
Design, Synthesis, Anti-Tumor Activity and Molecular Docking Studies of Novel Triphenylphosphine-Containing Formononetin Derivatives



Abstract
1. Introduction
2. Results
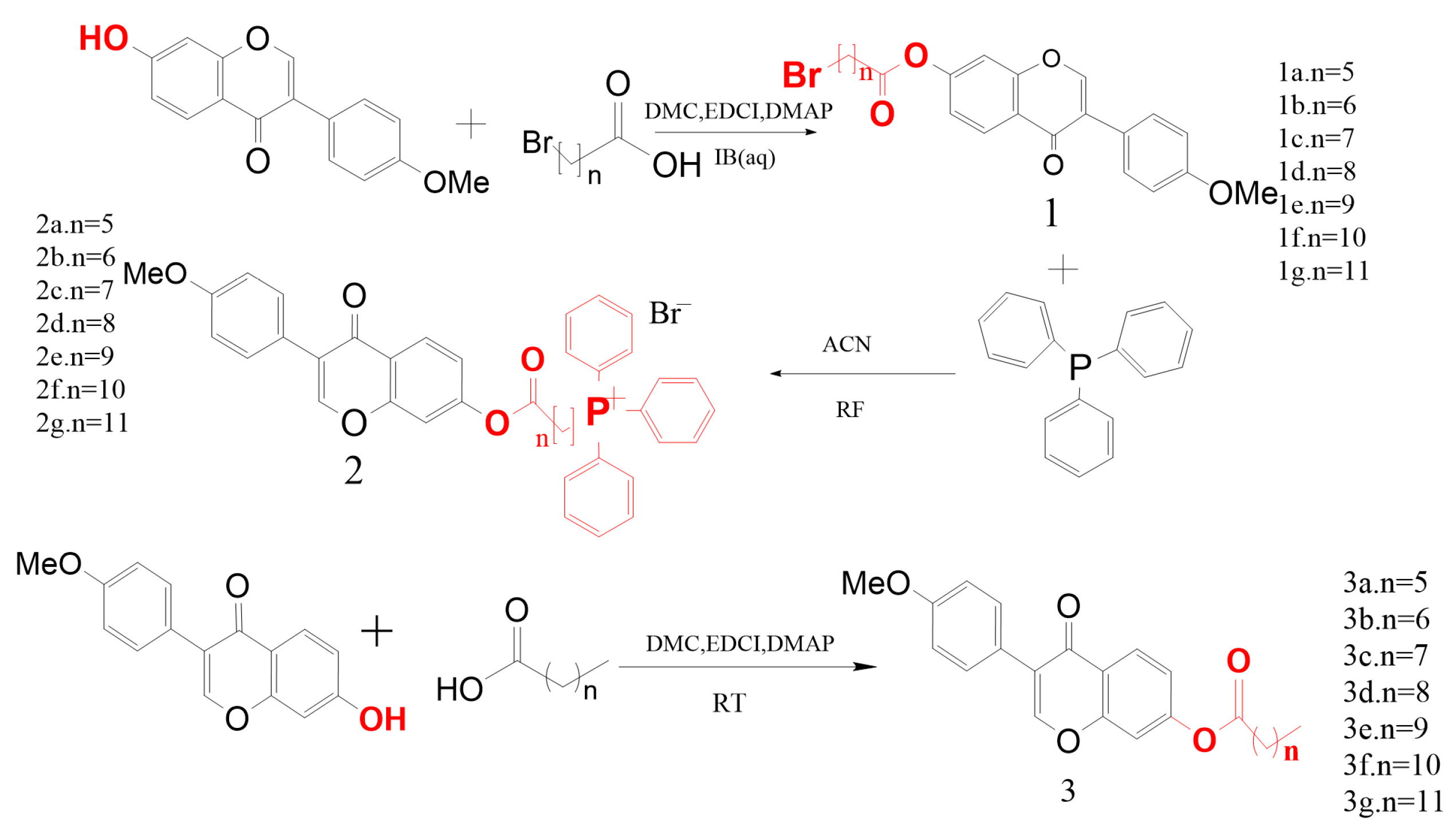
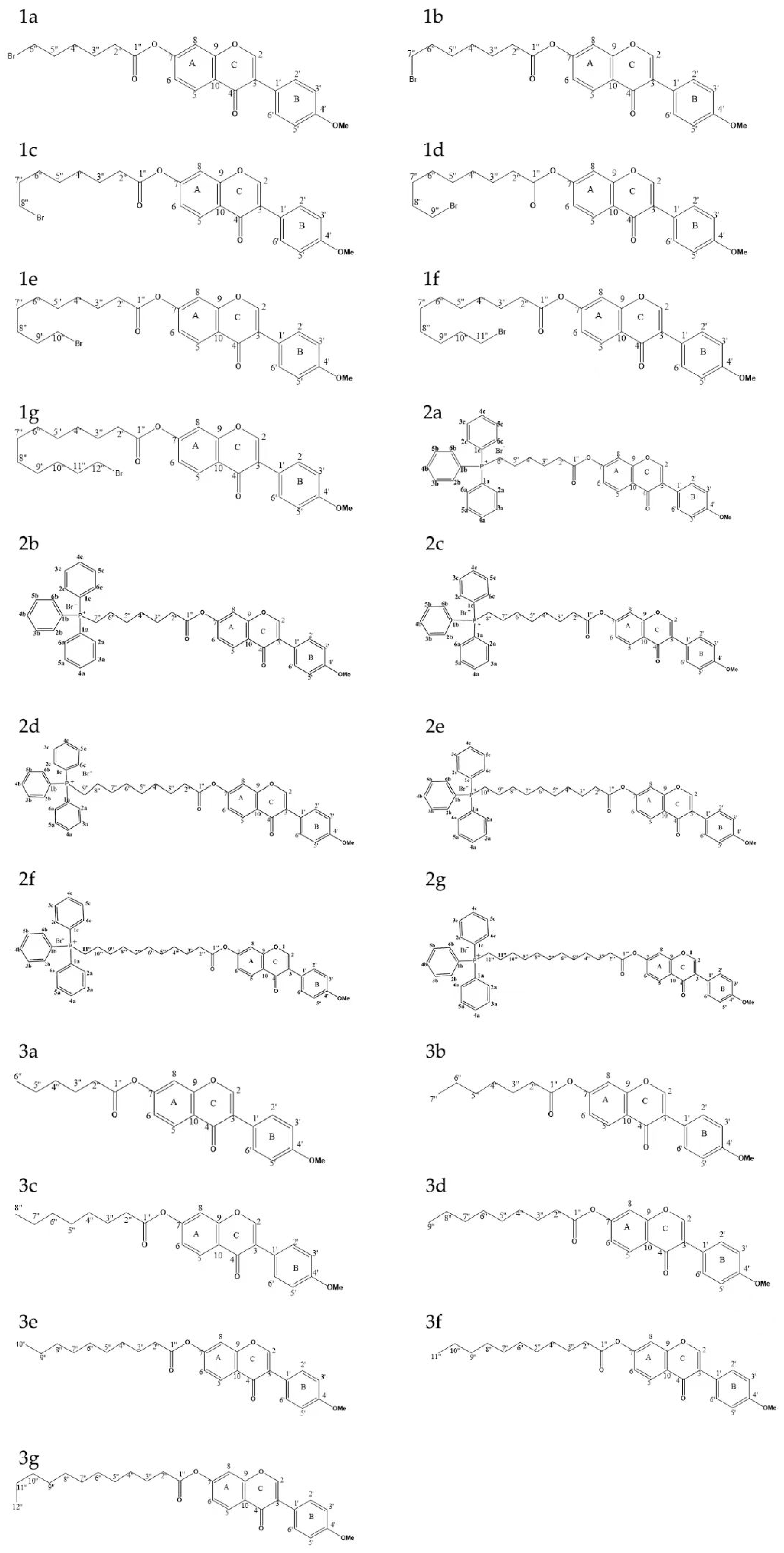
2.1. Synthesis
2.2. MTT Results
2.3. Network Pharmacology Results
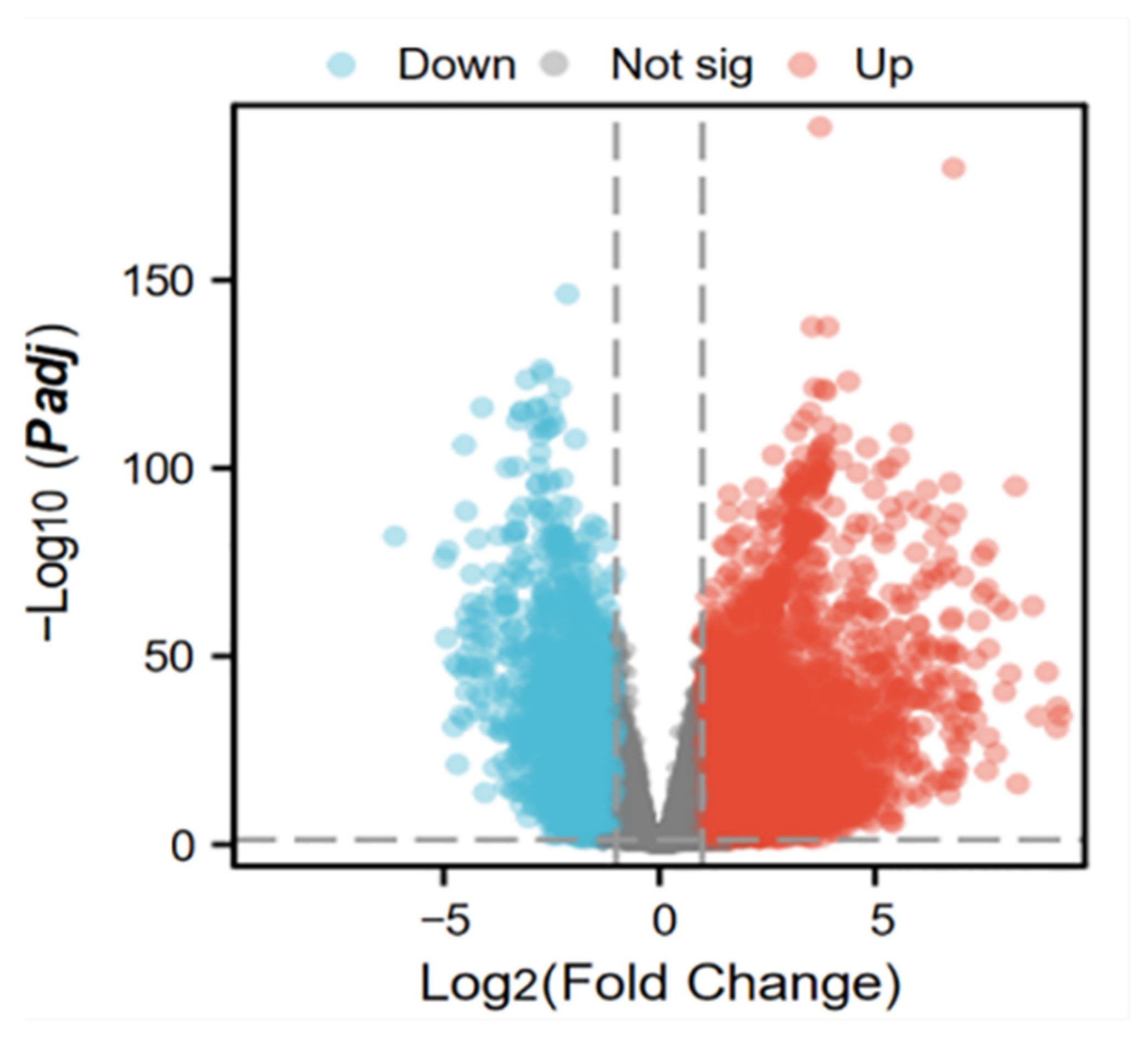
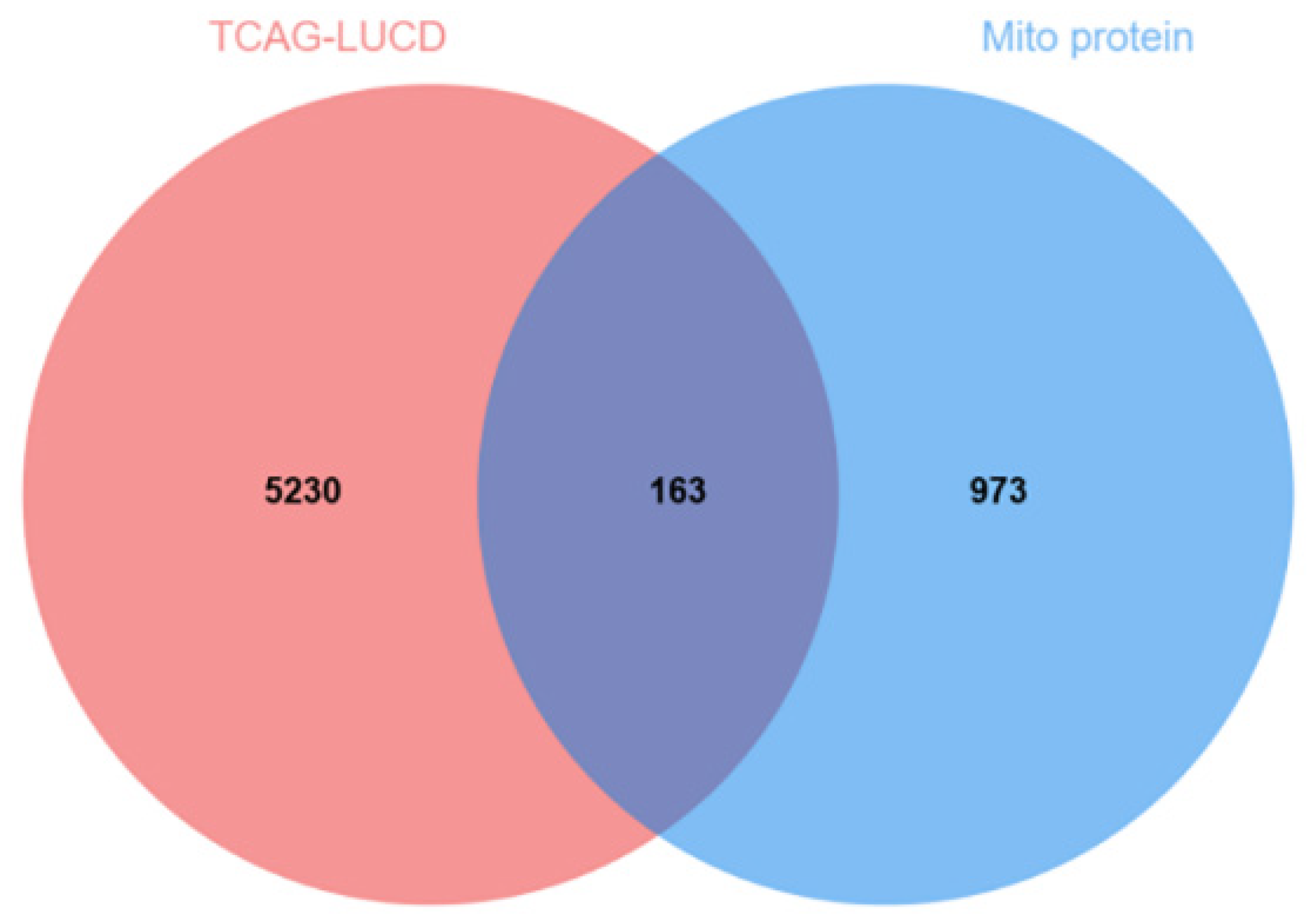
2.3.1. Tumor Mitochondrial Differentially Expressed Gene Analysis Results
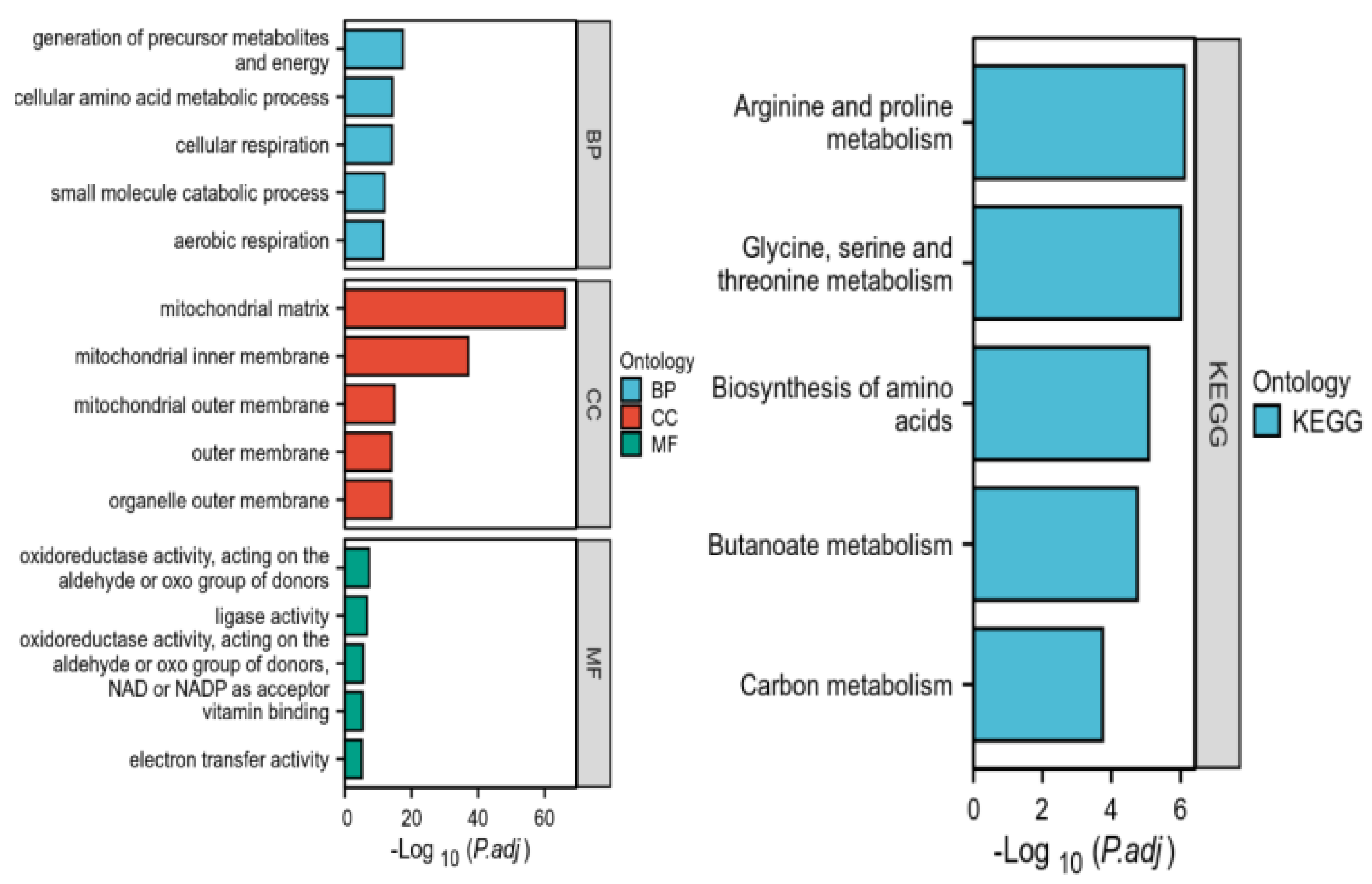
2.3.2. Results of GO and KEGG Pathway Enrichment Analysis of Intersected Genes
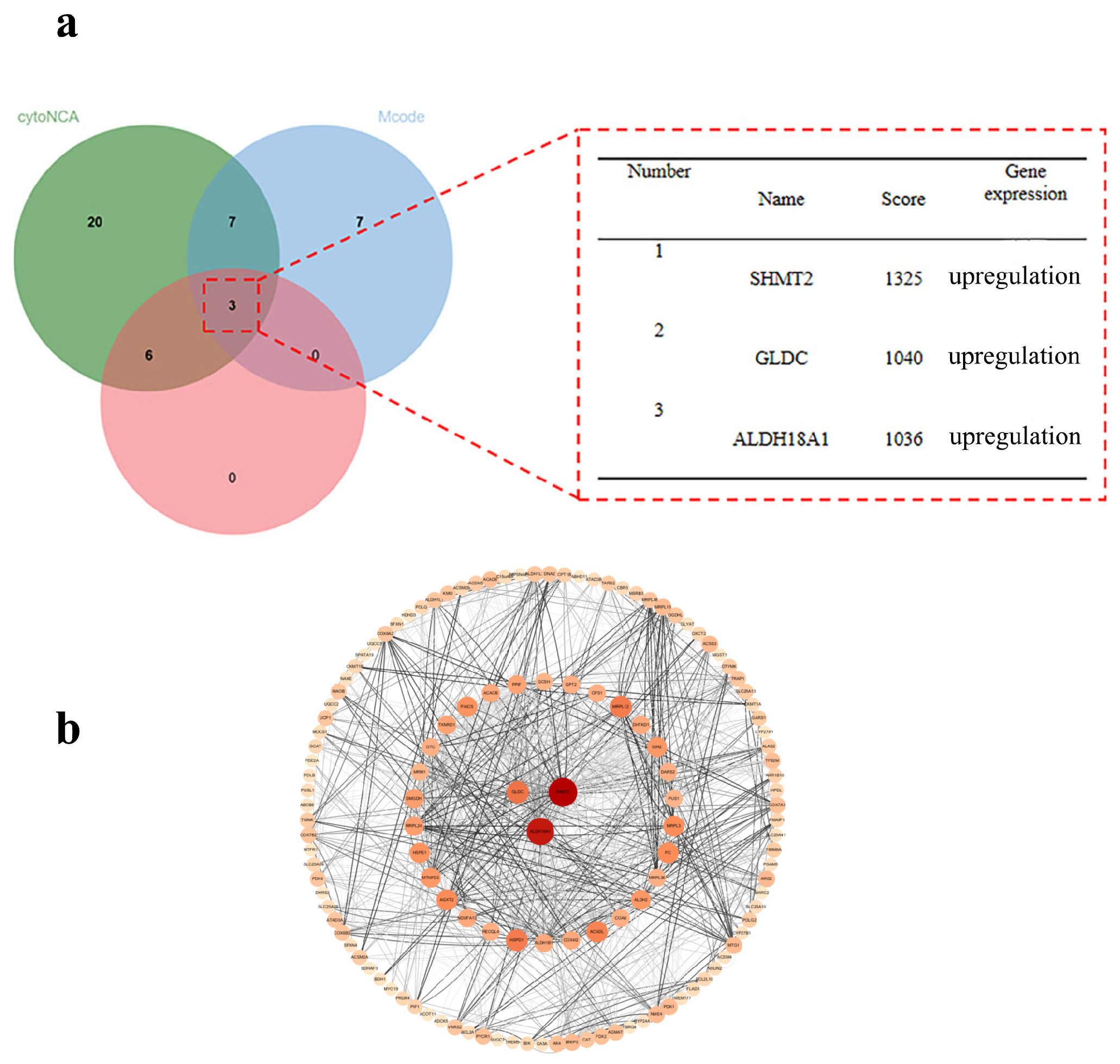
2.3.3. Core Gene Screening and Protein–Protein Interaction (PPI) Network Construction Results
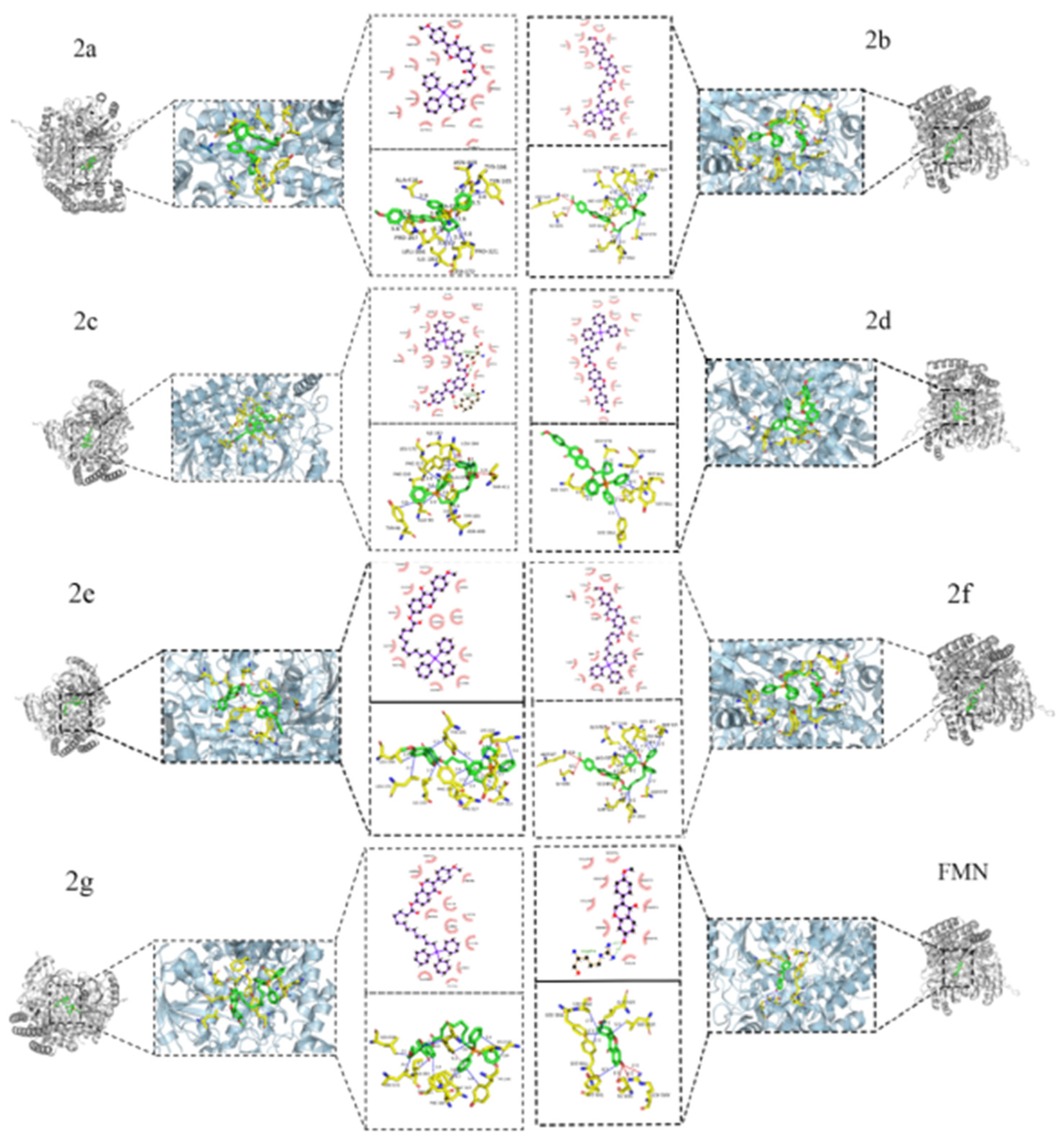
2.4. Molecular Docking Results
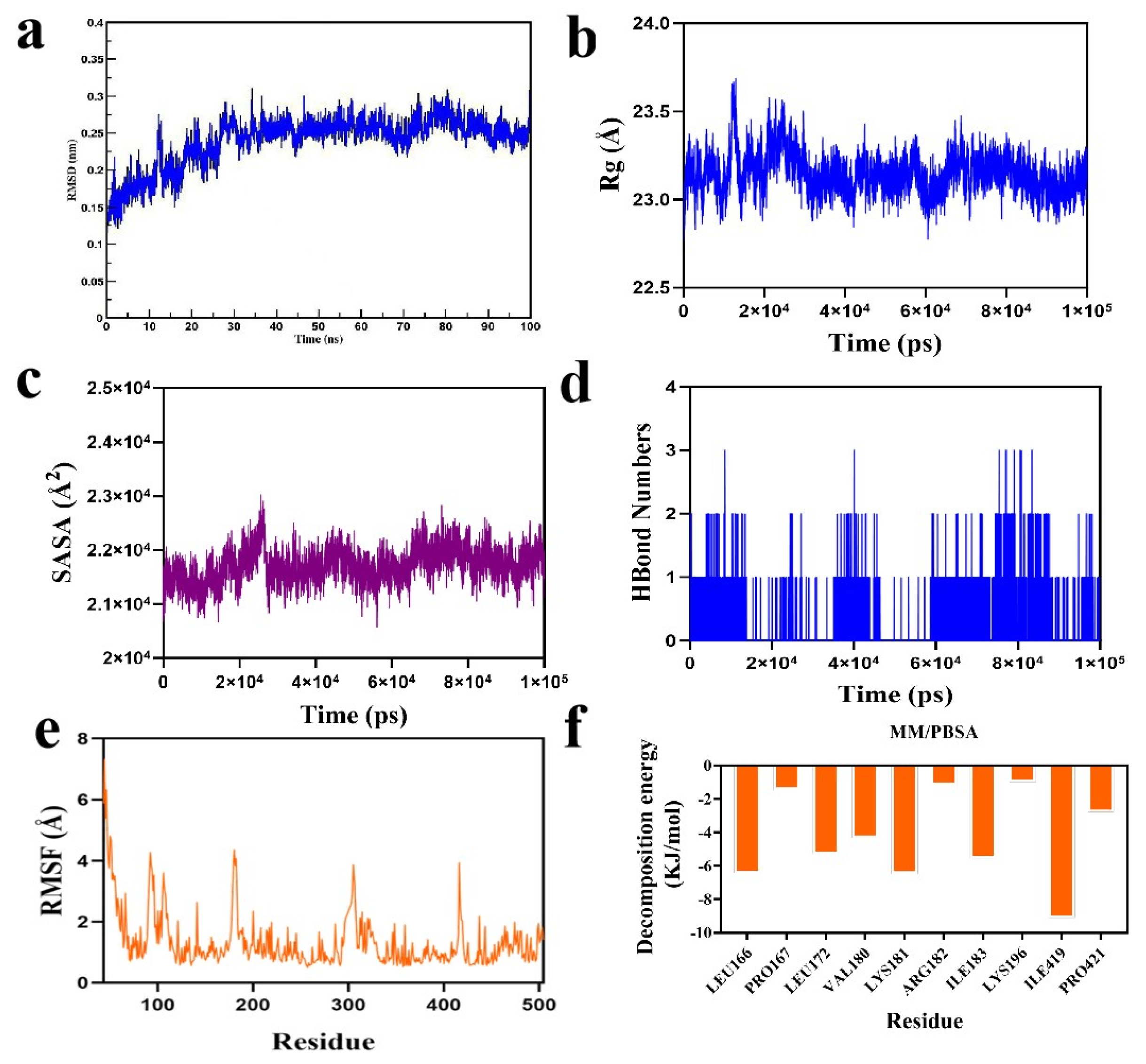
2.5. Molecular Dynamics Simulation Results
3. Discussion and Future Perspectives
4. Materials and Methods
4.1. Chemical Reagents
4.2. Design and Synthesis of the Derivatives
4.3. Cell Lines
4.4. Cell Viability Assay
4.5. Network Pharmacology
4.5.1. Tumor Cell and Mitochondrial Crossover Gene Screening
4.5.2. GO and KEGG Pathway Enrichment Analysis of Overlapping Genes
4.5.3. Core Gene Screening and Protein–Protein Interaction (PPI) Network Construction
4.6. Molecular Docking
4.7. Molecular Dynamics Simulations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhanasekaran, S.; Venugopal, D.; Al-Dayan, N.; Ravinayagam, V.; Ahmed Mohammed, A. Emerging insights into mitochondria-specific targeting and drug delivering strategies: Recent milestones and therapeutic implications. Saudi J. Biol. Sci. 2020, 27, 3581–3592. [Google Scholar] [CrossRef] [PubMed]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Hee, M.L.; Sessler, J.L.; Seung, J.K. Disulfide-based multifunctional conjugates for targeted theranostic drug delivery. Acc. Chem. Res. 2015, 48, 2935–2946. [Google Scholar]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 9352–9379. [Google Scholar] [CrossRef]
- Yu, G.; Xiang, J.; Shao, S.; Tang, J.; Shen, Y. Mitochondria-targeted polymer-celastrol conjugate with enhanced anticancer efficacy. J. Control. Release 2022, 342, 122–133. [Google Scholar]
- Das, S.; Zea, M.P.; Russon, M.P.; Xing, Z.; Torregrosa Allen, S.; Cervantes, H.E.; Harper, H.A.; Elzey, B.D.; Tran, E.J. Supinoxin blocks small cell lung cancer progression by inhibiting mitochondrial respiration through DDX5. iScience 2025, 28, 112219. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Patel, J.S.; Klebanoff, C.A.; Mehta, C.; Roychoudhuri, R.; Crompton, J.; Clever, D.; Gattinoni, L.; Muranski, P.; et al. Modulating immunometabolism of tumor specific mouse and human lymphocytes to enhance T cell based therapy for cancer. J. Immunother. Cancer 2015, 3, 325. [Google Scholar] [CrossRef]
- Šileikytė, J.; Forte, M. The mitochondrial permeability transition in mitochondrial disorders. Oxid. Med. Cell Longev. 2019, 2019, 3403075. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, R.; Wang, R.; Huang, J.; Xu, Y.; Wang, N.; Li, D.; Xu, C.; Wang, B.; Li, Y.; et al. Design, synthesis and antitumor activity of triphenylphosphonium-linked derivatives of quinazolinone. Nat. Prod. Res. 2024, 1–6. [Google Scholar] [CrossRef]
- Aguilar, F.A.; Bakker, G.A.; Jovanović, M.; Stojanov, S.J.; Puerta, A.; Gargano, A.; Dinić, J.; Vega-Báez, J.L.; Montiel, M.P.; Smith, M.S.; et al. Coumarins-lipophilic cations conjugates: Efficient mitocans targeting carbonic anhydrases. Bioorganic Chem. 2024, 145, 107168. [Google Scholar]
- Miao, H.; Cui, W.; Zhang, T.; Zhang, Y.; Zhang, J.; Lou, H.; Fan, P. Mitochondrial targeting derivatives of honokiol enhanced selective antitumor activity in NCI-H446 cells and decreased in vivo toxicity in Caenorhabditis elegans. Eur. J. Med. Chem. 2024, 264, 115996. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.G.; Kang, J.H.; Kang, S.J.; Truong, H.Q.; Kang, H.C.; Rhee, W.J.; Zhang, Y.S.; Ko, Y.T.; Shim, M.S. Brain endothelial cell-derived extracellular vesicles with a mitochondria-targeting photosensitizer effectively treat glioblastoma by hijacking the blood–brain barrier. Acta Pharm. Sin. B 2023, 13, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, Y.; Guo, J.; Chen, S.; Jin, J.; Li, P.; Pan, Y.; Lei, S.; Li, J.; Wu, S.; et al. Synthesis and anti-proliferative effect of novel 4-Aryl-1, 3-Thiazole-TPP conjugates via mitochondrial uncoupling process. Bioorganic Chem. 2024, 150, 107588. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’skii, Y.P.; Bel’skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitohondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. Med. Chem. Commun. 2017, 8, 1934–1945. [Google Scholar] [CrossRef]
- Jin, L.; Dai, L.; Ji, M.; Wang, H. Mitochondria-targeted triphenylphosphonium conjugated glycyrrhetinic acid derivatives as potent anticancer drugs. Bioorg. Chem. 2019, 85, 179–190. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, T.; Lou, H.; Song, H.; Li, C.; Fan, P. Anti-cancer effects of honokiol via mitochondrial dysfunction are strongly enhanced by the mitochondria-targeting carrier berberine. J. Med. Chem. 2020, 63, 11786–11800. [Google Scholar] [CrossRef]
- Ronis, M.J.; Little, J.M.; Barone, G.W.; Chen, G.; Pandya, R.A.; Badger, T.M. Sulfation of the isoflavones genistein and daidzein in human and rat liver and gastrointestinal tract. J. Med. Food 2006, 9, 348–355. [Google Scholar] [CrossRef]
- Selka, A.; Abidli, A.; Schiavo, L.; Jeanmart, L.; Hanquet, G.; Lubell, W.D. Recent Advances in Sustainable Total Synthesis and Chiral Pool Strategies with Emphasis on (−)-Sclareol in Natural Products Synthesis. Eur. J. Org. Chem. 2025, 28, e202400983. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Wu, X.C.; Negoita, S.; Stevens, J.; Coyle, L.; Zhao, J.; Mumphrey, B.J.; Jemal, A.; Ward, K.C. Association of the COVID-19 Pandemic with Patterns of Statewide Cancer Services. J. Natl. Cancer Inst. 2021, 114, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Bedi, M.; Ray, M.; Ghosh, A. Active mitochondrial respiration in cancer: A target for the drug. Mol. Cell Biochem. 2022, 477, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Jiang, Z. SLC25A19 is required for NADH homeostasis and mitochondrial respiration. Free. Radic. Biol. Med. 2024, 222, 317–330. [Google Scholar] [CrossRef]
- Komza, M.; Khatun, J.; Gelles, J.D.; Trotta, A.P.; Enachescu, A.I.; Henao, J.; Elsaadi, A.; Kotini, A.G.; Clementelli, C.; Arandela, J.; et al. Metabolic adaptations to acute glucose uptake inhibition converge upon mitochondrial respiration for leukemia cell survival. Cell Commun. Signal 2025, 23, 47. [Google Scholar] [CrossRef]
- Penjweini, R.; Link, A.K.; Qazi, S.; Mattu, N.; Zuchowski, A.; Vasta, A.; Sackett, D.L.; Knutson, J.R. Low dose Taxol causes mitochondrial dysfunction in actively respiring cancer cells. J. Biol. Chem. 2025, 301, 108450. [Google Scholar] [CrossRef]
- Hwa, Y.L.; In, H.P.; Woo-Suk, C.; Sig, G.C. Triphenylphosphonium-conjugated glycol chitosan microspheres for mitochondria-targeted drug delivery. Int. J. Biol. Macromol. 2021, 167, 35–45. [Google Scholar]
- Wang, J.Y.; Li, J.Q.; Xiao, Y.M.; Fu, B.; Qin, Z.H. Triphenylphosphonium (TPP)-Based Antioxidants: A New Perspective on Antioxidant Design. ChemMedChem 2020, 15, 404–410. [Google Scholar] [CrossRef]
- Bavo, F.; Pucci, S.; Fasoli, F.; Lammi, C.; Moretti, M.; Lammi, C.; Moretti, M.; Mucchietto, V.; Lattuada, D.; Viani, P.; et al. Potent Antiglioblastoma Agents by Hybridizing the Onium-Alkyloxy-StilBene Based Structures of an alpha7-nAChR, alpha9-nAChR Antagonist and of a Pro-Oxidant Mitocan. J. Med. Chem. 2018, 61, 10531–10544. [Google Scholar] [CrossRef]
- Singh, Y.; Viswanadham, K.; Pawar, V.K.; Meher, J.; Jajoriya, A.K.; Omer, A.; Jaiswal, S.; Dewangan, J.; Bora, H.K.; Singh, P.; et al. Induction of Mitochondrial Cell Death and Reversal of Anticancer Drug Resistance via Nanocarriers Composed of a Triphenylphosphonium Derivative of Tocopheryl Polyethylene Glycol Succinate. Mol. Pharm. 2019, 16, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Sikora, A.; Pięta, J.; Zielonka, M.; Cheng, G.; Kalyanaraman, B. Targeting redox-active pyridinium cations to mitochondria to inhibit proliferation of colon cancer cells. Free Radic. Biol. Med. 2018, 128 (Suppl. S1), S78. [Google Scholar] [CrossRef]
- Huang, X.; Hou, W.; Quan, Q.; Wang, J.; Wang, M.; Liu, X.; Jin, C. Pharmacological Action and Molecular Mechanism of Formononetin. Asian Agric. Res. 2023, 15, 35–37,41. [Google Scholar]
- Jiang, D.J.; Azhar, R.; Rabia, B.; Sarfraz, I.; Hussain, G.; Mateen, T.M.; Qin, T.; Selamoglu, Z.; Ali, M.; Li, J.; et al. Potential Anticancer Properties and Mechanisms of Action of Formononetin. BioMed Res. Int. 2019, 2019, 5854315. [Google Scholar] [CrossRef]
- Yan, H.L.; Xue, W.S.; Sai, C.Z.; Yan, H.L.; Xue, W.S.; Sai, C.Z.; Wei, H.H.; Xue, W.; Han, Y.Z.; Yue, H.Q.; et al. Synthesis, characterization and biological evaluation of formononetin derivatives as novel EGFR inhibitors via inhibiting growth, migration and inducing apoptosis in breast cancer cell line. RSC Adv. 2017, 7, 48404–48419. [Google Scholar]
- Liu, F.; Shu, H.; Liu, D.; Zhang, X.; Zhu, Y.; Zhao, Y.; Cai, E.; Han, J. Inhibitory Effects and Mechanisms of Arctigenin Derivatives on Tumor Cells. Chin. J. Cell Biol. 2024, 46, 1225–1234. [Google Scholar]
- Zhao, Y. Synthesis and Anti-Severe Acute Pancreatitis Activity of 7-(3,4-Dihydroxy) Phenyl Lactic Acid Acyloxy Formononetin. Master’s Thesis, Binzhou Medical College, Yantai, China, 2020. [Google Scholar] [CrossRef]
- Li, B. Synthesis of Chickpea Shootin A,, Prickly Aristolochicin and Genistein Derivatives from Chickpea and Screening Studies on Their Hypoglycaemic Activities on Insulin-Resistant HepG2 Cells. Master’s Thesis, Beijing University of Traditional Chinese Medicine, Beijing, China, 2017. [Google Scholar]
- Kong, J. Synthesis and Activity Evaluation of Sulfonamide Derivatives Containing Pyrazoles and Triazole. Master’s Thesis, Qingdao University of Science and Technology, Qingdao, China, 2024. [Google Scholar]
- Shu, F. Design, Synthesis and Preliminary Study of Antitumour Effects of β-anhydroepimedin Derivatives. Master’s Thesis, Jilin Agricultural University, Changchun, China, 2024. [Google Scholar] [CrossRef]
- Song, H. Synthesis of Mitochondria-Targeted Tretinoin Derivatives and Their Antitumour Activities. Master’s Thesis, Shandong University, Jinan, China, 2020. [Google Scholar] [CrossRef]
- Cuthbertson, C.R.; Arabzada, Z.; Bankhead, A.; Kyani, A.; Neamati, N. A Review of Small-Molecule Inhibitors of One-Carbon Enzymes: SHMT2 and MTHFD2 in the Spotlight. ACS Pharmacol. Transl. Sci. 2021, 4, 624–646. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Rubini, E.; Paglia, G.; Chichiarelli, S.; Giamogante, F.; Macone, A.; Perugia, G.; Magliocca, F.M.; Gurtner, A.; et al. Shmt2: A Stat3 Signaling New Player in Prostate Cancer Energy Metabolism. Cells 2019, 8, 1048. [Google Scholar] [CrossRef]
- Wenqi, M.; Ronghan, L.; Kai, Z.; Zhong, J. Vital role of SHMT2 in diverse disease. Biochem. Biophys. Res. Commun. 2023, 671, 160–165. [Google Scholar]
- Liu, Z.; Fan, M.; Hou, J.; Pan, S.; Xu, Y.; Zhang, H.; Liu, C.; Hao, X.; Li, X.; Wang, H. Serine hydroxymethyltransferase 2 knockdown induces apoptosis in ccRCC by causing lysosomal membrane permeabilization via metabolic reprogramming. Cell Death Dis. 2023, 14, 144. [Google Scholar] [CrossRef]
- Han, T.; Wang, Y.; Cheng, M.; Han, T.; Wang, Y.; Cheng, M.; Hu, Q.; Wan, X.; Huang, M.; Liu, Y.; et al. Phosphorylated SHMT2 Regulates Oncogenesis Through m6 A Modification in Lung Adenocarcinoma. Adv. Sci. 2024, 11, e2307834. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, M.; Du, T.; Hou, Z.; You, S.; Zhang, S.; Ji, M.; Xue, N.; Chen, X. SHMT2 Promotes Gastric Cancer Development through Regulation of HIF1α/VEGF/STAT3 Signaling. Int. J. Mol. Sci. 2023, 24, 7150. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhao, M.; Li, R.; Zhang, Y.; Shi, X.; Ding, C.; Ma, C.; Lu, J.; Yue, X. SHMT2 promotes papillary thyroid cancer metastasis through epigenetic activation of AKT signaling. Cell Death Dis. 2024, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Vosooghi, M.; Firoozpour, L.; Rodaki, A.; Pordeli, M.; Safavi, M.; Ardestani, S.K.; Dadgar, A.; Asadipour, A.; Moshafi, M.H.; Foroumadi, A. Design, synthesis, docking study and cytotoxic activity evaluation of some novel letrozole analogs. Daru J. Fac. Pharm. Tehran Univ. Med. Sci. 2014, 22, 83. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, R.D. GNINA 1.0: Molecular docking with deep learning. J. Chem. Inf. 2021, 13, 43. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Cell Name | ||||
|---|---|---|---|---|---|
| HGC-27 | MCF-7 | A549 | PC-3M | HEK-293 | |
| 1a | 83.34 ± 8.32 | - | - | - | 83.43 ± 8.61 |
| 1b | 79.14 ± 5.65 | - | - | - | 70.72 ± 5.61 |
| 1c | 31.43 ± 4.93 | - | >100 | - | 67.96 ± 5.33 |
| 1d | 79.25 ± 9.27 | - | 86.20 ± 6.10 | - | 74.38 ± 4.28 |
| 1e | 66.06 ± 7.33 | - | 65.37 ± 5.01 | 83.99 ± 11.0 | 95.07 ± 7.97 |
| 1f | 55.58 ± 2.43 | - | 74.59 ± 11.44 | - | 95.72 ± 8.09 |
| 1g | 95.45 ± 6.23 | - | 53.57 ± 5.15 | >100 | 59.94 ± 7.79 |
| 2a | 13.39 ± 2.77 | 72.83 ± 6.25 | 28.79 ± 2.45 | 55.16 ± 4.40 | 44.82 ± 2.39 |
| 2b | 32.12 ± 5.96 | 46.49 ± 4.94 | 42.22 ± 3.69 | 69.18 ± 3.98 | 46.22 ± 3.99 |
| 2c | 18.62 ± 1.60 | 42.61 ± 9.44 | 12.19 ± 1.52 | 21.73 ± 1.25 | 76.99 ± 8.26 |
| 2d | 29.14 ± 2.58 | 56.75 ± 4.04 | 31.21 ± 4.40 | 66.99 ± 6.97 | 46.99 ± 8.41 |
| 2e | 39.16 ± 2.68 | 85.51 ± 4.99 | 38.96 ± 1.65 | 97.94 ± 6.33 | 64.05 ± 3.47 |
| 2f | 30.43 ± 2.12 | 52.28 ± 6.85 | 20.61 ± 1.21 | 27.86 ± 2.69 | 51.69 ± 5.48 |
| 2g | 29.36 ± 2.52 | 46.58 ± 4.95 | 23.64 ± 2.25 | 41.16 ± 4.40 | 59.57 ± 5.65 |
| 3a | 49.50 ± 3.35 | - | - | 61.97 ± 8.71 | - |
| 3b | 65.36 ± 3.88 | - | 96.44 ± 7.37 | - | - |
| 3c | 59.98 ± 7.82 | - | 82.96 ± 5.89 | - | - |
| 3d | 80.22 ± 4.80 | - | 78.08 ± 6.21 | 58.34 ± 5.36 | 78.28 ± 9.74 |
| 3e | 48.59 ± 5.30 | - | 74.02 ± 11.92 | - | - |
| 3f | 45.39 ± 5.24 | 84.99 ± 5.74 | 70.13 ± 9.67 | 46.71 ± 9.24 | 74.96 ± 5.49 |
| 3g | - | 67.75 ± 8.92 | 66.94 ± 9.70 | 50.35 ± 5.78 | 50.35 ± 6.13 |
| FMN | 29.55 ± 1.17 | - | 83.02 ± 6.25 | 76.39 ± 6.47 | 39.60 ± 3.26 |
| DOX | 5.97 ± 0.43 | 13.21 ± 0.43 | 15.41 ± 1.06 | 0.168 ± 0.06 | - |
| 5-FU | 36.24 ± 3.02 | - | 22.92 ± 3.54 | 48.05 ± 3.76 | >100 |
| NO. | Name | Synergy | Hydrophobicity |
|---|---|---|---|
| 2a | 7-O-(6-Bromohexanoyl)-formononetin sapogenins-triphenylphosphine couples | −9.3 Kcal/mol | ASN-408, ALA-227, ALA-418, TYR-106, TYR-105, PRO-321, PRO-167, LEU-172, LEU-166, ILE-183 |
| 2b | 7-O-(7-Bromoheptanoyl)-formononetin sapogenins-triphenylphosphine couples | −8.6 Kcal/mol | LYS-409, TYR-105, PRO-421, ALA-418, LYS-181, PHE-317, LEU-172, LEU-166, ILE-183, TYR-176 |
| 2c | 7-O-(8-Bromooctanoyl)-formononetin sapogenins-triphenylphosphine couples | −9.4 Kcal/mol | PHE-320, PHE-317, ILE-183, LEU-166, LEU-172, ALA-227, ASN-408, GLU-98, TYR-96, TYR-105 |
| 2d | 7-O-(9-Bromononanoyl)-formononetin sapogenins-triphenylphosphine couples | −9.2 Kcal/mol | LEU-166, TYR-106, ALA-418, PHE-320, ASN-408, TYR-105 |
| 2e | 7-O-(10-Bromodecanoyl)-formononetin sapogenins-triphenylphosphine couples | −9.0 Kcal/mol | LEU-172, PHE-320, LEU-166, PHE-317, ILE-183, ASP-313, TYR-105, LYS-103 |
| 2f | 7-O-(11-Bromoundecanoyl)-formononetin sapogenins-triphenylphosphine couples | −9.1 Kcal/mol | ALA-418, PHE-320, ASN-408, TYR-105, LYS-409, PRO-167, LEU-166, ALA-227 |
| 2g | 7-O-(12-Bromododecanoyl)-formononetin sapogenins-triphenylphosphine couplings | −8.9 Kcal/mol | TYR-100, LYS-103, PHE-317, LEU-172, PHE-320, PRO-321, ILE-183, LEU-166, TYR-105 |
| FMN | formononetin | −7.9 Kcal/mol | TYR-106, TYR-105, PHE-320, PRO-321, LEU-172, LEU-166 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.; Zhao, Y.; Li, W.; Cui, H.; Han, J.; Cai, E. Design, Synthesis, Anti-Tumor Activity and Molecular Docking Studies of Novel Triphenylphosphine-Containing Formononetin Derivatives. Int. J. Mol. Sci. 2025, 26, 5280. https://doi.org/10.3390/ijms26115280
Cui H, Zhao Y, Li W, Cui H, Han J, Cai E. Design, Synthesis, Anti-Tumor Activity and Molecular Docking Studies of Novel Triphenylphosphine-Containing Formononetin Derivatives. International Journal of Molecular Sciences. 2025; 26(11):5280. https://doi.org/10.3390/ijms26115280
Chicago/Turabian StyleCui, Hongjuan, Yan Zhao, Wei Li, Huanjie Cui, Jiahong Han, and Enbo Cai. 2025. "Design, Synthesis, Anti-Tumor Activity and Molecular Docking Studies of Novel Triphenylphosphine-Containing Formononetin Derivatives" International Journal of Molecular Sciences 26, no. 11: 5280. https://doi.org/10.3390/ijms26115280
APA StyleCui, H., Zhao, Y., Li, W., Cui, H., Han, J., & Cai, E. (2025). Design, Synthesis, Anti-Tumor Activity and Molecular Docking Studies of Novel Triphenylphosphine-Containing Formononetin Derivatives. International Journal of Molecular Sciences, 26(11), 5280. https://doi.org/10.3390/ijms26115280