch, R.M.; et al. APOE4 moderates effects of cortical iron on synchronized default mode network activity in cognitively healthy old-aged adults. Alzheimer’s Dement. (Amst.) 2020, 12, e12002. [Google Scholar] [CrossRef]
- Yin, X.; Qiu, Y.; Zhao, C.; Zhou, Z.; Bao, J.; Qian, W. The Role of Amyloid-Beta and Tau in the Early Pathogenesis of Alzheimer’s Disease. Med. Sci. Monit. 2021, 27, e933084. [Google Scholar] [CrossRef]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef]
- Tibodeau, J.D.; Fox, P.M.; Ropp, P.A.; Theil, E.C.; Thorp, H.H. The up-regulation of ferritin expression using a small-molecule ligand to the native mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 253–257. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Cahill, C.; Balleidier, A.; Huang, C.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: Implications for down syndrome and Alzheimer’s disease. PLoS ONE 2013, 8, e65978. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Rogers, J.T. Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: Maintenance of brain iron homeostasis. Biochem. Pharmacol. 2014, 88, 486–494. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Nwankwo, E.I.; Nussbaum, R.L.; Rogers, J.; Maccecchini, M.L. Translational inhibition of α-synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am. J. Neurodegener. Dis. 2019, 8, 1–15. [Google Scholar]
- Teich, A.F.; Sharma, E.; Barnwell, E.; Zhang, H.; Staniszewski, A.; Utsuki, T.; Padmaraju, V.; Mazell, C.; Tzekou, A.; Sambamurti, K.; et al. Translational inhibition of APP by Posiphen: Efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimer’s Dement. 2018, 4, 37–45. [Google Scholar] [CrossRef]
- Li, H.; Tan, Y.; Cheng, X.; Zhang, Z.; Huang, J.; Hui, S.; Zhu, L.; Liu, Y.; Zhao, D.; Liu, Z.; et al. Untargeted metabolomics analysis of the hippocampus and cerebral cortex identified the neuroprotective mechanisms of Bushen Tiansui formula in an aβ25-35-induced rat model of Alzheimer’s disease. Front. Pharmacol. 2022, 13, 990307. [Google Scholar] [CrossRef]
- Xiang, Q.; Xiang, Y.; Liu, Y.; Chen, Y.; He, Q.; Chen, T.; Tang, L.; He, B.; Li, J. Revealing the potential therapeutic mechanism of Lonicerae Japonicae Flos in Alzheimer’s disease: A computational biology approach. Front. Med. 2024, 11, 1468561. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Li, G.; Han, L.; Qin, X. Apelin-13 Suppresses Neuroinflammation Against Cognitive Deficit in a Streptozotocin-Induced Rat Model of Alzheimer’s Disease Through Activation of BDNF-TrkB Signaling Pathway. Front. Pharmacol. 2019, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kumarasamy, R.V.; Jayaraman, S.; Kanniappan, G.V.; Long, Q.; Palanisamy, C.P. Quercetin-functionalized nanomaterials: Innovative therapeutic avenues for Alzheimer’s disease management. Ageing Res. Rev. 2025, 104, 102665. [Google Scholar] [CrossRef]
- Rekha, A.; Afzal, M.; Babu, M.A.; Menon, S.V.; Nathiya, D.; Supriya, S.; Mishra, S.B.; Gupta, S.; Goyal, K.; Rana, M.; et al. GSK-3β dysregulation in aging: Implications for tau pathology and Alzheimer’s disease progression. Mol. Cell. Neurosci. 2025, 133, 104005. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Wang, Q.; Fu, J.X.; Xu, X.X.; Guo, D.S.; Pan, Y.C.; Zhang, T.; Wang, H. Multi Targeted Therapy for Alzheimer’s Disease by Guanidinium-Modified Calixarene and Cyclodextrin Co-Assembly Loaded with Insulin. ACS Nano 2024, 18, 33032–33041. [Google Scholar] [CrossRef]
- Yu, Y.-B.; Zhang, Q.; Guo, Y.-H.; Wu, J.-J.; Zhao, L.-X.; Wang, Z.; Gong, Z.-Q.; Kan, Y.-K.; Wang, Z.-Y. ROS-related nanoparticles for the management of Alzheimer’s disease: Wielding the double-edged sword. Chem. Eng. J. 2025, 511, 161784. [Google Scholar] [CrossRef]
- Dhariwal, R.; Jain, M.; Mir, Y.R.; Singh, A.; Jain, B.; Kumar, P.; Tariq, M.; Verma, D.; Deshmukh, K.; Yadav, V.K.; et al. Targeted drug delivery in neurodegenerative diseases: The role of nanotechnology. Front. Med. 2025, 12, 1522223. [Google Scholar] [CrossRef]
- Singh, A.; Maheshwari, S.; Yadav, J.P.; Kumar, R.; Verma, A.; Singh, S.; Prajapati, B.G. Bioactive Compound-Fortified Nanocarriers in the Management of Neurodegenerative Disease: A Review. Chem. Biodivers. 2025, 72, e202402018. [Google Scholar] [CrossRef]
- Rogers, J.T.; Lahiri, D.K. Metal and inflammatory targets for Alzheimer’s disease. Curr. Drug Targets 2004, 5, 535–551. [Google Scholar] [CrossRef]
- Pramanik, D.; Ghosh, C.; Dey, S.G. Heme-Cu bound aβ peptides: Spectroscopic characterization, reactivity, and relevance to Alzheimer’s disease. J. Am. Chem. Soc. 2011, 133, 15545–15552. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Fan, Y.G.; Zhao, L.X.; Zhang, Q.; Wang, Z.Y. The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorganic Chem. 2023, 131, 106301. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Lee, J.; Ahn, B.; Han, J.; Lim, M.H. Multi-target-directed therapeutic strategies for Alzheimer’s disease: Controlling amyloid-beta aggregation, metal ion homeostasis, and enzyme inhibition. Chem. Sci. 2025, 16, 2105–2135. [Google Scholar] [CrossRef]
- Rogers, J.T.; Randall, J.D.; Eder, P.S.; Huang, X.; Bush, A.I.; Tanzi, R.E.; Venti, A.; Payton, S.M.; Giordano, T.; Nagano, S.; et al. Alzheimer’s disease drug discovery targeted to the APP mRNA 5′untranslated region. J. Mol. Neurosci. 2002, 19, 77–82. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Raymick, J.; Sarkar, S.; Lahiri, D.K.; Ray, B.; Holtzman, D.; Dumas, M.; Schmued, L.C. Efficacy and toxicity of clioquinol treatment and A-beta42 inoculation in the APP/PSI mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Bar-Am, O.; Amit, T.; Kupershmidt, L.; Aluf, Y.; Mechlovich, D.; Kabha, H.; Danovitch, L.; Zurawski, V.R.; Youdim, M.B.; Weinreb, O. Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol. Aging 2015, 36, 1529–1542. [Google Scholar] [CrossRef]
- Das, B.; Kandegedara, A.; Xu, L.; Antonio, T.; Stemmler, T.; Reith, M.E.A.; Dutta, A.K. A Novel Iron(II) Preferring Dopamine Agonist Chelator as Potential Symptomatic and Neuroprotective Therapeutic Agent for Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 723–730. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Z.; Lin, T.M.; Chanana, S.; Xiong, M.P. Nanogel-DFO conjugates as a model to investigate pharmacokinetics, biodistribution and iron chelation in vivo. Int. J. Pharm. 2018, 538, 79–86. [Google Scholar] [CrossRef]
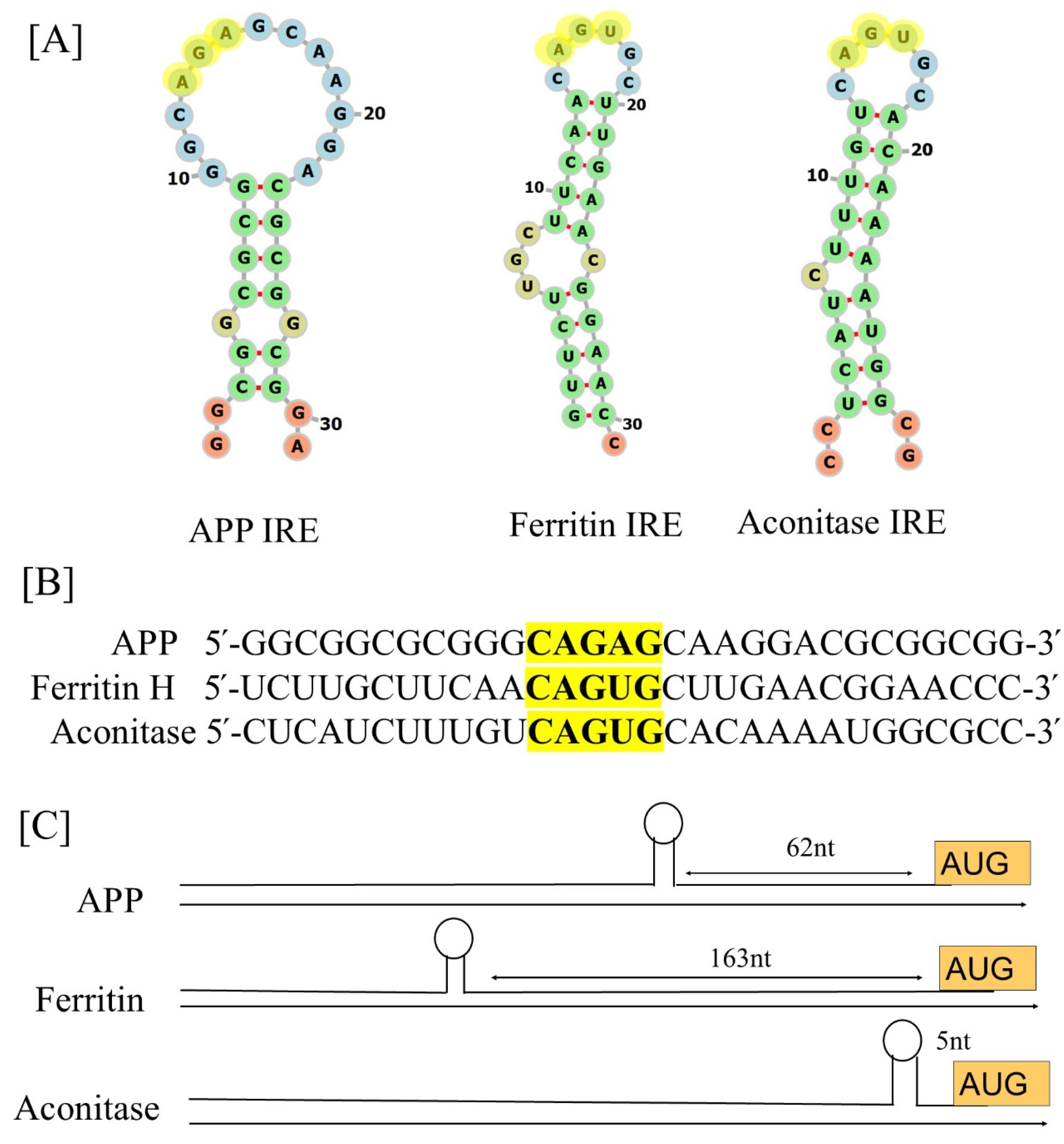

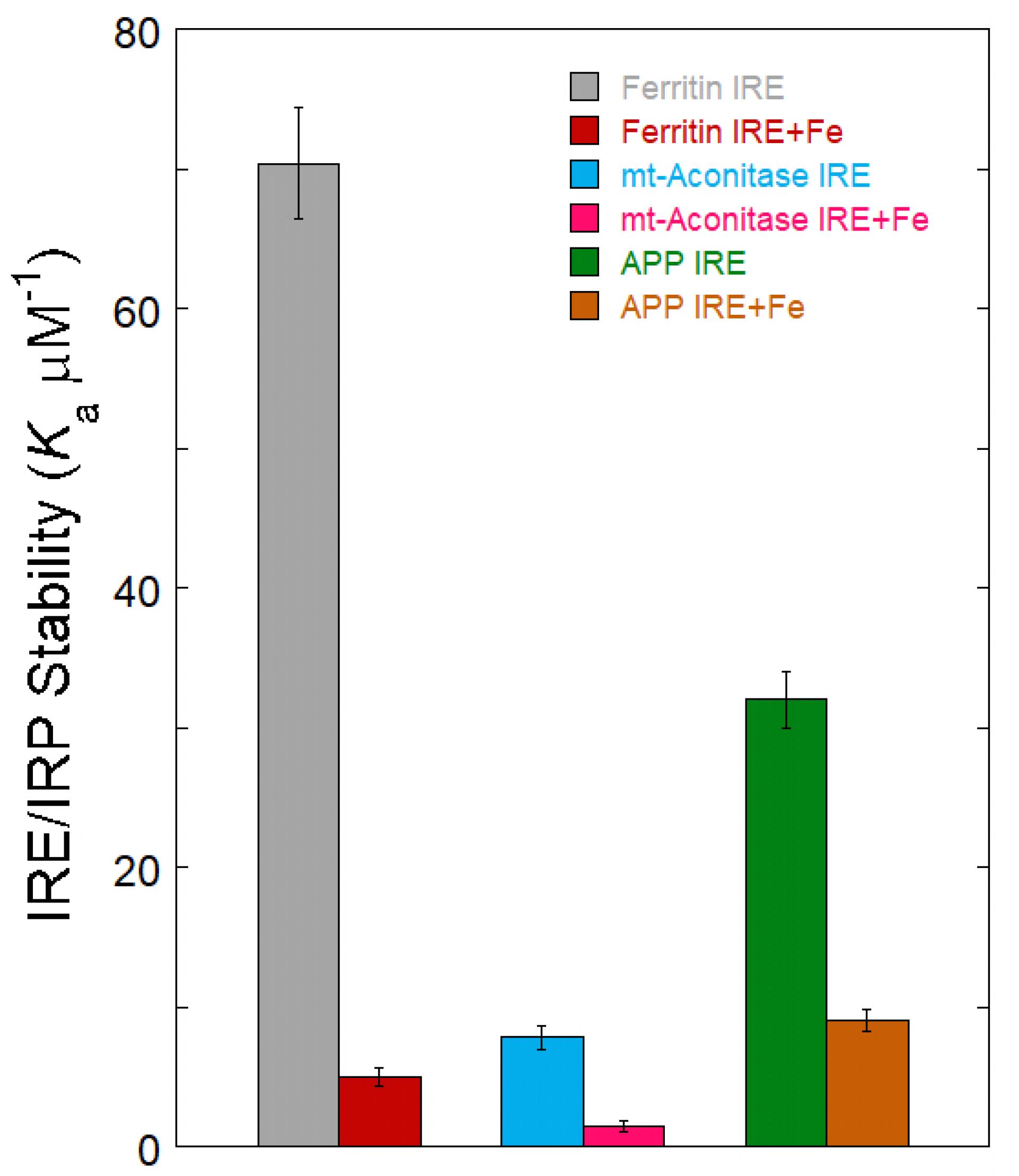
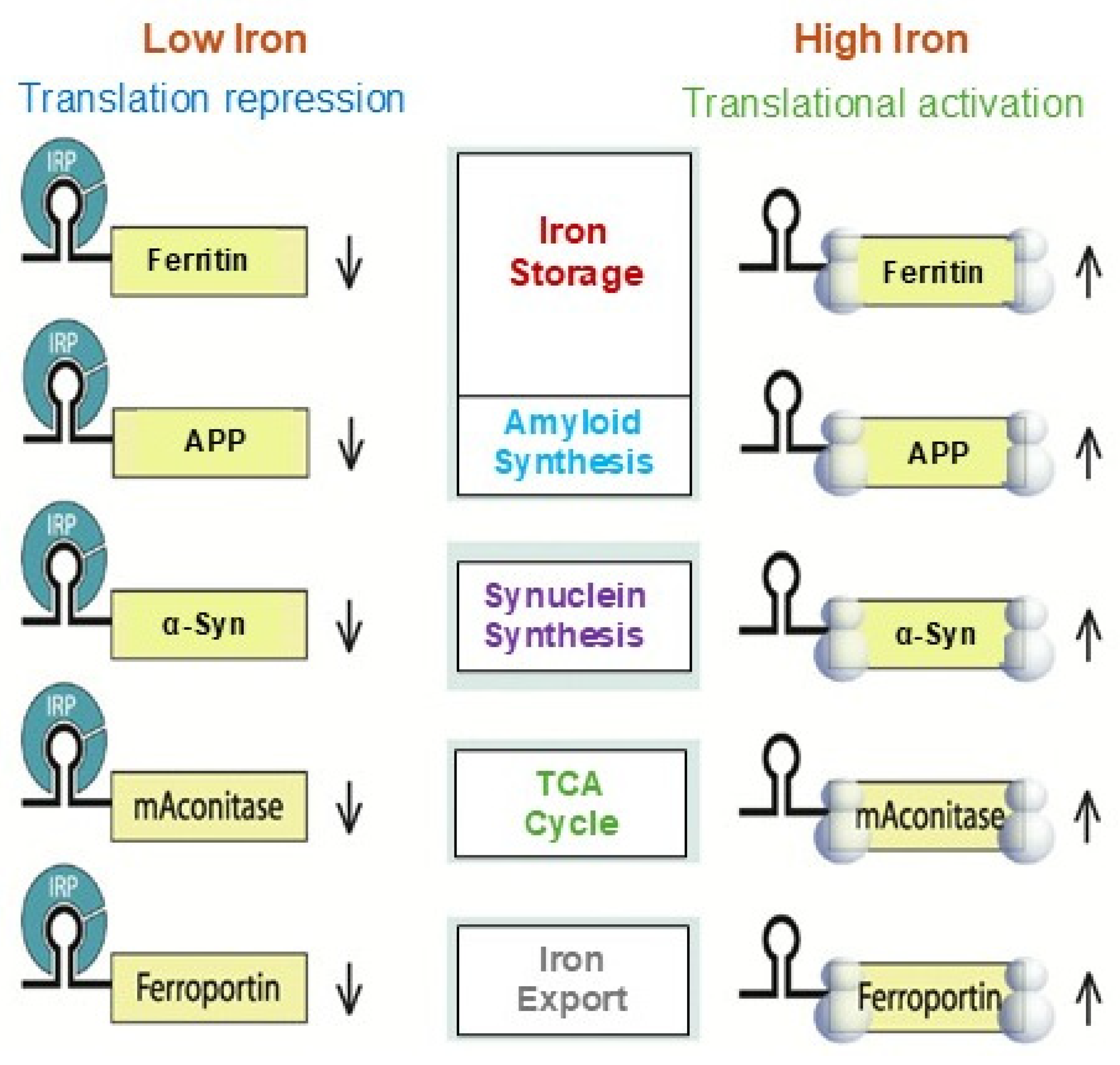
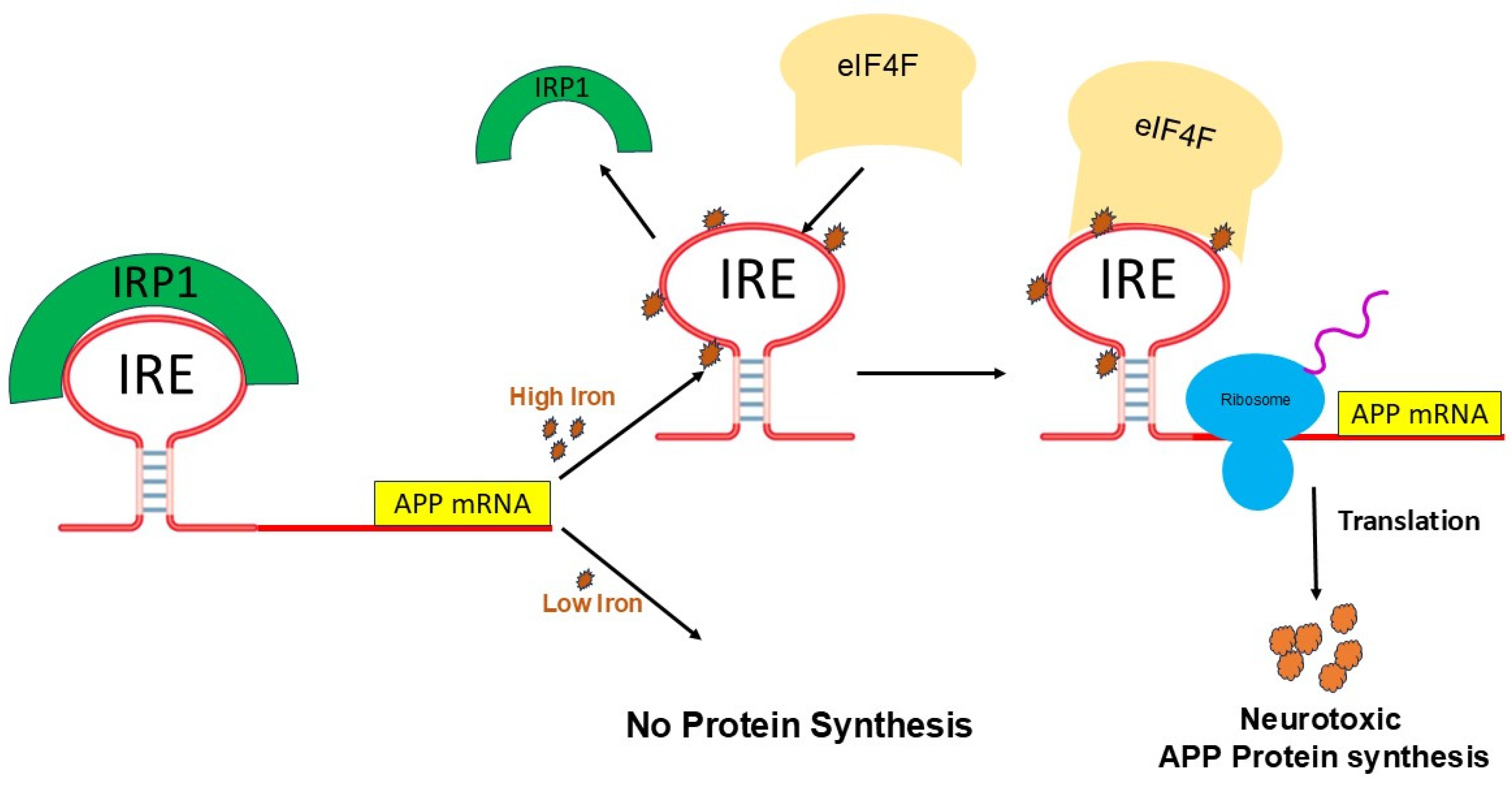
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A. Iron-Mediated Overexpression of Amyloid Precursor Protein via Iron Responsive mRNA in Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 5283. https://doi.org/10.3390/ijms26115283
Khan MA. Iron-Mediated Overexpression of Amyloid Precursor Protein via Iron Responsive mRNA in Alzheimer’s Disease. International Journal of Molecular Sciences. 2025; 26(11):5283. https://doi.org/10.3390/ijms26115283
Chicago/Turabian StyleKhan, Mateen A. 2025. "Iron-Mediated Overexpression of Amyloid Precursor Protein via Iron Responsive mRNA in Alzheimer’s Disease" International Journal of Molecular Sciences 26, no. 11: 5283. https://doi.org/10.3390/ijms26115283
APA StyleKhan, M. A. (2025). Iron-Mediated Overexpression of Amyloid Precursor Protein via Iron Responsive mRNA in Alzheimer’s Disease. International Journal of Molecular Sciences, 26(11), 5283. https://doi.org/10.3390/ijms26115283