
Evolutionary Origins and Functional Diversification of 2′-O-Methyltransferases: Insights from Phylogenetic and Structural Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Literature-Based Collection of 2′-O-MTases Reveals 11 Distinct MTase Domains for Nm Catalyzation
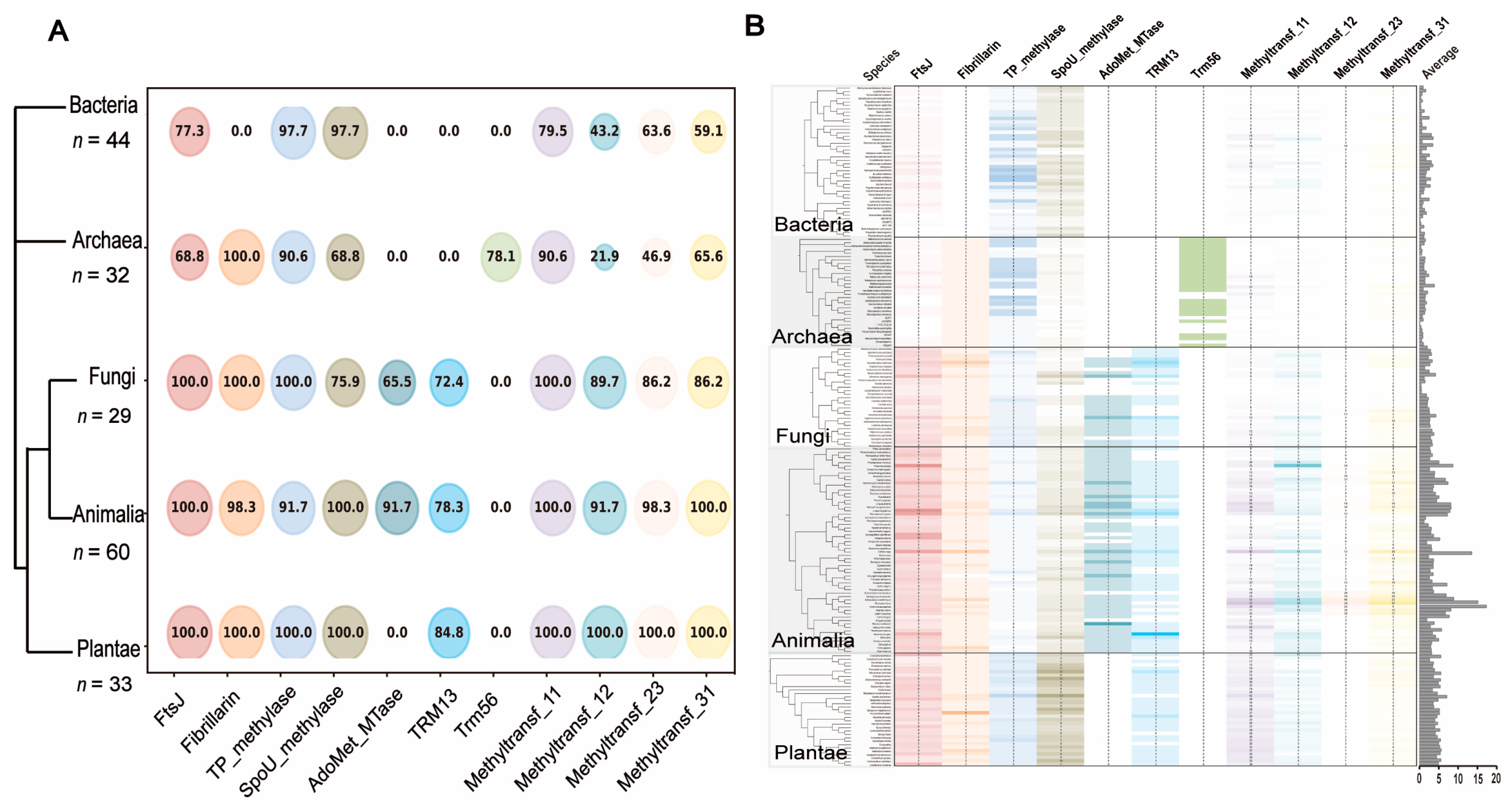
2.2. Phylogenetic Distribution of the 11 MTase Domain-Containing Proteins Suggests Ancient and Life Kingdom-Specific Origins of 2′-O-MTases
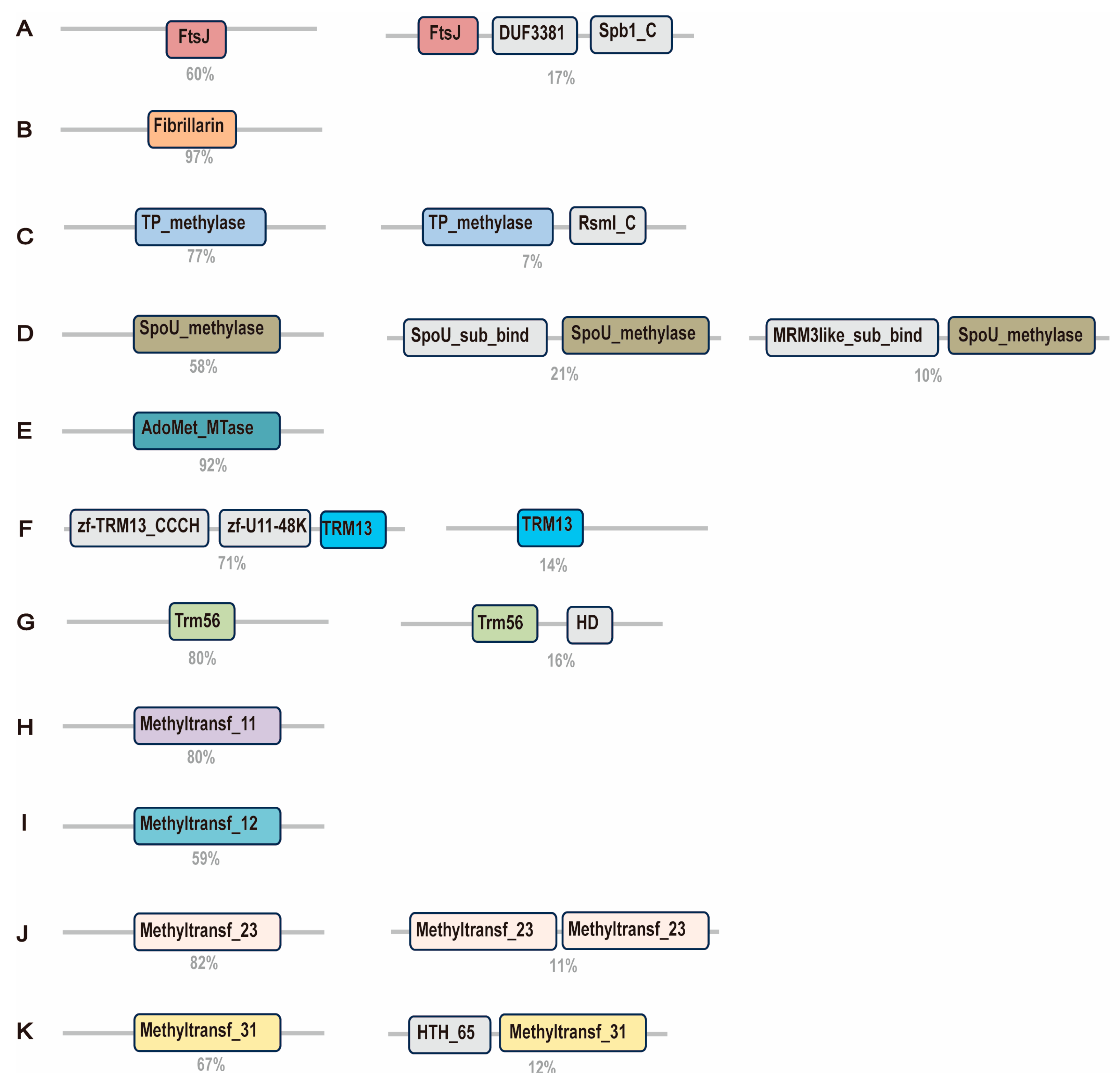
2.3. Diverse Domain Architectures of 2′-O-MTases
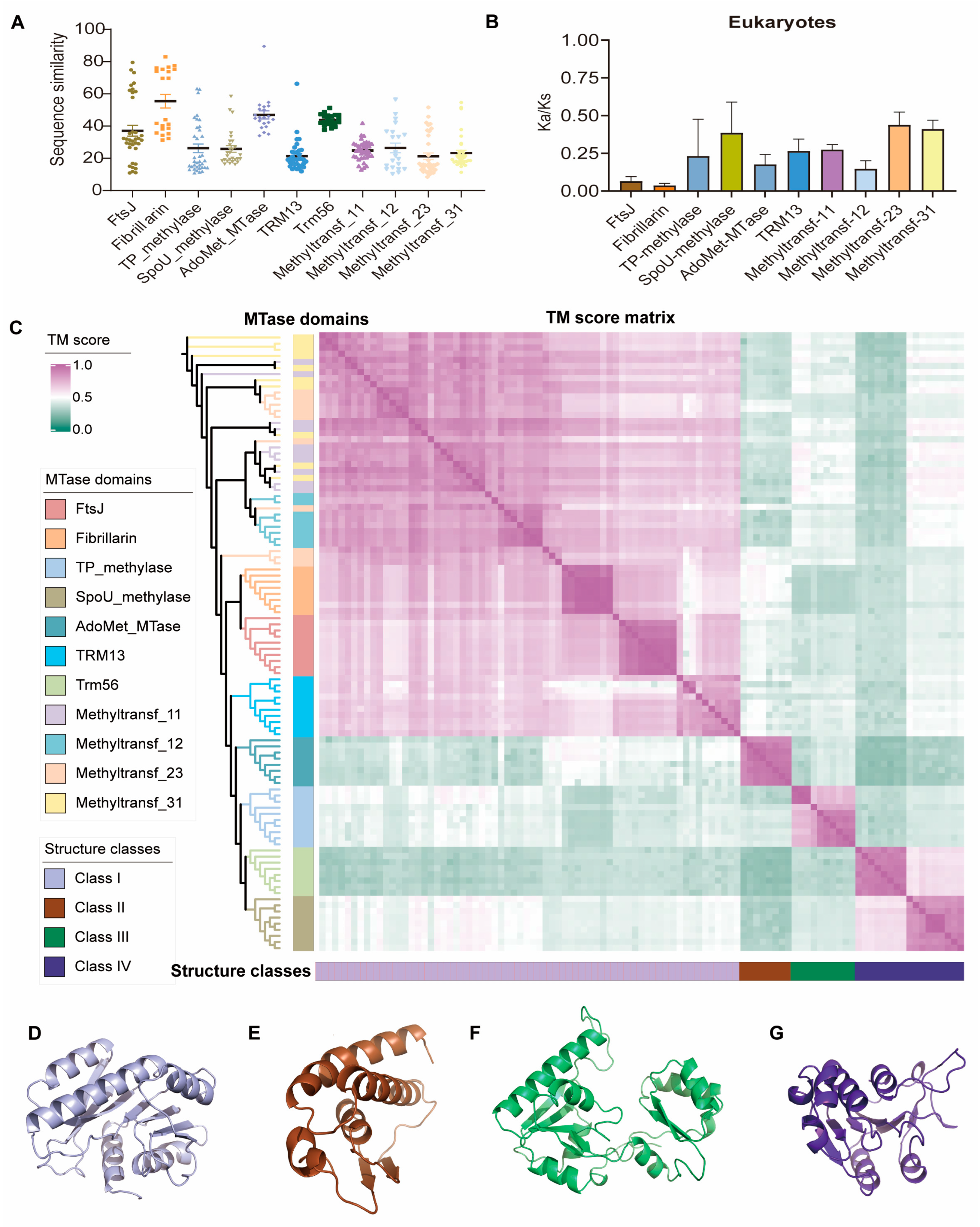
2.4. Evolutionary Features of Sequence and Structure of 2′-O-MTases
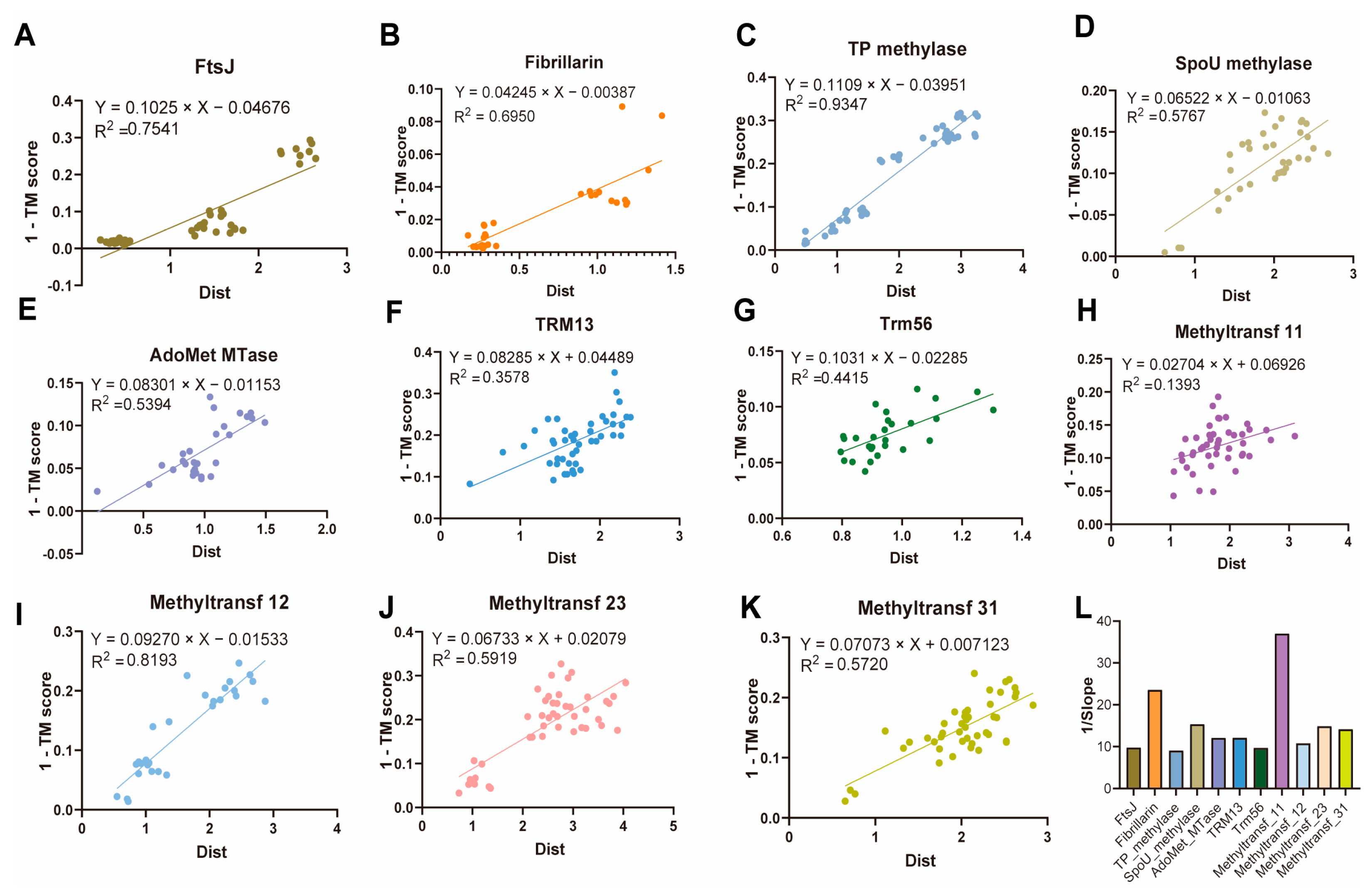
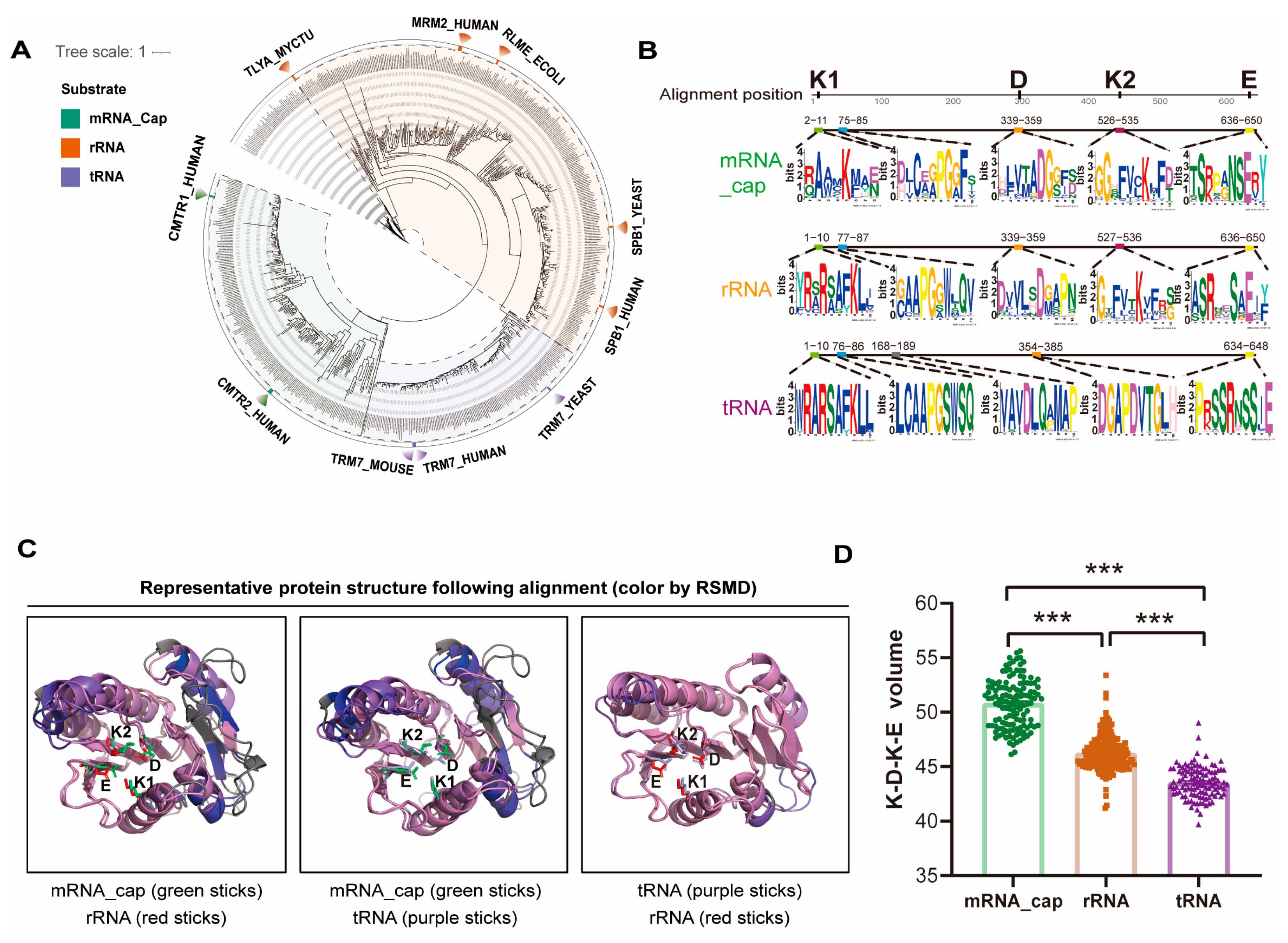
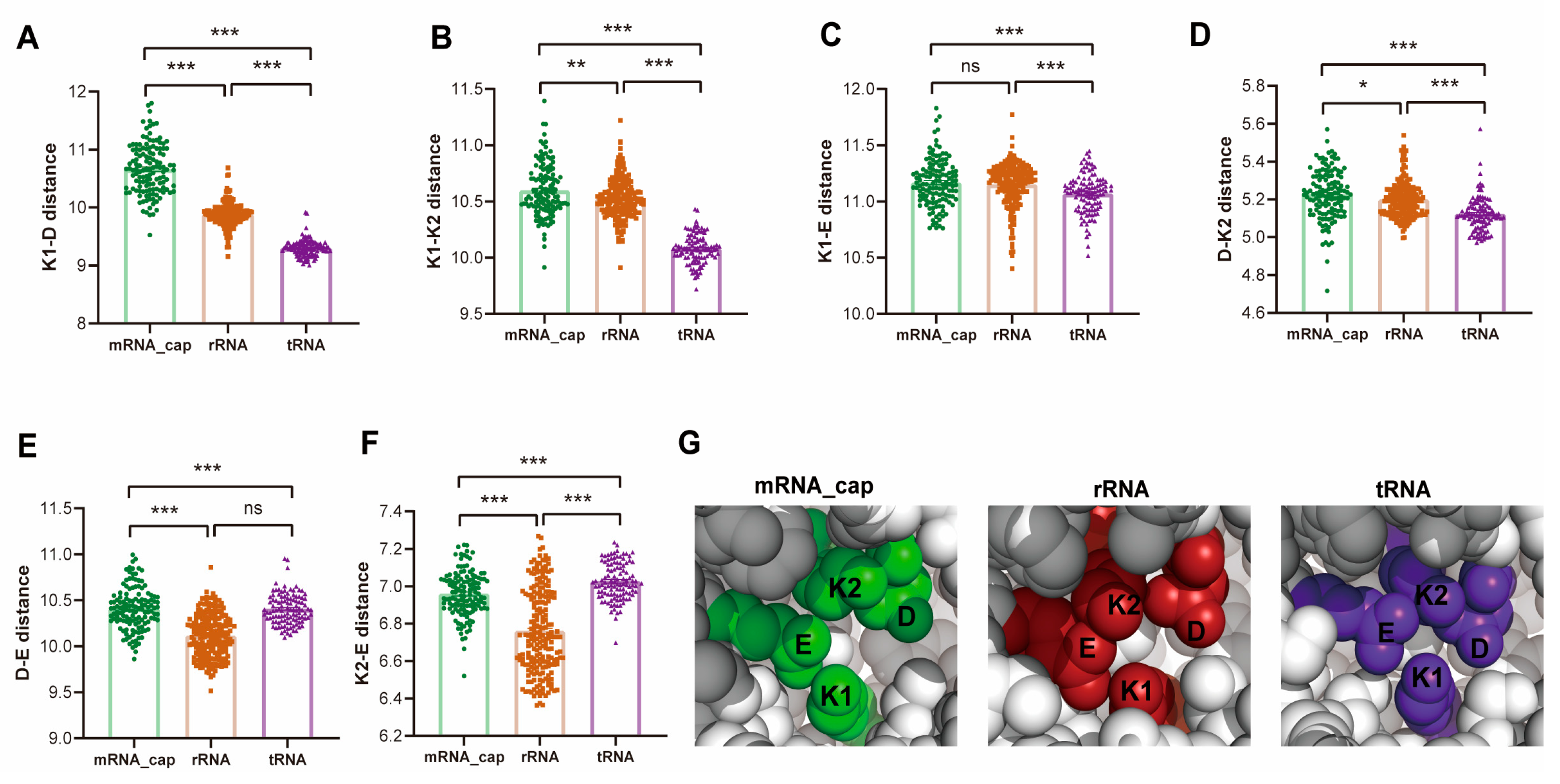
2.5. Ancient Divergence and Substrate-Adaptive Evolution of the FtsJ Protein Family
3. Discussion
4. Materials and Methods
4.1. Identification of 2′-O-MTase-Conserved, Methyltransferase-Associated Domains
4.2. Identification of 2′-O-MTases Across the Tree of Life
4.3. Domain Architecture Pattern Analysis
4.4. Multiple-Sequence Alignment and Phylogenetic Analysis
4.5. Ka/Ks Analysis
4.6. Structural Similarity Analysis and Clustering
4.7. Polyphony Analysis
4.8. Motif Identification
4.9. Tetrahedron KDKE Volume Calculation and Pairwise Distance Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Nm | 2′-O-methylation |
| 2′-O-MTase | 2′-O-methyltransferase |
| MTase | Methyltransferase |
| LUCA | Last Universal Common Ancestor |
| Ka | Nonsynonymous substitution rate |
| Ks | Synonymous substitution rate |
| m6A | N6-methyladenosine |
| m5C | 5-methylcytosine |
| m1A | N1-methyladenosine |
| m7G | N7-methylguanosine |
| ac4C | N4-acetylcytosine |
| Ψ | pseudouridine |
| SAM | S-adenosylmethionine |
| piRNA | Piwi-interacting RNA |
| miRNA | MicroRNA |
| snoRNP | Small nucleolar ribonucleoprotein |
References
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Pecot, C.V.; Holley, C.L. 2′-O-methylation (Nm) in RNA: Progress, challenges, and future directions. RNA 2024, 30, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Khoshnevis, S.; Dreggors-Walker, R.E.; Marchand, V.; Motorin, Y.; Ghalei, H. Ribosomal RNA 2′-O-methylations regulate translation by impacting ribosome dynamics. Proc. Natl. Acad. Sci. USA 2022, 119, e2117334119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, Z. tRNA modifications: Greasing the wheels of translation and beyond. RNA Biol. 2025, 22, 1–25. [Google Scholar] [CrossRef]
- Li, J.; Yang, Z.; Yu, B.; Liu, J.; Chen, X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005, 15, 1501–1507. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Ge, J.; Liu, H.; Yu, Y.T. Regulation of pre-mRNA splicing in Xenopus oocytes by targeted 2′-O-methylation. RNA 2010, 16, 1078–1085. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, Y.; Wang, S.; Lu, X.; Gu, X.; Li, Y.; Yang, F.; Xu, R.; Wang, T.; Jiao, Z.; et al. An integrative platform for detection of RNA 2′-O-methylation reveals its broad distribution on mRNA. Cell Rep. Methods 2024, 4, 100721. [Google Scholar] [CrossRef]
- Rodriguez-Corona, U.; Sobol, M.; Rodriguez-Zapata, L.C.; Hozak, P.; Castano, E. Fibrillarin from Archaea to human. Biol. Cell 2015, 107, 159–174. [Google Scholar] [CrossRef]
- Pintard, L.; Lecointe, F.; Bujnicki, J.M.; Bonnerot, C.; Grosjean, H.; Lapeyre, B. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J. 2002, 21, 1811–1820. [Google Scholar] [CrossRef]
- Drazkowska, K.; Tomecki, R.; Warminski, M.; Baran, N.; Cysewski, D.; Depaix, A.; Kasprzyk, R.; Kowalska, J.; Jemielity, J.; Sikorski, P.J. 2′-O-Methylation of the second transcribed nucleotide within the mRNA 5′ cap impacts the protein production level in a cell-specific manner and contributes to RNA immune evasion. Nucleic Acids Res. 2022, 50, 9051–9071. [Google Scholar] [CrossRef] [PubMed]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef]
- Decombe, A.; El Kazzi, P.; Decroly, E. Interplay of RNA 2′-O-methylations with viral replication. Curr. Opin. Virol. 2023, 59, 101302. [Google Scholar] [CrossRef] [PubMed]
- Park, G.J.; Osinski, A.; Hernandez, G.; Eitson, J.L.; Majumdar, A.; Tonelli, M.; Henzler-Wildman, K.; Pawłowski, K.; Chen, Z.; Li, Y.; et al. The mechanism of RNA capping by SARS-CoV-2. Nature 2022, 609, 793–800. [Google Scholar] [CrossRef]
- Deng, J.; Yang, S.; Li, Y.; Tan, X.; Liu, J.; Yu, Y.; Ding, Q.; Fan, C.; Wang, H.; Chen, X.; et al. Natural evidence of coronaviral 2′-O-methyltransferase activity affecting viral pathogenesis via improved substrate RNA binding. Signal Transduct. Target. Ther. 2024, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Bergant, V.; Yamada, S.; Grass, V.; Tsukamoto, Y.; Lavacca, T.; Krey, K.; Mühlhofer, M.T.; Wittmann, S.; Ensser, A.; Herrmann, A.; et al. Attenuation of SARS-CoV-2 replication and associated inflammation by concomitant targeting of viral and host cap 2′-O-ribose methyltransferases. EMBO J. 2022, 41, e111608. [Google Scholar] [CrossRef]
- Pereira-Santana, A.; Gamboa-Tuz, S.D.; Zhao, T.; Schranz, M.E.; Vinuesa, P.; Bayona, A.; Rodríguez-Zapata, L.C.; Castano, E. Fibrillarin evolution through the Tree of Life: Comparative genomics and microsynteny network analyses provide new insights into the evolutionary history of Fibrillarin. PLoS Comput. Biol. 2020, 16, e1008318. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Wang, Y.; Wang, H.; Yang, J.; Pan, X.; Zhao, Y.; Xu, H.; Jiang, P.; Qian, P.; et al. Fibrillarin homologs regulate translation in divergent cell lineages during planarian homeostasis and regeneration. EMBO J. 2024, 43, 6591–6625. [Google Scholar] [CrossRef]
- Hajieghrari, B.; Niazi, A. Phylogenetic and Evolutionary Analysis of Plant Small RNA 2′-O-Methyltransferase (HEN1) Protein Family. J. Mol. Evol. 2023, 91, 424–440. [Google Scholar] [CrossRef]
- Tkaczuk, K.L.; Dunin-Horkawicz, S.; Purta, E.; Bujnicki, J.M. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinform. 2007, 8, 73. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.N.; Xu, B.S.; Liu, Y.P.; Zhou, M.; Long, T.; Li, H.; Dong, H.; Nie, Y.; Chen, P.R.; et al. Intellectual disability-associated gene ftsj1 is responsible for 2′-O-methylation of specific tRNAs. EMBO Rep. 2020, 21, e50095. [Google Scholar] [CrossRef] [PubMed]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 2019, 565, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.L.; Crary, S.M.; Jackman, J.E.; Grayhack, E.J.; Phizicky, E.M. The 2′-O-methyltransferase responsible for modification of yeast tRNA at position 4. RNA 2007, 13, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Suzuki, T. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 2010, 38, 1341–1352. [Google Scholar] [CrossRef]
- Persson, B.C.; Jäger, G.; Gustafsson, C. The spoU gene of Escherichia coli, the fourth gene of the spoT operon, is essential for tRNA (Gm18) 2′-O-methyltransferase activity. Nucleic Acids Res. 1997, 25, 4093–4097. [Google Scholar] [CrossRef]
- Cavaillé, J.; Chetouani, F.; Bachellerie, J.P. The yeast Saccharomyces cerevisiae YDL112w ORF encodes the putative 2′-O-ribose methyltransferase catalyzing the formation of Gm18 in tRNAs. RNA 1999, 5, 66–81. [Google Scholar] [CrossRef]
- Purta, E.; van Vliet, F.; Tkaczuk, K.L.; Dunin-Horkawicz, S.; Mori, H.; Droogmans, L.; Bujnicki, J.M. The yfhQ gene of Escherichia coli encodes a tRNA:Cm32/Um32 methyltransferase. BMC Mol. Biol. 2006, 7, 23. [Google Scholar] [CrossRef]
- Benítez-Páez, A.; Villarroya, M.; Douthwaite, S.; Gabaldón, T.; Armengod, M.E. YibK is the 2′-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNA(Leu) isoacceptors. RNA 2010, 16, 2131–2143. [Google Scholar] [CrossRef]
- Kressler, D.; Rojo, M.; Linder, P.; Cruz, J. Spb1p is a putative methyltransferase required for 60S ribosomal subunit biogenesis in Saccharomyces cerevisiae. Nucleic Acids Res. 1999, 27, 4598–4608. [Google Scholar] [CrossRef]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins 2009, 77, 499–508. [Google Scholar] [CrossRef]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Smietanski, M.; Werner, M.; Purta, E.; Kaminska, K.H.; Stepinski, J.; Darzynkiewicz, E.; Nowotny, M.; Bujnicki, J.M. Structural analysis of human 2′-O-ribose methyltransferases involved in mRNA cap structure formation. Nat. Commun. 2014, 5, 3004. [Google Scholar] [CrossRef]
- Punekar, A.S.; Shepherd, T.R.; Liljeruhm, J.; Forster, A.C.; Selmer, M. Crystal structure of RlmM, the 2′O-ribose methyltransferase for C2498 of Escherichia coli 23S rRNA. Nucleic Acids Res. 2012, 40, 10507–10520. [Google Scholar] [CrossRef]
- Werner, M.; Purta, E.; Kaminska, K.H.; Cymerman, I.A.; Campbell, D.A.; Mittra, B.; Zamudio, J.R.; Sturm, N.R.; Jaworski, J.; Bujnicki, J.M. 2′-O-ribose methylation of cap2 in human: Function and evolution in a horizontally mobile family. Nucleic Acids Res. 2011, 39, 4756–4768. [Google Scholar] [CrossRef]
- Häfner, S.J.; Jansson, M.D.; Altinel, K.; Andersen, K.L.; Abay-Nørgaard, Z.; Ménard, P.; Fontenas, M.; Sørensen, D.M.; Gay, D.M.; Arendrup, F.S.; et al. Ribosomal RNA 2′-O-methylation dynamics impact cell fate decisions. Dev. Cell 2023, 58, 1593–1609.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, B.; Xu, H.; Li, J.; Ma, C.; Ge, W.; Lu, C.; Cao, X. Identification of FBLL1 as a neuron-specific RNA 2′-O-methyltransferase mediating neuronal differentiation. Proc. Natl. Acad. Sci. USA 2024, 121, e2406961121. [Google Scholar] [CrossRef] [PubMed]
- Vaysset, H.; Meers, C.; Cury, J.; Bernheim, A.; Sternberg, S.H. Evolutionary origins of archaeal and eukaryotic RNA-guided RNA modification in bacterial IS110 transposons. Nat. Microbiol. 2025, 10, 20–27. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Ghaly, T.M.; Tetu, S.G.; Penesyan, A.; Qi, Q.; Rajabal, V.; Gillings, M.R. Discovery of integrons in Archaea: Platforms for cross-domain gene transfer. Sci. Adv. 2022, 8, eabq6376. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. Camb. Philos. Soc. 2019, 94, 2101–2137. [Google Scholar] [CrossRef] [PubMed]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The Fungal Tree of Life: From Molecular Systematics to Genome-Scale Phylogenies. Microbiol. Spectr. 2017, 5, 10.1128. [Google Scholar] [CrossRef] [PubMed]
- Simakov, O.; Kawashima, T.; Marlétaz, F.; Jenkins, J.; Koyanagi, R.; Mitros, T.; Hisata, K.; Bredeson, J.; Shoguchi, E.; Gyoja, F.; et al. Hemichordate genomes and deuterostome origins. Nature 2015, 527, 459–465. [Google Scholar] [CrossRef]
- Telford, M.J.; Budd, G.E.; Philippe, H. Phylogenomic Insights into Animal Evolution. Curr. Biol. 2015, 25, R876–R887. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, X.; Liu, Y.; Yang, L.; Shi, X.; Wang, H.; Diao, R.; Zhong, B. Origin and evolution of green plants in the light of key evolutionary events. J. Integr. Plant Biol. 2022, 64, 516–535. [Google Scholar] [CrossRef]
- Li, S.X.; Liu, Y.; Zhang, Y.M.; Chen, J.Q.; Shao, Z.Q. Convergent reduction of immune receptor repertoires during plant adaptation to diverse special lifestyles and habitats. Nat. Plants 2025, 11, 248–262. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-align: Universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Pitt, W.R.; Montalvão, R.W.; Blundell, T.L. Polyphony: Superposition independent methods for ensemble-based drug discovery. BMC Bioinform. 2014, 15, 324. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.-N.; Liu, X.-X.; Lei, L.-J.; Wang, Q.; Shao, Z.-Q.; Liu, Y. Evolutionary Origins and Functional Diversification of 2′-O-Methyltransferases: Insights from Phylogenetic and Structural Analysis. Int. J. Mol. Sci. 2025, 26, 5260. https://doi.org/10.3390/ijms26115260
Wang S-N, Liu X-X, Lei L-J, Wang Q, Shao Z-Q, Liu Y. Evolutionary Origins and Functional Diversification of 2′-O-Methyltransferases: Insights from Phylogenetic and Structural Analysis. International Journal of Molecular Sciences. 2025; 26(11):5260. https://doi.org/10.3390/ijms26115260
Chicago/Turabian StyleWang, Sai-Nan, Xiao-Xia Liu, Ling-Jie Lei, Qiang Wang, Zhu-Qing Shao, and Yang Liu. 2025. "Evolutionary Origins and Functional Diversification of 2′-O-Methyltransferases: Insights from Phylogenetic and Structural Analysis" International Journal of Molecular Sciences 26, no. 11: 5260. https://doi.org/10.3390/ijms26115260
APA StyleWang, S.-N., Liu, X.-X., Lei, L.-J., Wang, Q., Shao, Z.-Q., & Liu, Y. (2025). Evolutionary Origins and Functional Diversification of 2′-O-Methyltransferases: Insights from Phylogenetic and Structural Analysis. International Journal of Molecular Sciences, 26(11), 5260. https://doi.org/10.3390/ijms26115260