
Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets
 , , , , ,
, , , , ,  , and
, and 
Abstract
1. Introduction
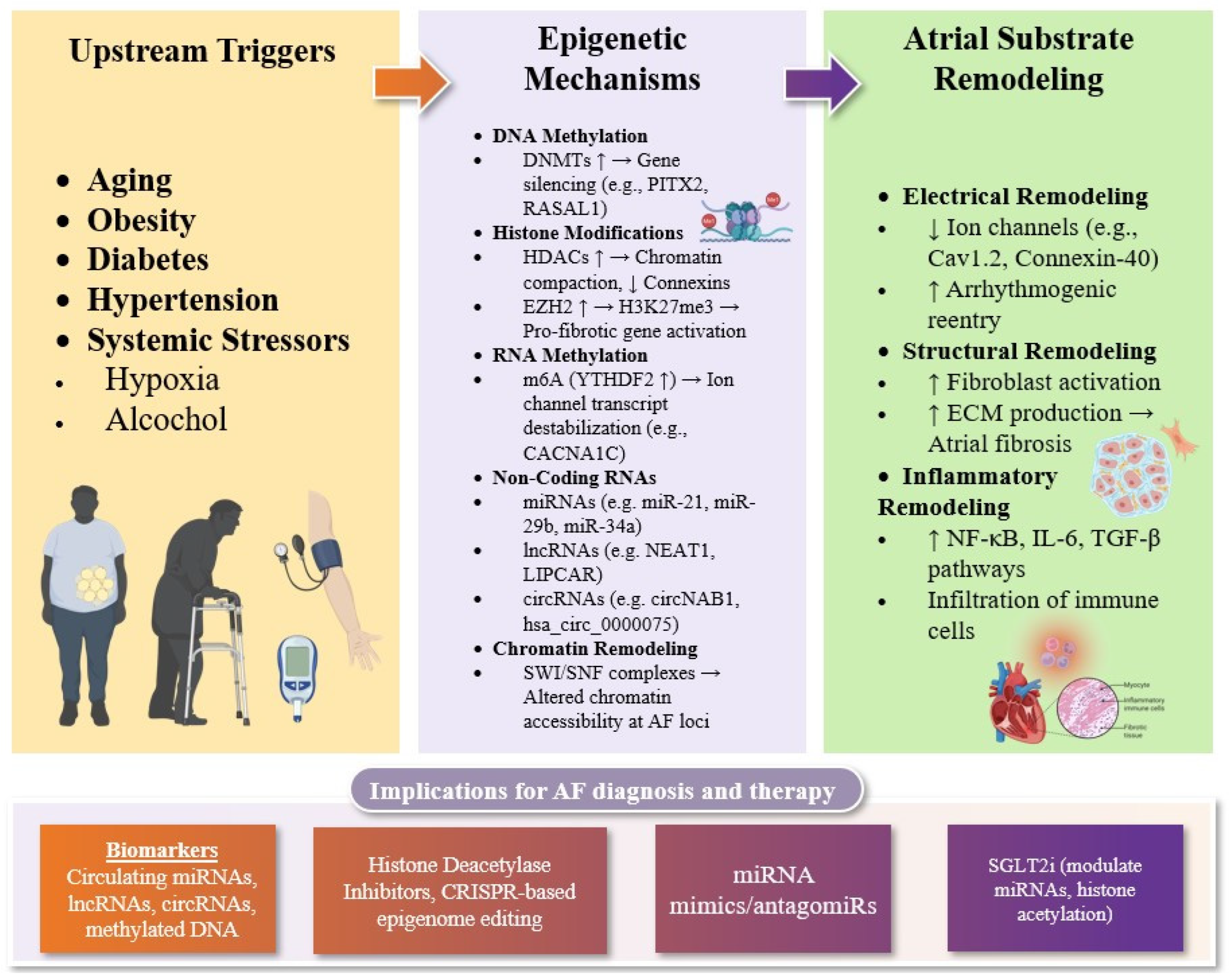
2. Epigenetic Regulation of Structural and Electrical Remodeling in AF
2.1. DNA Methylation and Histone Modifications in AF
2.2. RNA Methylation in AF
2.3. Chromatin Remodeling in AF
2.4. Non-Coding RNAs in AF
2.4.1. MicroRNAs
2.4.2. Long Non-Coding RNAs
2.4.3. Circular RNAs
3. Modifiers of Atrial Epigenetics: The Role of Systemic Stressors
3.1. Aging
3.2. Obesity
3.3. Diabetes Mellitus
3.4. Hypertension
3.5. Hypoxia
3.6. Alcohol
4. Emerging Epigenetic Therapies for AF
4.1. Histone Deacetylase Inhibitors
4.2. miRNA-Based Therapies
4.3. CRISPR-Based Epigenome Editing
4.4. Sodium-Glucose Cotransporter 2 Inhibitors
5. Limitations and Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tan, S.; Zhou, J.; Veang, T.; Lin, Q.; Liu, Q. Global, Regional, and National Burden of Atrial Fibrillation and Atrial Flutter from 1990 to 2021: Sex Differences and Global Burden Projections to 2046—A Systematic Analysis of the Global Burden of Disease Study 2021. Europace 2025, 27, euaf027. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef]
- Linz, D.; Gawalko, M.; Betz, K.; Hendriks, J.M.; Lip, G.Y.H.; Vinter, N.; Guo, Y.; Johnsen, S. Atrial Fibrillation: Epidemiology, Screening and Digital Health. Lancet Reg. Heal. Eur. 2024, 37, 100786. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Bengtson, L.G.S. A Rising Tide: The Global Epidemic of Atrial Fibrillation. Circulation 2014, 129, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Andrade, J.G.; Arbelo, E.; Boriani, G.; Breithardt, G.; Camm, A.J.; Caso, V.; Nielsen, J.C.; De Melis, M.; De Potter, T.; et al. Longer and Better Lives for Patients with Atrial Fibrillation: The 9th AFNET/EHRA Consensus Conference. Europace 2024, 26, euae070. [Google Scholar] [CrossRef]
- Karakasis, P.; Pamporis, K.; Siontis, K.C.; Theofilis, P.; Samaras, A.; Patoulias, D.; Stachteas, P.; Karagiannidis, E.; Stavropoulos, G.; Tzikas, A.; et al. Major Clinical Outcomes in Symptomatic vs. Asymptomatic Atrial Fibrillation: A Meta-Analysis. Eur. Heart J. 2024, 46, 1189–1202. [Google Scholar] [CrossRef]
- Pamporis, K.; Karakasis, P.; Sagris, M.; Theofilis, P.; Milaras, N.; Pantelidaki, A.; Mourouzis, I.; Fragakis, N.; Vlachos, K.; Kordalis, A.; et al. Prevalence of Asymptomatic Atrial Fibrillation and Risk Factors Associated with Asymptomatic Status: A Systematic Review and Meta-Analysis. Eur. J. Prev. Cardiol. 2025, zwaf138. [Google Scholar] [CrossRef]
- Packer, D.L.; Mark, D.B.; Robb, R.A.; Monahan, K.H.; Bahnson, T.D.; Poole, J.E.; Noseworthy, P.A.; Rosenberg, Y.D.; Jeffries, N.; Mitchell, L.B.; et al. Effect of Catheter Ablation vs Antiarrhythmic Drug Therapy on Mortality, Stroke, Bleeding, and Cardiac Arrest Among Patients With Atrial Fibrillation: The CABANA Randomized Clinical Trial. JAMA 2019, 321, 1261–1274. [Google Scholar] [CrossRef]
- Verma, A.; Jiang, C.; Betts, T.R.; Chen, J.; Deisenhofer, I.; Mantovan, R.; Macle, L.; Morillo, C.A.; Haverkamp, W.; Weerasooriya, R.; et al. Approaches to Catheter Ablation for Persistent Atrial Fibrillation. N. Engl. J. Med. 2015, 372, 1812–1822. [Google Scholar] [CrossRef]
- Karakasis, P.; Fragakis, N.; Patoulias, D.; Theofilis, P.; Kassimis, G.; Karamitsos, T.; El-Tanani, M.; Rizzo, M. Effects of Glucagon-Like Peptide 1 Receptor Agonists on Atrial Fibrillation Recurrence After Catheter Ablation: A Systematic Review and Meta-Analysis. Adv. Ther. 2024, 41, 3749–3756. [Google Scholar] [CrossRef]
- Goette, A.; Corradi, D.; Dobrev, D.; Aguinaga, L.; Cabrera, J.-A.; Chugh, S.S.; de Groot, J.R.; Soulat-Dufour, L.; Fenelon, G.; Hatem, S.N.; et al. Atrial Cardiomyopathy Revisited-Evolution of a Concept: A Clinical Consensus Statement of the European Heart Rhythm Association (EHRA) of the ESC, the Heart Rhythm Society (HRS), the Asian Pacific Heart Rhythm Society (APHRS), and the Latin American Hear. Europace 2024, 26, euae204. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Arora, R.; Jalife, J. Atrial Myopathy. JACC Basic Transl. Sci. 2019, 4, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Gerra, L.; Mantovani, M.; Tartaglia, E.; Mei, D.A.; Imberti, J.F.; Vitolo, M.; Bonini, N. Atrial Cardiomyopathy: An Entity of Emerging Interest in the Clinical Setting. Eur. J. Intern. Med. 2023, 118, 14–21. [Google Scholar] [CrossRef]
- Karakasis, P.; Patoulias, D.; Popovic, D.S.; Pamporis, K.; Theofilis, P.; Nasoufidou, A.; Stachteas, P.; Samaras, A.; Tzikas, A.; Giannakoulas, G.; et al. Effects of Mineralocorticoid Receptor Antagonists on New-Onset or Recurrent Atrial Fibrillation: A Bayesian and Frequentist Network Meta-Analysis of Randomized Trials. Curr. Probl. Cardiol. 2024, 49, 102742. [Google Scholar] [CrossRef]
- Wilber, D.J.; Pappone, C.; Neuzil, P.; De Paola, A.; Marchlinski, F.; Natale, A.; Macle, L.; Daoud, E.G.; Calkins, H.; Hall, B.; et al. Comparison of Antiarrhythmic Drug Therapy and Radiofrequency Catheter Ablation in Patients with Paroxysmal Atrial Fibrillation: A Randomized Controlled Trial. JAMA 2010, 303, 333–340. [Google Scholar] [CrossRef]
- Razzack, A.A.; Lak, H.M.; Pothuru, S.; Rahman, S.; Hassan, S.A.; Hussain, N.; Najeeb, H.; Reddy, K.T.; Syeda, H.; Yasmin, F.; et al. Efficacy and Safety of Catheter Ablation vs Antiarrhythmic Drugs as Initial Therapy for Management of Symptomatic Paroxysmal Atrial Fibrillation: A Meta-Analysis. Rev. Cardiovasc. Med. 2022, 23, 112. [Google Scholar] [CrossRef]
- Sanchez-Somonte, P.; Kittichamroen, N.; Gao-Kang, J.; Azizi, Z.; Alipour, P.; Kahykin, Y.; Pantano, A.; Verma, A. Incremental Efficacy for Repeat Ablation Procedures for Catheter Ablation of Atrial Fibrillation: 5-Year Follow-Up. JACC Adv. 2024, 3, 101200. [Google Scholar] [CrossRef]
- Ngo, L.; Lee, X.W.; Elwashahy, M.; Arumugam, P.; Yang, I.A.; Denman, R.; Haqqani, H.; Ranasinghe, I. Freedom from Atrial Arrhythmia and Other Clinical Outcomes at 5 Years and beyond after Catheter Ablation of Atrial Fibrillation: A Systematic Review and Meta-Analysis. Eur. Heart J.-Qual. Care Clin. Outcomes 2023, 9, 447–458. [Google Scholar] [CrossRef]
- Van Gelder, I.C.; Rienstra, M.; Bunting, K.V.; Casado-Arroyo, R.; Caso, V.; Crijns, H.J.G.M.; De Potter, T.J.R.; Dwight, J.; Guasti, L.; Hanke, T.; et al. 2024 ESC Guidelines for the Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2024, 45, 3314–3414. [Google Scholar] [CrossRef]
- Joglar, J.A.; Chung, M.K.; Armbruster, A.L.; Benjamin, E.J.; Chyou, J.Y.; Cronin, E.M.; Deswal, A.; Eckhardt, L.L.; Goldberger, Z.D.; Gopinathannair, R.; et al. 2023 ACC/AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1–e156. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Ktenopoulos, N.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial Cardiomyopathy in Atrial Fibrillation: Mechanistic Pathways and Emerging Treatment Concepts. J. Clin. Med. 2025, 14, 3250. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Vlachakis, P.K.; Theofilis, P.; Ktenopoulos, N.; Patoulias, D.; Fyntanidou, B.; Antoniadis, A.P.; Fragakis, N. Atrial Cardiomyopathy in Atrial Fibrillation: A Multimodal Diagnostic Framework. Diagnostics 2025, 15, 1207. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Korantzopoulos, P.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial Fibrosis in Atrial Fibrillation: Mechanistic Insights, Diagnostic Challenges, and Emerging Therapeutic Targets. Int. J. Mol. Sci. 2024, 26, 209. [Google Scholar] [CrossRef] [PubMed]
- Donniacuo, M.; De Angelis, A.; Telesca, M.; Bellocchio, G.; Riemma, M.A.; Paolisso, P.; Scisciola, L.; Cianflone, E.; Torella, D.; Castaldo, G.; et al. Atrial Fibrillation: Epigenetic Aspects and Role of Sodium-Glucose Cotransporter 2 Inhibitors. Pharmacol. Res. 2023, 188, 106591. [Google Scholar] [CrossRef]
- Molina, C.E.; Abu-Taha, I.H.; Wang, Q.; Roselló-Díez, E.; Kamler, M.; Nattel, S.; Ravens, U.; Wehrens, X.H.T.; Hove-Madsen, L.; Heijman, J.; et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients With and Without Atrial Fibrillation. Front. Physiol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Deans, C.; Maggert, K.A. What Do You Mean, “Epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The Impact of Epigenetics on Cardiovascular Disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y.; et al. EZH2 as a Novel Therapeutic Target for Atrial Fibrosis and Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef]
- Gillette, T.G.; Hill, J.A. Readers, Writers, and Erasers: Chromatin as the Whiteboard of Heart Disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef]
- Chiarella, A.M.; Lu, D.; Hathaway, N.A. Epigenetic Control of a Local Chromatin Landscape. Int. J. Mol. Sci. 2020, 21, 943. [Google Scholar] [CrossRef]
- van Ouwerkerk, A.F.; Bosada, F.M.; van Duijvenboden, K.; Hill, M.C.; Montefiori, L.E.; Scholman, K.T.; Liu, J.; de Vries, A.A.F.; Boukens, B.J.; Ellinor, P.T.; et al. Identification of Atrial Fibrillation Associated Genes and Functional Non-Coding Variants. Nat. Commun. 2019, 10, 4755. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.G.; Cavarretta, E. MicroRNAs in Cardiovascular Diseases: Current Knowledge and the Road Ahead. J. Am. Coll. Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Han, M.-R.; Jeong, J.H.; Kim, Y.G.; Yang, H.-H.; Seo, C.-O.; Kim, Y.; Lee, H.S.; Shim, J.; Kim, Y.-H.; Choi, J.-I. Epigenetic Regulation on Left Atrial Function and Disease Recurrence after Catheter Ablation in Atrial Fibrillation. Clin. Epigenetics 2024, 16, 183. [Google Scholar] [CrossRef]
- Brundel, B.J.J.M.; Li, J.; Zhang, D. Role of HDACs in Cardiac Electropathology: Therapeutic Implications for Atrial Fibrillation. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118459. [Google Scholar] [CrossRef]
- Auguste, G.; Rouhi, L.; Matkovich, S.J.; Coarfa, C.; Robertson, M.J.; Czernuszewicz, G.; Gurha, P.; Marian, A.J. BET Bromodomain Inhibition Attenuates Cardiac Phenotype in Myocyte-Specific Lamin A/C-Deficient Mice. J. Clin. Investig. 2020, 130, 4740–4758. [Google Scholar] [CrossRef]
- Creemers, E.E.; Wilde, A.A.; Pinto, Y.M. Heart Failure: Advances through Genomics. Nat. Rev. Genet. 2011, 12, 357–362. [Google Scholar] [CrossRef]
- Sweat, M.E.; Pu, W.T. Genetic and Molecular Underpinnings of Atrial Fibrillation. NPJ Cardiovasc. Health 2024, 1, 35. [Google Scholar] [CrossRef]
- Paludan-Müller, C.; Svendsen, J.H.; Olesen, M.S. The Role of Common Genetic Variants in Atrial Fibrillation. J. Electrocardiol. 2016, 49, 864–870. [Google Scholar] [CrossRef]
- Lee, J.; Lee, H.; El Sherbini, A.; Baghaie, L.; Leroy, F.; Abdel-Qadir, H.; Szewczuk, M.R.; El-Diasty, M. Epigenetic MicroRNAs as Prognostic Markers of Postoperative Atrial Fibrillation: A Systematic Review. Curr. Probl. Cardiol. 2024, 49, 102106. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Arnar, D.O.; Helgadottir, A.; Gretarsdottir, S.; Holm, H.; Sigurdsson, A.; Jonasdottir, A.; Baker, A.; Thorleifsson, G.; Kristjansson, K.; et al. Variants Conferring Risk of Atrial Fibrillation on Chromosome 4q25. Nature 2007, 448, 353–357. [Google Scholar] [CrossRef]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpón, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 Insufficiency Leads to Atrial Electrical and Structural Remodeling Linked to Arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Velasco, E.; Hernández-Torres, F.; Daimi, H.; Serra, S.A.; Herraiz, A.; Hove-Madsen, L.; Aránega, A.; Franco, D. Pitx2 Impairs Calcium Handling in a Dose-Dependent Manner by Modulating Wnt Signalling. Cardiovasc. Res. 2016, 109, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Chung, M.K. Genomics of Atrial Fibrillation. Curr. Cardiol. Rep. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Kahr, P.C.; Kaese, S.; Piccini, I.; Vokshi, I.; Scheld, H.-H.; Rotering, H.; Fortmueller, L.; Laakmann, S.; Verheule, S.; et al. PITX2c Is Expressed in the Adult Left Atrium, and Reducing Pitx2c Expression Promotes Atrial Fibrillation Inducibility and Complex Changes in Gene Expression. Circ. Cardiovasc. Genet. 2011, 4, 123–133. [Google Scholar] [CrossRef]
- Taryma-Leśniak, O.; Bińkowski, J.; Przybylowicz, P.K.; Sokolowska, K.E.; Borowski, K.; Wojdacz, T.K. Methylation Patterns at the Adjacent CpG Sites within Enhancers Are a Part of Cell Identity. Epigenetics Chromatin 2024, 17, 30. [Google Scholar] [CrossRef]
- Stefansson, O.A.; Sigurpalsdottir, B.D.; Rognvaldsson, S.; Halldorsson, G.H.; Juliusson, K.; Sveinbjornsson, G.; Gunnarsson, B.; Beyter, D.; Jonsson, H.; Gudjonsson, S.A.; et al. The Correlation between CpG Methylation and Gene Expression Is Driven by Sequence Variants. Nat. Genet. 2024, 56, 1624–1631. [Google Scholar] [CrossRef]
- Ambrosi, C.; Manzo, M.; Baubec, T. Dynamics and Context-Dependent Roles of DNA Methylation. J. Mol. Biol. 2017, 429, 1459–1475. [Google Scholar] [CrossRef]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- Lin, H.; Yin, X.; Xie, Z.; Lunetta, K.L.; Lubitz, S.A.; Larson, M.G.; Ko, D.; Magnani, J.W.; Mendelson, M.M.; Liu, C.; et al. Methylome-Wide Association Study of Atrial Fibrillation in Framingham Heart Study. Sci. Rep. 2017, 7, 40377. [Google Scholar] [CrossRef]
- Felisbino, M.B.; McKinsey, T.A. Epigenetics in Cardiac Fibrosis: Emphasis on Inflammation and Fibroblast Activation. JACC Basic Transl. Sci. 2018, 3, 704–715. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, L.; Toolan-Kerr, P.; Bannister, A.J.; Millán-Zambrano, G. Dynamic Histone Modification Patterns Coordinating DNA Processes. Mol. Cell 2025, 85, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Tingare, A.; Thienpont, B.; Roderick, H.L. Epigenetics in the Heart: The Role of Histone Modifications in Cardiac Remodelling. Biochem. Soc. Trans. 2013, 41, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, S.; Sun, Z. Targeting Histone Deacetylase in Cardiac Diseases. Front. Physiol. 2024, 15, 1405569. [Google Scholar] [CrossRef]
- Backs, J.; Olson, E.N. Control of Cardiac Growth by Histone Acetylation/Deacetylation. Circ. Res. 2006, 98, 15–24. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-Driven Genomic Discovery Yields New Insight into Atrial Fibrillation Biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef]
- Lkhagva, B.; Chang, S.-L.; Chen, Y.-C.; Kao, Y.-H.; Lin, Y.-K.; Chiu, C.T.-H.; Chen, S.-A.; Chen, Y.-J. Histone Deacetylase Inhibition Reduces Pulmonary Vein Arrhythmogenesis through Calcium Regulation. Int. J. Cardiol. 2014, 177, 982–989. [Google Scholar] [CrossRef]
- Victorino, J.; Alvarez-Franco, A.; Manzanares, M. Functional Genomics and Epigenomics of Atrial Fibrillation. J. Mol. Cell. Cardiol. 2021, 157, 45–55. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Henning, R.H.; Brundel, B.J.J.M. Keeping up the Balance: Role of HDACs in Cardiac Proteostasis and Therapeutic Implications for Atrial Fibrillation. Cardiovasc. Res. 2016, 109, 519–526. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The Role of M6A Modification in the Biological Functions and Diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Shen, H.; Lan, Y.; Zhao, Y.; Shi, Y.; Jin, J.; Xie, W. The Emerging Roles of N6-Methyladenosine RNA Methylation in Human Cancers. Biomark. Res. 2020, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, G.; Zou, Q.; Xiong, K.; Chen, Z.; Shao, B.; Liu, Y.; Xie, D.; Ji, Y. M(6)A Reader YTHDF2 Governs the Onset of Atrial Fibrillation by Modulating Cacna1c Translation. Sci. China. Life Sci. 2025, 68, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-F.; Zhou, S.-Y.; Zhong, C.-Q.; Zheng, Z.-F.; Liu, Z.-Y.; Pan, H.-W.; Peng, J.-Q. Identification of M6A Regulator-Mediated RNA Methylation Modification Patterns and Key Immune-Related Genes Involved in Atrial Fibrillation. Aging 2023, 15, 1371–1393. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Doñate Puertas, R.; Arora, R.; Rome, S.; Asatryan, B.; Roderick, H.L.; Chevalier, P. Epigenetics in Atrial Fibrillation: A Reappraisal. Hear. Rhythm 2021, 18, 824–832. [Google Scholar] [CrossRef]
- Nie, Y.; Song, C.; Huang, H.; Mao, S.; Ding, K.; Tang, H. Chromatin Modifiers in Human Disease: From Functional Roles to Regulatory Mechanisms. Mol. Biomed. 2024, 5, 12. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Foo, R.; Anene-Nzelu, C.G.; Travers, J.G.; Vagnozzi, R.J.; Weber, N.; Thum, T. Emerging Epigenetic Therapies of Cardiac Fibrosis and Remodelling in Heart Failure: From Basic Mechanisms to Early Clinical Development. Cardiovasc. Res. 2023, 118, 3482–3498. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating High Confidence MicroRNAs Using Deep Sequencing Data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Ha, T.-Y. MicroRNAs in Human Diseases: From Cancer to Cardiovascular Disease. Immune Netw. 2011, 11, 135–154. [Google Scholar] [CrossRef]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The Role of MicroRNA-1 and MicroRNA-133 in Skeletal Muscle Proliferation and Differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zheng, S.; Xie, X.; Zhang, Y.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, J.; Gao, M.; Hou, Y. MicroRNA-1 Accelerates the Shortening of Atrial Effective Refractory Period by Regulating KCNE1 and KCNB2 Expression: An Atrial Tachypacing Rabbit Model. PLoS ONE 2013, 8, e85639. [Google Scholar] [CrossRef]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in MicroRNA-1 Expression and IK1 up-Regulation in Human Atrial Fibrillation. Hear. Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef]
- Li, Y.-D.; Hong, Y.-F.; Yusufuaji, Y.; Tang, B.-P.; Zhou, X.-H.; Xu, G.-J.; Li, J.-X.; Sun, L.; Zhang, J.-H.; Xin, Q.; et al. Altered Expression of Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels and MicroRNA-1 and -133 in Patients with Age-Associated Atrial Fibrillation. Mol. Med. Rep. 2015, 12, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, S.; Huang, D.; Luo, X.; Zhang, J.; Chen, J.; Xu, W. Expression Profile Analysis of Circulating MicroRNAs and Their Effects on Ion Channels in Chinese Atrial Fibrillation Patients. Int. J. Clin. Exp. Med. 2015, 8, 845–853. [Google Scholar]
- Barana, A.; Matamoros, M.; Dolz-Gaitón, P.; Pérez-Hernández, M.; Amorós, I.; Núñez, M.; Sacristán, S.; Pedraz, Á.; Pinto, Á.; Fernández-Avilés, F.; et al. Chronic Atrial Fibrillation Increases MicroRNA-21 in Human Atrial Myocytes Decreasing L-Type Calcium Current. Circ. Arrhythm. Electrophysiol. 2014, 7, 861–868. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 Contributes to Adverse Electrical Remodeling in Atrial Fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef]
- Ling, T.-Y.; Wang, X.-L.; Chai, Q.; Lu, T.; Stulak, J.M.; Joyce, L.D.; Daly, R.C.; Greason, K.L.; Wu, L.-Q.; Shen, W.-K.; et al. Regulation of Cardiac CACNB2 by MicroRNA-499: Potential Role in Atrial Fibrillation. BBA Clin. 2017, 7, 78–84. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Kongchan, N.; Beavers, D.L.; Alsina, K.M.; Voigt, N.; Neilson, J.R.; Jakob, H.; Martin, J.F.; Dobrev, D.; Wehrens, X.H.T.; et al. Loss of MicroRNA-106b-25 Cluster Promotes Atrial Fibrillation by Enhancing Ryanodine Receptor Type-2 Expression and Calcium Release. Circ. Arrhythm. Electrophysiol. 2014, 7, 1214–1222. [Google Scholar] [CrossRef]
- Cañón, S.; Caballero, R.; Herraiz-Martínez, A.; Pérez-Hernández, M.; López, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpón, E.; Bernad, A. MiR-208b Upregulation Interferes with Calcium Handling in HL-1 Atrial Myocytes: Implications in Human Chronic Atrial Fibrillation. J. Mol. Cell. Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef]
- Adam, O.; Löhfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.-L.; Schäfers, H.-J.; Böhm, M.; Laufs, U. Role of MiR-21 in the Pathogenesis of Atrial Fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Shi, P.; Ge, J.-J. MiR-21 Enhances Cardiac Fibrotic Remodeling and Fibroblast Proliferation via CADM1/STAT3 Pathway. BMC Cardiovasc. Disord. 2017, 17, 88. [Google Scholar] [CrossRef] [PubMed]
- Dawson, K.; Wakili, R.; Ordög, B.; Clauss, S.; Chen, Y.; Iwasaki, Y.; Voigt, N.; Qi, X.Y.; Sinner, M.F.; Dobrev, D.; et al. MicroRNA29: A Mechanistic Contributor and Potential Biomarker in Atrial Fibrillation. Circulation 2013, 127, 1466–1475. [Google Scholar] [CrossRef]
- Goren, Y.; Meiri, E.; Hogan, C.; Mitchell, H.; Lebanony, D.; Salman, N.; Schliamser, J.E.; Amir, O. Relation of Reduced Expression of MiR-150 in Platelets to Atrial Fibrillation in Patients with Chronic Systolic Heart Failure. Am. J. Cardiol. 2014, 113, 976–981. [Google Scholar] [CrossRef]
- McManus, D.D.; Tanriverdi, K.; Lin, H.; Esa, N.; Kinno, M.; Mandapati, D.; Tam, S.; Okike, O.N.; Ellinor, P.T.; Keaney, J.F.J.; et al. Plasma MicroRNAs Are Associated with Atrial Fibrillation and Change after Catheter Ablation (the MiRhythm Study). Hear. Rhythm 2015, 12, 3–10. [Google Scholar] [CrossRef]
- Harling, L.; Lambert, J.; Ashrafian, H.; Darzi, A.; Gooderham, N.J.; Athanasiou, T. Elevated Serum MicroRNA 483-5p Levels May Predict Patients at Risk of Post-Operative Atrial Fibrillation. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2017, 51, 73–78. [Google Scholar] [CrossRef]
- Shen, X.-B.; Zhang, S.-H.; Li, H.-Y.; Chi, X.-D.; Jiang, L.; Huang, Q.-L.; Xu, S.-H. Rs12976445 Polymorphism Is Associated with Post-Ablation Recurrence of Atrial Fibrillation by Modulating the Expression of MicroRNA-125a and Interleukin-6R. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 6349–6358. [Google Scholar] [CrossRef]
- Wei, X.J.; Han, M.; Yang, F.Y.; Wei, G.C.; Liang, Z.G.; Yao, H.; Ji, C.W.; Xie, R.S.; Gong, C.L.; Tian, Y. Biological Significance of MiR-126 Expression in Atrial Fibrillation and Heart Failure. Braz. J. Med. Biol. Res. 2015, 48, 983–989. [Google Scholar] [CrossRef]
- Ren, B.; Cai, S.; Wang, M. Risk Factors for Recurrence of Persistent Atrial Fibrillation after Radiofrequency Ablation and Correlation with Plasma MiRNA Expression. Minerva Cardiol. Angiol. 2025. [Google Scholar] [CrossRef]
- Balan, A.I.; Halaţiu, V.B.; Comșulea, E.; Mutu, C.C.; Cozac, D.A.; Aspru, I.; Păcurar, D.; Bănescu, C.; Perian, M.; Scridon, A. The Diagnostic and Predictive Potential of MiR-328 in Atrial Fibrillation: Insights from a Spontaneously Hypertensive Rat Model. Int. J. Mol. Sci. 2025, 26, 3049. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial Extracellular Matrix Remodeling and the Maintenance of Atrial Fibrillation. Circulation 2004, 109, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Farhat, K.; Po, S.S.; Armoundas, A.A.; Stavrakis, S. Autonomic Nervous System and Cardiac Metabolism: Links Between Autonomic and Metabolic Remodeling in Atrial Fibrillation. JACC Clin. Electrophysiol. 2023, 9, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Hu, J.; Zhang, Y.; Gao, F.; Zhang, F.; Yang, Z.; Zhang, X.; Hou, Y. Time-Dependent Cervical Vagus Nerve Stimulation and Frequency-Dependent Right Atrial Pacing Mediates Induction of Atrial Fibrillation. Anatol. J. Cardiol. 2018, 20, 206–212. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, S.; Geng, Y.; Xue, J.; Wang, Z.; Xie, X.; Wang, J.; Zhang, S.; Hou, Y. MicroRNA Profiling of Atrial Fibrillation in Canines: MiR-206 Modulates Intrinsic Cardiac Autonomic Nerve Remodeling by Regulating SOD1. PLoS ONE 2015, 10, e0122674. [Google Scholar] [CrossRef]
- Srivastava, K.; Tyagi, K. Single Nucleotide Polymorphisms of MicroRNA in Cardiovascular Diseases. Clin. Chim. Acta 2018, 478, 101–110. [Google Scholar] [CrossRef]
- Króliczewski, J.; Sobolewska, A.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. MicroRNA Single Polynucleotide Polymorphism Influences on MicroRNA Biogenesis and MRNA Target Specificity. Gene 2018, 640, 66–72. [Google Scholar] [CrossRef]
- Su, Y.-M.; Li, J.; Guo, Y.-F.; Cai, F.; Cai, X.-X.; Pan, H.-Y.; Deng, X.-T.; Pan, M. A Functional Single-Nucleotide Polymorphism in Pre-MicroRNA-196a2 Is Associated with Atrial Fibrillation in Han Chinese. Clin. Lab. 2015, 61, 1179–1185. [Google Scholar] [CrossRef]
- Chodurska, B.; Kunej, T. Long Non-Coding RNAs in Humans: Classification, Genomic Organization and Function. Non-Coding RNA Res. 2025, 11, 313–327. [Google Scholar] [CrossRef]
- Babapoor-Farrokhran, S.; Gill, D.; Rasekhi, R.T. The Role of Long Noncoding RNAs in Atrial Fibrillation. Hear. Rhythm 2020, 17, 1043–1049. [Google Scholar] [CrossRef]
- Dai, H.; Zhao, N.; Liu, H.; Zheng, Y.; Zhao, L. LncRNA Nuclear-Enriched Abundant Transcript 1 Regulates Atrial Fibrosis via the MiR-320/NPAS2 Axis in Atrial Fibrillation. Front. Pharmacol. 2021, 12, 647124. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Li, Z.; Wang, X.; Li, J.; Liu, D.; Wang, X.; Wei, J.; Ma, S.; Zhang, Y.; Hou, Y. Long Noncoding RNA TCONS-00106987 Promotes Atrial Electrical Remodelling during Atrial Fibrillation by Sponging MiR-26 to Regulate KCNJ2. J. Cell. Mol. Med. 2020, 24, 12777–12788. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Wang, W.; Du, J.; Wei, J.; Zhang, Y.; Wang, J.; Hou, Y. Altered Long Non-Coding RNA Expression Profile in Rabbit Atria with Atrial Fibrillation: TCONS_00075467 Modulates Atrial Electrical Remodeling by Sponging MiR-328 to Regulate CACNA1C. J. Mol. Cell. Cardiol. 2017, 108, 73–85. [Google Scholar] [CrossRef]
- Ramos, K.S.; Li, J.; Wijdeveld, L.F.J.; van Schie, M.S.; Taverne, Y.J.H.J.; Boon, R.A.; de Groot, N.M.S.; Brundel, B.J.J.M. Long Noncoding RNA UCA1 Correlates With Electropathology in Patients With Atrial Fibrillation. JACC Clin. Electrophysiol. 2023, 9, 1097–1107. [Google Scholar] [CrossRef]
- Wang, H.; Song, T.; Zhao, Y.; Zhao, J.; Wang, X.; Fu, X. Long Non-Coding RNA LICPAR Regulates Atrial Fibrosis via TGF-β/Smad Pathway in Atrial Fibrillation. Tissue Cell 2020, 67, 101440. [Google Scholar] [CrossRef]
- Ruan, Z.; Sun, X.; Sheng, H.; Zhu, L. Long Non-Coding RNA Expression Profile in Atrial Fibrillation. Int. J. Clin. Exp. Pathol. 2015, 8, 8402–8410. [Google Scholar]
- Xu, Y.; Huang, R.; Gu, J.; Jiang, W. Identification of Long Non-Coding RNAs as Novel Biomarker and Potential Therapeutic Target for Atrial Fibrillation in Old Adults. Oncotarget 2016, 7, 10803–10811. [Google Scholar] [CrossRef]
- Mei, B.; Liu, H.; Yang, S.; Liang, M.-Y.; Yue, Y.; Huang, S.-Q.; Hou, J.; Chen, G.-X.; Wu, Z.-K. Long Non-Coding RNA Expression Profile in Permanent Atrial Fibrillation Patients with Rheumatic Heart Disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6940–6947. [Google Scholar] [CrossRef]
- Su, Y.; Li, L.; Zhao, S.; Yue, Y.; Yang, S. The Long Noncoding RNA Expression Profiles of Paroxysmal Atrial Fibrillation Identified by Microarray Analysis. Gene 2018, 642, 125–134. [Google Scholar] [CrossRef]
- Chen, G.; Guo, H.; Song, Y.; Chang, H.; Wang, S.; Zhang, M.; Liu, C. Long Non-coding RNA AK055347 Is Upregulated in Patients with Atrial Fibrillation and Regulates Mitochondrial Energy Production in Myocardiocytes. Mol. Med. Rep. 2016, 14, 5311–5317. [Google Scholar] [CrossRef]
- Qian, C.; Li, H.; Chang, D.; Wei, B.; Wang, Y. Identification of Functional LncRNAs in Atrial Fibrillation by Integrative Analysis of the LncRNA-MRNA Network Based on Competing Endogenous RNAs Hypothesis. J. Cell. Physiol. 2019, 234, 11620–11630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ma, Z.; Guo, Z.; Zheng, M.; Li, K.; Yang, X. Analysis of Long Non-Coding RNA and MRNA Profiles in Epicardial Adipose Tissue of Patients with Atrial Fibrillation. Biomed. Pharmacother. 2020, 121, 109634. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, F.-Q.; Guo, J.-J.; Lin, P.-L.; Meng, Z.; Hu, L.-G.; Li, J.; Li, D.; Lu, X.-H.; An, Y. Long Noncoding RNA GAS5 Attenuates Cardiac Fibroblast Proliferation in Atrial Fibrillation via Repressing ALK5. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7605–7610. [Google Scholar] [CrossRef]
- Sun, F.; Guo, Z.; Zhang, C.; Che, H.; Gong, W.; Shen, Z.; Shi, Y.; Ge, S. LncRNA NRON Alleviates Atrial Fibrosis through Suppression of M1 Macrophages Activated by Atrial Myocytes. Biosci. Rep. 2019, 39, BSR20192215. [Google Scholar] [CrossRef]
- Yu, X.-J.; Zou, L.-H.; Jin, J.-H.; Xiao, F.; Li, L.; Liu, N.; Yang, J.-F.; Zou, T. Long Noncoding RNAs and Novel Inflammatory Genes Determined by RNA Sequencing in Human Lymphocytes Are Up-Regulated in Permanent Atrial Fibrillation. Am. J. Transl. Res. 2017, 9, 2314–2326. [Google Scholar]
- Shen, C.; Kong, B.; Liu, Y.; Xiong, L.; Shuai, W.; Wang, G.; Quan, D.; Huang, H. YY1-Induced Upregulation of LncRNA KCNQ1OT1 Regulates Angiotensin II-Induced Atrial Fibrillation by Modulating MiR-384b/CACNA1C Axis. Biochem. Biophys. Res. Commun. 2018, 505, 134–140. [Google Scholar] [CrossRef]
- Wen, J.; Ruan, Z.-B.; Wang, F.; Chen, G.-C.; Zhu, J.-G.; Ren, Y.; Zhu, L. Construction of Atrial Fibrillation-Related CircRNA/LncRNA-MiRNA-MRNA Regulatory Network and Analysis of Potential Biomarkers. J. Clin. Lab. Anal. 2023, 37, e24833. [Google Scholar] [CrossRef]
- Feng, X.-Y.; Zhu, S.-X.; Pu, K.-J.; Huang, H.-J.; Chen, Y.-Q.; Wang, W.-T. New Insight into CircRNAs: Characterization, Strategies, and Biomedical Applications. Exp. Hematol. Oncol. 2023, 12, 91. [Google Scholar] [CrossRef]
- Zhou, W.-Y.; Cai, Z.-R.; Liu, J.; Wang, D.-S.; Ju, H.-Q.; Xu, R.-H. Circular RNA: Metabolism, Functions and Interactions with Proteins. Mol. Cancer 2020, 19, 172. [Google Scholar] [CrossRef]
- Xue, Z.; Zhu, J.; Liu, J.; Wang, L.; Ding, J. Circular RNAs in Atrial Fibrillation: From Bioinformatics Analysis of CircRNA-MiRNA-MRNA Network to Serum Expression. Biochem. Biophys. Rep. 2023, 36, 101577. [Google Scholar] [CrossRef]
- Liu, S.-S.; Guo, H.-Y.; Zhu, J.; Ma, J.-L.; Liu, S.-Z.; He, K.-L.; Bian, S.-Y. Circulating CircRNA Expression Profile and Its Potential Role in Late Recurrence of Paroxysmal Atrial Fibrillation Post Catheter Ablation. J. Geriatr. Cardiol. 2023, 20, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wei, X.; Li, M.; Tao, L.; Wei, L.; Zhang, M.; Cheng, H.; Yuan, Y. Circular RNA Expression Profiles of Persistent Atrial Fibrillation in Patients with Rheumatic Heart Disease. Anatol. J. Cardiol. 2019, 21, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Cortez-Dias, N.; Gabriel, A.; de Sousa, J.; Fiúza, M.; Gallego, J.; Nobre, Â.; Pinto, F.J.; Enguita, F.J. CircRNA-MiRNA Cross-Talk in the Transition from Paroxysmal to Permanent Atrial Fibrillation. Int. J. Cardiol. 2019, 290, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.-B.; Wang, F.; Bao, T.-T.; Yu, Q.-P.; Chen, G.-C.; Zhu, L. Genome-Wide Analysis of Circular RNA Expression Profiles in Patients with Atrial Fibrillation. Int. J. Clin. Exp. Pathol. 2020, 13, 1933–1950. [Google Scholar]
- Zhu, X.; Tang, X.; Chong, H.; Cao, H.; Fan, F.; Pan, J.; Wang, D.; Zhou, Q. Expression Profiles of Circular RNA in Human Atrial Fibrillation With Valvular Heart Diseases. Front. Cardiovasc. Med. 2020, 7, 597932. [Google Scholar] [CrossRef]
- Du, W.W.; Rafiq, M.; Yuan, H.; Li, X.; Wang, S.; Wu, J.; Wei, J.; Li, R.-K.; Guo, H.; Yang, B.B. A Novel Protein NAB1-356 Encoded by CircRNA CircNAB1 Mitigates Atrial Fibrillation by Reducing Inflammation and Fibrosis. Adv. Sci. 2025, e2411959. [Google Scholar] [CrossRef]
- Shangguan, W.; Liang, X.; Shi, W.; Liu, T.; Wang, M.; Li, G. Identification and Characterization of Circular RNAs in Rapid Atrial Pacing Dog Atrial Tissue. Biochem. Biophys. Res. Commun. 2018, 506, 1–6. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; Wang, P.; Min, J.; Yu, Y.; Wang, Q.; Wang, S.; Xi, W.; Nguyen, Q.M.; Xiao, J.; et al. Identification and Characterization of Circular RNAs in Atrial Appendage of Patients with Atrial Fibrillation. Exp. Cell Res. 2020, 389, 111821. [Google Scholar] [CrossRef]
- Jiang, S.; Guo, C.; Zhang, W.; Che, W.; Zhang, J.; Zhuang, S.; Wang, Y.; Zhang, Y.; Liu, B. The Integrative Regulatory Network of CircRNA, MicroRNA, and MRNA in Atrial Fibrillation. Front. Genet. 2019, 10, 526. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, G.; Wang, Y.; Rao, M.; Zhang, Y.; Guo, A.; Wang, M. Identification of Circular RNA-MicroRNA-Messenger RNA Regulatory Network in Atrial Fibrillation by Integrated Analysis. Biomed Res. Int. 2020, 2020, 8037273. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B. Circular RNA in Diseased Heart. Cells 2020, 9, 1240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-P.; Sun, J.; Li, W. Genome-Wide Profiling Reveals Atrial Fibrillation-Related Circular RNAs in Atrial Appendages. Gene 2020, 728, 144286. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, L.; Wu, S.; Xu, K.; Jiang, W.; Qin, M.; Zhang, Y.; Liu, X. Integrative Analysis Reveals Key Circular RNA in Atrial Fibrillation. Front. Genet. 2019, 10, 108. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Franco, D.; Aranega, A.; Daimi, H. Genetics and Epigenetics of Atrial Fibrillation. Int. J. Mol. Sci. 2020, 21, 5717. [Google Scholar] [CrossRef]
- Li, D.; Nie, J.; Han, Y.; Ni, L. Epigenetic Mechanism and Therapeutic Implications of Atrial Fibrillation. Front. Cardiovasc. Med. 2021, 8, 763824. [Google Scholar] [CrossRef]
- Dai, D.-F.; Rabinovitch, P.S. Cardiac Aging in Mice and Humans: The Role of Mitochondrial Oxidative Stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Kolettis, T.M.; Galaris, D.; Goudevenos, J.A. The Role of Oxidative Stress in the Pathogenesis and Perpetuation of Atrial Fibrillation. Int. J. Cardiol. 2007, 115, 135–143. [Google Scholar] [CrossRef]
- Lv, L.; Chen, Q.; Lu, J.; Zhao, Q.; Wang, H.; Li, J.; Yuan, K.; Dong, Z. Potential Regulatory Role of Epigenetic Modifications in Aging-Related Heart Failure. Int. J. Cardiol. 2024, 401, 131858. [Google Scholar] [CrossRef]
- Grzeczka, A.; Graczyk, S.; Kordowitzki, P. DNA Methylation and Telomeres-Their Impact on the Occurrence of Atrial Fibrillation during Cardiac Aging. Int. J. Mol. Sci. 2023, 24, 15699. [Google Scholar] [CrossRef]
- Roberts, J.D.; Vittinghoff, E.; Lu, A.T.; Alonso, A.; Wang, B.; Sitlani, C.M.; Mohammadi-Shemirani, P.; Fornage, M.; Kornej, J.; Brody, J.A.; et al. Epigenetic Age and the Risk of Incident Atrial Fibrillation. Circulation 2021, 144, 1899–1911. [Google Scholar] [CrossRef]
- Doñate Puertas, R.; Meugnier, E.; Romestaing, C.; Rey, C.; Morel, E.; Lachuer, J.; Gadot, N.; Scridon, A.; Julien, C.; Tronc, F.; et al. Atrial Fibrillation Is Associated with Hypermethylation in Human Left Atrium, and Treatment with Decitabine Reduces Atrial Tachyarrhythmias in Spontaneously Hypertensive Rats. Transl. Res. 2017, 184, 57–67.e5. [Google Scholar] [CrossRef]
- Fatima, N.; Schooley, J.F.J.; Claycomb, W.C.; Flagg, T.P. Promoter DNA Methylation Regulates Murine SUR1 (Abcc8) and SUR2 (Abcc9) Expression in HL-1 Cardiomyocytes. PLoS ONE 2012, 7, e41533. [Google Scholar] [CrossRef]
- Shen, K.; Tu, T.; Yuan, Z.; Yi, J.; Zhou, Y.; Liao, X.; Liu, Q.; Zhou, X. DNA Methylation Dysregulations in Valvular Atrial Fibrillation. Clin. Cardiol. 2017, 40, 686–691. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J. Epigenetics and Obesity Cardiomyopathy: From Pathophysiology to Prevention and Management. Pharmacol. Ther. 2016, 161, 52–66. [Google Scholar] [CrossRef]
- Takahashi, K.; Sasano, T.; Sugiyama, K.; Kurokawa, J.; Tamura, N.; Soejima, Y.; Sawabe, M.; Isobe, M.; Furukawa, T. High-Fat Diet Increases Vulnerability to Atrial Arrhythmia by Conduction Disturbance via MiR-27b. J. Mol. Cell. Cardiol. 2016, 90, 38–46. [Google Scholar] [CrossRef]
- Guedes, E.C.; França, G.S.; Lino, C.A.; Koyama, F.C.; Moreira, L.D.; Alexandre, J.G.; Barreto-Chaves, M.L.M.; Galante, P.A.F.; Diniz, G.P. MicroRNA Expression Signature Is Altered in the Cardiac Remodeling Induced by High Fat Diets. J. Cell. Physiol. 2016, 231, 1771–1783. [Google Scholar] [CrossRef]
- Nalliah, C.J.; Sanders, P.; Kottkamp, H.; Kalman, J.M. The Role of Obesity in Atrial Fibrillation. Eur. Heart J. 2016, 37, 1565–1572. [Google Scholar] [CrossRef]
- Kantharia, B.K.; Tabary, M.; Wu, L.; Wang, X.; Narasimhan, B.; Linz, D.; Heijman, J.; Wehrens, X.H.T. Diabetes and Atrial Fibrillation: Insight From Basic to Translational Science Into the Mechanisms and Management. J. Cardiovasc. Electrophysiol. 2025. [Google Scholar] [CrossRef]
- Ma, X.; Mei, S.; Wuyun, Q.; Zhou, L.; Sun, D.; Yan, J. Epigenetics in Diabetic Cardiomyopathy. Clin. Epigenetics 2024, 16, 52. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic Vascular Diseases: Molecular Mechanisms and Therapeutic Strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef]
- Chavali, V.; Tyagi, S.C.; Mishra, P.K. MicroRNA-133a Regulates DNA Methylation in Diabetic Cardiomyocytes. Biochem. Biophys. Res. Commun. 2012, 425, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic Balance of Aberrant Rasal1 Promoter Methylation and Hydroxymethylation Regulates Cardiac Fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef]
- Pan, J.-A.; Lin, H.; Yu, J.-Y.; Zhang, H.-L.; Zhang, J.-F.; Wang, C.-Q.; Gu, J. MiR-21-3p Inhibits Adipose Browning by Targeting FGFR1 and Aggravates Atrial Fibrosis in Diabetes. Oxid. Med. Cell. Longev. 2021, 2021, 9987219. [Google Scholar] [CrossRef] [PubMed]
- Kantharia, B.K.; Zhao, S.; Linz, D.; Heijman, J.; Wehrens, X.H.T. Hypertension and Atrial Fibrillation: Insight From Basic to Translational Science Into the Mechanisms and Management. J. Cardiovasc. Electrophysiol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Antoun, I.; Layton, G.R.; Nizam, A.; Barker, J.; Abdelrazik, A.; Eldesouky, M.; Koya, A.; Lau, E.Y.M.; Zakkar, M.; Somani, R.; et al. Hypertension and Atrial Fibrillation: Bridging the Gap Between Mechanisms, Risk, and Therapy. Medicina 2025, 61, 362. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic Regulation in Cardiovascular Disease: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2022, 7, 200. [Google Scholar] [CrossRef]
- Kao, Y.-H.; Chen, Y.-C.; Chung, C.-C.; Lien, G.-S.; Chen, S.-A.; Kuo, C.-C.; Chen, Y.-J. Heart Failure and Angiotensin II Modulate Atrial Pitx2c Promotor Methylation. Clin. Exp. Pharmacol. Physiol. 2013, 40, 379–384. [Google Scholar] [CrossRef]
- Shafi, O.; Zahra, K.; Shah, H.H. Dysregulations in Cardiogenic Mechanisms by TGF-Beta and Angiotensin II in Cardiac Remodeling Post-Ischemic Injury: A Systematic Review. medRxiv 2024. medRxiv:2024.07.11.24310260. [Google Scholar] [CrossRef]
- Yuntao, F.; Jinjun, L.; Hua Fen, L.; Huiyu, C.; Dishiwen, L.; Zhen, C.; Wang, Y.; Wang, X.; Ke, Y.; Yanni, C.; et al. Atrial Fibroblast-Derived Exosomal MiR-21 Upregulate Myocardial KCa3.1 via the PI3K-Akt Pathway during Rapid Pacing. Heliyon 2024, 10, e33059. [Google Scholar] [CrossRef]
- Watanabe, K.; Narumi, T.; Watanabe, T.; Otaki, Y.; Takahashi, T.; Aono, T.; Goto, J.; Toshima, T.; Sugai, T.; Wanezaki, M.; et al. The Association between MicroRNA-21 and Hypertension-Induced Cardiac Remodeling. PLoS ONE 2020, 15, e0226053. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic Inhibition of MiR-208a Improves Cardiac Function and Survival during Heart Failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef]
- Park, H. Hypoxia Suffocates Histone Demethylases to Change Gene Expression: A Metabolic Control of Histone Methylation. BMB Rep. 2017, 50, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiong, W.; Li, C.; Zhao, R.; Lu, H.; Song, S.; Zhou, Y.; Hu, Y.; Shi, B.; Ge, J. Hypoxia-Induced Signaling in the Cardiovascular System: Pathogenesis and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 431. [Google Scholar] [CrossRef]
- Zhou, F.; Zhou, J.-B.; Wei, T.-P.; Wu, D.; Wang, R.-X. The Role of HIF-1α in Atrial Fibrillation: Recent Advances and Therapeutic Potentials. Rev. Cardiovasc. Med. 2025, 26, 26787. [Google Scholar] [CrossRef]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-Induced Epigenetic Modifications Are Associated with Cardiac Tissue Fibrosis and the Development of a Myofibroblast-like Phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef]
- Grimaldi, V.; De Pascale, M.R.; Zullo, A.; Soricelli, A.; Infante, T.; Mancini, F.P.; Napoli, C. Evidence of Epigenetic Tags in Cardiac Fibrosis. J. Cardiol. 2017, 69, 401–408. [Google Scholar] [CrossRef]
- Shao, J.; Liu, J.; Zuo, S. Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis. Cells 2022, 11, 2347. [Google Scholar] [CrossRef]
- Hancock, R.L.; Dunne, K.; Walport, L.J.; Flashman, E.; Kawamura, A. Epigenetic Regulation by Histone Demethylases in Hypoxia. Epigenomics 2015, 7, 791–811. [Google Scholar] [CrossRef]
- Varrias, D.; Kossack, A.; Leavitt, J.; Chhetri, C.; Roselli, V.; Velichkovikj, S.; Altschul, E.; Bhasin, K.; Mina, B.; Oks, M.; et al. Adherence to CPAP for Patients with Atrial Fibrillation Undergoing Catheter Ablation: A “Real-World” Analysis. Hear. Rhythm 2025. [Google Scholar] [CrossRef]
- Li, F.; He, C.-J.; Ding, C.-H.; Wang, R.-X.; Li, H. Continuous Positive Airway Pressure Therapy Might Be an Effective Strategy on Reduction of Atrial Fibrillation Recurrence after Ablation in Patients with Obstructive Sleep Apnea: Insights from the Pooled Studies. Front. Neurol. 2023, 14, 1269945. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Roh, S.-Y.; Yoon, W.-S.; Kim, J.; Jo, E.; Bae, D.-H.; Kim, M.; Lee, J.-H.; Kim, S.M.; Choi, W.G.; et al. Changes in Alcohol Consumption Habits and Risk of Atrial Fibrillation: A Nationwide Population-Based Study. Eur. J. Prev. Cardiol. 2024, 31, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Pachunka, J.M.; Mott, J.L. Role of MicroRNAs in Alcohol-Induced Multi-Organ Injury. Biomolecules 2015, 5, 3309–3338. [Google Scholar] [CrossRef] [PubMed]
- Yeligar, S.; Tsukamoto, H.; Kalra, V.K. Ethanol-Induced Expression of ET-1 and ET-BR in Liver Sinusoidal Endothelial Cells and Human Endothelial Cells Involves Hypoxia-Inducible Factor-1alpha and MicrorNA-199. J. Immunol. 2009, 183, 5232–5243. [Google Scholar] [CrossRef]
- Wan, Y.; Slevin, E.; Koyama, S.; Huang, C.-K.; Shetty, A.K.; Li, X.; Harrison, K.; Li, T.; Zhou, B.; Lorenzo, S.R.; et al. MiR-34a Regulates Macrophage-Associated Inflammation and Angiogenesis in Alcohol-Induced Liver Injury. Hepatol. Commun. 2023, 7, e0089. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. MiR-34a Repression of SIRT1 Regulates Apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nitta, J.; Kobori, A.; Sakamoto, Y.; Nagata, Y.; Tanimoto, K.; Matsuo, S.; Yamane, T.; Morita, N.; Satomi, K.; et al. Alcohol Consumption Reduction and Clinical Outcomes of Catheter Ablation for Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2021, 14, e009770. [Google Scholar] [CrossRef]
- Xie, M.; Hill, J.A. HDAC-Dependent Ventricular Remodeling. Trends Cardiovasc. Med. 2013, 23, 229–235. [Google Scholar] [CrossRef]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.-J.; Kim, N.; et al. Inhibition of Histone Deacetylation Blocks Cardiac Hypertrophy Induced by Angiotensin II Infusion and Aortic Banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, C.-T.; Qi, X.; Meijering, R.A.M.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.M.; et al. Activation of Histone Deacetylase-6 Induces Contractile Dysfunction through Derailment of α-Tubulin Proteostasis in Experimental and Human Atrial Fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, Q.; Ruan, H.; Guo, C.; Wu, S.; Liu, Q.; Zhang, D.; Long, S.; Wang, W.; Wu, Z.; et al. Pazopanib Attenuated Bleomycin-Induced Pulmonary Fibrosis via Suppressing TGF-Β1 Signaling Pathway. J. Thorac. Dis. 2024, 16, 2244–2258. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β Signal Transduction for Fibrosis and Cancer Therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.-J.; Hu, W.; Shi, K.-H.; Li, J. HDAC6 Promotes Cardiac Fibrosis Progression through Suppressing RASSF1A Expression. Cardiology 2016, 133, 18–26. [Google Scholar] [CrossRef]
- McKinsey, T.A. Isoform-Selective HDAC Inhibitors: Closing in on Translational Medicine for the Heart. J. Mol. Cell. Cardiol. 2011, 51, 491–496. [Google Scholar] [CrossRef]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs Regulate Angiotensin II-Dependent Cardiac Fibrosis via Fibroblasts and Circulating Fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef]
- Fang, J.; Shu, S.; Dong, H.; Yue, X.; Piao, J.; Li, S.; Hong, L.; Cheng, X.W. Histone Deacetylase 6 Controls Cardiac Fibrosis and Remodelling through the Modulation of TGF-Β1/Smad2/3 Signalling in Post-Infarction Mice. J. Cell. Mol. Med. 2024, 28, e70063. [Google Scholar] [CrossRef]
- Vardas, E.P.; Theofilis, P.; Oikonomou, E.; Vardas, P.E.; Tousoulis, D. MicroRNAs in Atrial Fibrillation: Mechanisms, Vascular Implications, and Therapeutic Potential. Biomedicines 2024, 12, 811. [Google Scholar] [CrossRef]
- van den Berg, N.W.E.; Kawasaki, M.; Berger, W.R.; Neefs, J.; Meulendijks, E.; Tijsen, A.J.; de Groot, J.R. MicroRNAs in Atrial Fibrillation: From Expression Signatures to Functional Implications. Cardiovasc. Drugs Ther. 2017, 31, 345–365. [Google Scholar] [CrossRef]
- Garreau, M.; Weidner, J.; Hamilton, R.; Kolosionek, E.; Toki, N.; Stavenhagen, K.; Paris, C.; Bonetti, A.; Czechtizky, W.; Gnerlich, F.; et al. Chemical Modification Patterns for MicroRNA Therapeutic Mimics: A Structure-Activity Relationship (SAR) Case-Study on MiR-200c. Nucleic Acids Res. 2024, 52, 2792–2807. [Google Scholar] [CrossRef]
- Khalaji, A.; Mehrtabar, S.; Jabraeilipour, A.; Doustar, N.; Rahmani Youshanlouei, H.; Tahavvori, A.; Fattahi, P.; Alavi, S.M.A.; Taha, S.R.; Fazlollahpour-Naghibi, A.; et al. Inhibitory Effect of MicroRNA-21 on Pathways and Mechanisms Involved in Cardiac Fibrosis Development. Ther. Adv. Cardiovasc. Dis. 2024, 18, 17539447241253134. [Google Scholar] [CrossRef]
- Raucci, A.; Macrì, F.; Castiglione, S.; Badi, I.; Vinci, M.C.; Zuccolo, E. MicroRNA-34a: The Bad Guy in Age-Related Vascular Diseases. Cell. Mol. Life Sci. 2021, 78, 7355–7378. [Google Scholar] [CrossRef] [PubMed]
- Chioccioli, M.; Roy, S.; Newell, R.; Pestano, L.; Dickinson, B.; Rigby, K.; Herazo-Maya, J.; Jenkins, G.; Ian, S.; Saini, G.; et al. A Lung Targeted MiR-29 Mimic as a Therapy for Pulmonary Fibrosis. EBioMedicine 2022, 85, 104304. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gao, Y.; Zhang, X.; Lughmani, H.Y.; Kennedy, D.J.; Haller, S.T.; Pierre, S.V.; Shapiro, J.I.; Tian, J. A Strategic Expression Method of MiR-29b and Its Anti-Fibrotic Effect Based on RNA-Sequencing Analysis. PLoS ONE 2020, 15, e0244065. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Soleymani, M.; Shahriyary, F.; Amirzargar, M.R.; Ofoghi, M.; Fattahi, M.D.; Safa, M. Viral Vectors and Extracellular Vesicles: Innate Delivery Systems Utilized in CRISPR/Cas-Mediated Cancer Therapy. Cancer Gene Ther. 2023, 30, 936–954. [Google Scholar] [CrossRef]
- Iqbal, Z.; Rehman, K.; Mahmood, A.; Shabbir, M.; Liang, Y.; Duan, L.; Zeng, H. Exosome for MRNA Delivery: Strategies and Therapeutic Applications. J. Nanobiotechnol. 2024, 22, 395. [Google Scholar] [CrossRef]
- Kang, J.-Y.; Park, H.; Kim, H.; Mun, D.; Park, H.; Yun, N.; Joung, B. Human Peripheral Blood-derived Exosomes for MicroRNA Delivery. Int. J. Mol. Med. 2019, 43, 2319–2328. [Google Scholar] [CrossRef]
- Policarpi, C.; Munafò, M.; Tsagkris, S.; Carlini, V.; Hackett, J.A. Systematic Epigenome Editing Captures the Context-Dependent Instructive Function of Chromatin Modifications. Nat. Genet. 2024, 56, 1168–1180. [Google Scholar] [CrossRef]
- Nishiga, M.; Liu, C.; Qi, L.S.; Wu, J.C. The Use of New CRISPR Tools in Cardiovascular Research and Medicine. Nat. Rev. Cardiol. 2022, 19, 505–521. [Google Scholar] [CrossRef]
- Qian, J.; Liu, S.X. CRISPR/DCas9-Tet1-Mediated DNA Methylation Editing. Bio-Protocol 2024, 14, e4976. [Google Scholar] [CrossRef]
- Cai, R.; Lv, R.; Shi, X.; Yang, G.; Jin, J. CRISPR/DCas9 Tools: Epigenetic Mechanism and Application in Gene Transcriptional Regulation. Int. J. Mol. Sci. 2023, 24, 14865. [Google Scholar] [CrossRef]
- Kang, J.G.; Park, J.S.; Ko, J.-H.; Kim, Y.-S. Regulation of Gene Expression by Altered Promoter Methylation Using a CRISPR/Cas9-Mediated Epigenetic Editing System. Sci. Rep. 2019, 9, 11960. [Google Scholar] [CrossRef] [PubMed]
- Tarifa, C.; Serra, S.A.; Herraiz-Martínez, A.; Lozano-Velasco, E.; Benítez, R.; Aranega, A.; Franco, D.; Hove-Madsen, L. Pitx2c Deficiency Confers Cellular Electrophysiological Hallmarks of Atrial Fibrillation to Isolated Atrial Myocytes. Biomed. Pharmacother. 2023, 162, 114577. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Blackwell, D.J.; Yuen, S.L.; Thorpe, M.P.; Johnston, J.N.; Cornea, R.L.; Knollmann, B.C. The Selective RyR2 Inhibitor Ent-Verticilide Suppresses Atrial Fibrillation Susceptibility Caused by Pitx2 Deficiency. J. Mol. Cell. Cardiol. 2023, 180, 1–9. [Google Scholar] [CrossRef]
- Schulz, C.; Lemoine, M.D.; Mearini, G.; Koivumäki, J.; Sani, J.; Schwedhelm, E.; Kirchhof, P.; Ghalawinji, A.; Stoll, M.; Hansen, A.; et al. PITX2 Knockout Induces Key Findings of Electrical Remodeling as Seen in Persistent Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2023, 16, e011602. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.-F.; Yu, T. CRISPR/Cas9 Therapeutics: Progress and Prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Pamporis, K.; Sagris, M.; Ktenopoulos, N.; Kassimis, G.; Antoniadis, A.P.; Fragakis, N. Sodium–Glucose Cotransporter 2 Inhibitors in Aortic Stenosis: Toward a Comprehensive Cardiometabolic Approach. Int. J. Mol. Sci. 2025, 26, 4494. [Google Scholar] [CrossRef]
- Mylonas, N.; Nikolaou, P.E.; Karakasis, P.; Stachteas, P.; Fragakis, N.; Andreadou, I. Endothelial Protection by Sodium-Glucose Cotransporter 2 Inhibitors: A Literature Review of In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2024, 25, 7274. [Google Scholar] [CrossRef]
- Karakasis, P.; Pamporis, K.; Stachteas, P.; Patoulias, D.; Bougioukas, K.I.; Fragakis, N. Efficacy and Safety of Sodium-Glucose Cotransporter-2 Inhibitors in Heart Failure with Mildly Reduced or Preserved Ejection Fraction: An Overview of 36 Systematic Reviews. Heart Fail. Rev. 2023, 28, 1033–1051. [Google Scholar] [CrossRef]
- Stachteas, P.; Nasoufidou, A.; Patoulias, D.; Karakasis, P.; Karagiannidis, E.; Mourtzos, M.-A.; Samaras, A.; Apostolidou, X.; Fragakis, N. The Role of Sodium-Glucose Co-Transporter-2 Inhibitors on Diuretic Resistance in Heart Failure. Int. J. Mol. Sci. 2024, 25, 3122. [Google Scholar] [CrossRef]
- Stachteas, P.; Nasoufidou, A.; Karagiannidis, E.; Patoulias, D.; Karakasis, P.; Alexiou, S.; Samaras, A.; Zormpas, G.; Stavropoulos, G.; Tsalikakis, D.; et al. The Role of Sodium Glucose Co-Transporter 2 Inhibitors in Atrial Fibrillation: A Comprehensive Review. J. Clin. Med. 2024, 13, 5408. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Schuermans, A.; Vlachakis, P.K.; Klisic, A.; Rizzo, M.; Fragakis, N. Sodium-Glucose Cotransporter 2 Inhibitors and Outcomes in Transthyretin Amyloid Cardiomyopathy: Systematic Review and Meta-Analysis. Eur. J. Clin. Investig. 2025, 55, e14392. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Oikonomou, E.; K Vlachakis, P.; Karakasis, P.; Dimitriadis, K.; Sagris, M.; Pamporis, K.; Drakopoulou, M.; Siasos, G.; Tsioufis, K.; et al. Sodium-Glucose Cotransporter 2 Inhibitors and Changes in Epicardial Adipose Tissue: A Systematic Literature Review And Meta-Analysis. Curr. Vasc. Pharmacol. 2025, 23, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Patoulias, D.; Kassimis, G.; Koufakis, T.; Klisic, A.; Doumas, M.; Fragakis, N.; Rizzo, M. Therapeutic Potential of Sodium-Glucose Co-Transporter-2 Inhibitors and Glucagon-like Peptide-1 Receptor Agonists for Patients with Acute Coronary Syndrome: A Review of Clinical Evidence. Curr. Pharm. Des. 2024, 30, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Vizi, D.; Khammy, O.; Mariani, J.A.; Kaye, D.M. The Transcardiac Gradient of Cardio-MicroRNAs in the Failing Heart. Eur. J. Heart Fail. 2016, 18, 1000–1008. [Google Scholar] [CrossRef]
- Mone, P.; Lombardi, A.; Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Marzocco, S.; De Gennaro, S.; Famiglietti, M.; Macina, G.; et al. Empagliflozin Improves the MicroRNA Signature of Endothelial Dysfunction in Patients with Heart Failure with Preserved Ejection Fraction and Diabetes. J. Pharmacol. Exp. Ther. 2023, 384, 116–122. [Google Scholar] [CrossRef]
- Lai, L.; Chen, J.; Wang, N.; Zhu, G.; Duan, X.; Ling, F. MiRNA-30e Mediated Cardioprotection of ACE2 in Rats with Doxorubicin-Induced Heart Failure through Inhibiting Cardiomyocytes Autophagy. Life Sci. 2017, 169, 69–75. [Google Scholar] [CrossRef]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef]
- el Azzouzi, H.; Leptidis, S.; Dirkx, E.; Hoeks, J.; van Bree, B.; Brand, K.; McClellan, E.A.; Poels, E.; Sluimer, J.C.; van den Hoogenhof, M.M.G.; et al. The Hypoxia-Inducible MicroRNA Cluster MiR-199a∼214 Targets Myocardial PPARδ and Impairs Mitochondrial Fatty Acid Oxidation. Cell Metab. 2013, 18, 341–354. [Google Scholar] [CrossRef]
- Nishitani, S.; Fukuhara, A.; Shin, J.; Okuno, Y.; Otsuki, M.; Shimomura, I. Metabolomic and Microarray Analyses of Adipose Tissue of Dapagliflozin-Treated Mice, and Effects of 3-Hydroxybutyrate on Induction of Adiponectin in Adipocytes. Sci. Rep. 2018, 8, 8805. [Google Scholar] [CrossRef]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Guerrero-Mauvecin, J.; Goma-Garces, E.; Fernandez-Fernandez, B.; Carriazo, S.; Sanchez-Niño, M.D.; Ramos, A.M.; Ruiz-Ortega, M.; et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4113. [Google Scholar] [CrossRef]
- Morciano, C.; Gugliandolo, S.; Capece, U.; Di Giuseppe, G.; Mezza, T.; Ciccarelli, G.; Soldovieri, L.; Brunetti, M.; Avolio, A.; Splendore, A.; et al. SGLT2 Inhibition and Adipose Tissue Metabolism: Current Outlook and Perspectives. Cardiovasc. Diabetol. 2024, 23, 449. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Author (Year) | miRNA | Target(s) | Experimental Model | Mechanism | Epigenetic Impact | Functional Role in AF Pathogenesis |
|---|---|---|---|---|---|---|
| Jia et al. (2013) [72] | miR-1 | KCNE1, KCNB2 | Right atrial tachypacing rabbit model | miR-1 overexpression downregulates KCNE1 and KCNB2, increasing IKs current | Post-transcriptional repression via 3′UTR binding (confirmed by luciferase assay) | Shortened atrial effective refractory period (AERP), increased AF inducibility |
| Li et al. (2015) [74] | miR-1, miR-133 | HCN2, HCN4 | Human right atrial appendage samples (CABG patients with and without AF) | Age-associated downregulation of miR-1/133 correlates with upregulation of HCN2/4 | Post-transcriptional repression of HCN channels; inverse expression pattern supports regulatory role | Increased HCN2/4 expression enhances pacemaker current (If), contributing to AF pathogenesis in aged patients |
| Girmatsion et al. (2009) [73] | miR-1 | Kir2.1 (KCNJ2) | Human left atrial tissue (patients with persistent AF undergoing mitral valve surgery); ex vivo atrial slices | miR-1 downregulation associated with increased Kir2.1 expression and IK1 current; confirmed by pacing-induced downregulation in vitro | Post-transcriptional repression of KCNJ2 by miR-1; expression inversely correlated | Enhanced IK1 stabilizes atrial rotors and promotes AF maintenance |
| Lu et al. (2015) [75] | miR-1 | CACNB2 (β2-subunit of L-type Ca2+ channel) | Plasma from AF patients; neonatal rat cardiomyocytes (in vitro transfection) | miR-1 directly targets CACNB2, confirmed by transfection and protein expression assays | Post-transcriptional inhibition of CACNB2 protein expression | Reduced L-type Ca2+ current (ICaL), leading to shortened action potential duration and increased AF susceptibility |
| Barana et al. (2014) [76] | miR-21 | CACNA1C, CACNB2 | Human atrial myocytes (CAF vs. SR); HL-1 cells (transfection); CHO cells (luciferase assay) | miR-21 directly binds to 3′UTRs of CACNA1C and CACNB2, reducing mRNA and protein levels of L-type Ca2+ channels | Post-transcriptional repression of calcium channel subunits | Reduced ICa,L density, shortened APD, promoting electrical remodeling and AF maintenance |
| Lu et al. (2010) [77] | miR-328 | CACNA1C, CACNB1 | AF patients (rheumatic heart disease); canine AF model (atrial tachypacing); transgenic mice; rat atrial myocytes; HEK293 cells | miR-328 upregulation reduces L-type Ca2+ channel subunit expression; confirmed by luciferase assay and Western blot | Post-transcriptional silencing of CACNA1C and CACNB1 via 3′UTR interaction | Reduced ICaL, shortened APD, increased AF susceptibility and maintenance |
| Ling et al. (2017) [78] | miR-499 | CACNB2 | Human atrial tissue (permanent AF vs. SR); HL-1 cells (mouse atrial myocytes); luciferase assay; Argonaute pull-down | miR-499 binds to 3′UTR of CACNB2, repressing protein synthesis without degrading mRNA | Post-transcriptional translational repression of CACNB2 via miRISC recruitment | Reduced CACNB2 levels impair L-type Ca2+ channel function; long-term suppression decreases CACNA1C expression, promoting electrical remodeling in AF |
| Chiang et al. (2014) [79] | miR-93, miR-106b-25 cluster | RyR2 (ryanodine receptor type-2) | Human atrial tissue (pAF vs. SR); miR-106b-25 knockout mice; HEK293 cells (luciferase assay) | Downregulation of the miR-106b-25 cluster leads to loss of repression of RyR2; miR-93 confirmed to bind RyR2 3′UTR | Post-transcriptional derepression of RyR2 via miRNA silencing loss | Increased RyR2 expression, enhanced SR Ca2+ leak, increased atrial ectopy and AF inducibility |
| Cañón et al. (2016) [80] | miR-208b | CACNA1C, CACNB2, SERCA2, Sox6 | Human atrial myocytes (CAF vs. SR); HL-1 cells (transfection); CHO cells (luciferase assay); ovine AF model | miR-208b overexpression suppresses L-type Ca2+ channel subunits and SERCA2; represses transcriptional regulator Sox6 | Post-transcriptional repression via 3′UTR targeting of ion channel genes and Ca2+-handling proteins | Reduced ICa,L and SERCA2 expression; disrupted Ca2+ homeostasis; altered myosin isoform expression; promotes electrical remodeling and AF maintenance |
| Adam et al. (2012) [81] | miR-21 | Sprouty1 (Spry1) | Human left atrial tissue (AF vs. SR); neonatal rat fibroblasts; Rac1-transgenic mice; antagomir-21 treatment | miR-21 expression is upregulated by Rac1 and AngII via CTGF and LOX; miR-21 represses Spry1, promoting profibrotic signaling | Post-transcriptional silencing of Spry1 via 3′UTR targeting by miR-21 | Enhanced CTGF and collagen expression, increased fibrosis, structural remodeling contributing to AF maintenance |
| Cao et al. (2017) [82] | miR-21 | CADM1 | Human atrial tissue (AF vs. SR); SD rat model (ISO-induced fibrosis); neonatal rat cardiac fibroblasts | miR-21 overexpression represses CADM1, leading to activation of STAT3 signaling in cardiac fibroblasts | Post-transcriptional silencing of CADM1 by miR-21 | Increased fibroblast proliferation and STAT3 activation, promoting fibrotic remodeling in AF |
| Dawson et al. (2013) [83] | miR-29b | COL1A1, COL3A1, FBN1 | Canine CHF model (ventricular tachypacing); human plasma and atrial tissue; mouse AAV knockdown model; atrial fibroblasts | miR-29b downregulation leads to derepression of ECM genes; confirmed by sponge-mediated knockdown and overexpression studies | Post-transcriptional repression of fibrosis-related ECM genes | Increased collagen expression, fibrosis, and structural remodeling contributing to AF substrate; miR-29b reduction also observed in AF/CHF patients |
| Goren et al. (2014) [84] | miR-150 | Not specified (platelet-associated expression) | Platelets and serum from HF patients with and without AF; miRNA microarray and RT-PCR | Reduced miR-150 levels in platelets and serum correlate with AF; predictive independent of BNP, troponin, and age | miR-150 downregulation in platelets and serum; may influence inflammatory or fibrotic gene expression indirectly | Associated with AF presence in systolic HF; lower miR-150 may contribute to remodeling or thrombogenicity |
| McManus et al. (2015) [85] | miR-21, miR-150 | Not directly evaluated; associated with atrial remodeling pathways | Plasma from AF vs. non-AF patients (n = 211); right atrial tissue (n = 31); follow-up post-ablation (n = 47) | Plasma levels of miR-21 and miR-150 were significantly lower in AF; both increased after catheter ablation | Circulating downregulation of remodeling-associated miRNAs in AF; reversed post-ablation | Suggests miR-21 and miR-150 as potential biomarkers of AF burden and remodeling activity |
| Harling et al. (2017) [86] | miR-483-5p | Not specified | CABG patients (POAF vs. SR); right atrial biopsies and serial serum sampling | Upregulation of miR-483-5p in atrial myocardium and pre-operative serum of POAF patients; ROC AUC = 0.78 | Putative post-transcriptional modulation related to IGF2 transcription under cardiac stress | Elevated pre-op serum levels predict POAF risk; supports existence of arrhythmogenic substrate |
| Shen et al. (2018) [87] | miR-125a | IL-6R | Human atrial tissue (ERAF vs. LRAF); HL-1 and HEK293 cells (transfection); luciferase assay | miR-125a expression regulated by rs12976445 SNP; miR-125a binds IL-6R 3′UTR and suppresses its expression | Post-transcriptional silencing of IL-6R by miR-125a; rs12976445 affects miR-125a maturation | Reduced miR-125a in ERAF promotes IL-6R-mediated inflammation and increases AF recurrence risk |
| Wei et al. (2015) [88] | miR-126 | Not directly assessed; associated with vascular/endothelial function | Serum from AF, HF, and AF-HF patients vs. healthy controls | miR-126 downregulated in AF and HF; levels inversely correlated with NT-proBNP and LA diameter, positively with LVEF | Implied regulatory role on endothelial function and heart failure biomarkers | Low miR-126 correlates with worse cardiac function and AF/HF severity; potential circulating biomarker |
| Ren et al. (2025) [89] | miR-21, miR-27b | TGFβRIII, PTEN, MMP-2 (miR-21); Wnt/β-catenin pathway (miR-27b) | Plasma samples from PeAF patients post-RFCA | miR-21 upregulation promotes atrial fibrosis via TGF-β/Smad and PTEN-AKT pathways; miR-27b may modulate fibrosis via Wnt/β-catenin signaling | Post-transcriptional gene silencing via 3′UTR binding; both miRNAs influence transcriptional regulation of fibrotic genes | Higher levels associated with atrial fibrosis and increased recurrence after ablation |
| Balan et al. (2025) [90] | miR-328 | CACNA1C, CACNB1 | Spontaneously hypertensive rat (SHR) model with aging | Upregulation of miR-328 reduces L-type Ca2+ current by downregulating CACNA1C and CACNB1, shortening action potential duration and promoting re-entry | Post-transcriptional silencing; correlates with progressive electrical remodeling and arrhythmogenic substrate | Correlated with atrial fibrillation burden and predictive of AF onset in hypertensive rats |
| Author (Year) | lncRNA | Target(s) | Experimental Model | Mechanism | Epigenetic Impact | Functional Role in AF Pathogenesis |
|---|---|---|---|---|---|---|
| Dai et al. (2021) [101] | lncRNA NEAT1 | NPAS2 | Human atrial tissue (AF vs. SR); Ang II-induced murine atrial fibrosis model; cardiac fibroblasts; HEK293T luciferase assay | lncRNA NEAT1 functions as a ceRNA for miR-320, relieving its suppression of NPAS2 | Competing endogenous RNA (ceRNA) activity of NEAT1 against miR-320 | NEAT1 upregulation promotes NPAS2-mediated fibroblast proliferation, migration, and collagen synthesis; contributes to atrial fibrosis and AF substrate |
| Du et al. (2020) [102] | lncRNA TCONS-00106987 | KCNJ2 | Rabbit AF model; primary cardiomyocytes; HEK293T cells; lentiviral transfection | lncRNA TCONS-00106987 acts as a ceRNA, sponging miR-26 to derepress KCNJ2 expression | lncRNA-mediated ceRNA activity relieving miR-26–induced repression of KCNJ2 | Upregulation of KCNJ2 enhances IK1 current, shortens AERP, and increases AF inducibility |
| Li et al. (2017) [103] | lncRNA TCONS_00075467 | CACNA1C | Rabbit AF model (RA tachypacing); primary atrial myocytes; HEK293T cells; lentiviral infection | lncRNA TCONS_00075467 sponges miR-328, relieving suppression on CACNA1C; confirmed by luciferase assay and co-infection experiments | ceRNA mechanism: TCONS_00075467 binds and inhibits miR-328, indirectly restoring CACNA1C expression | Downregulation of TCONS_00075467 reduces ICaL, shortens AERP and APD, increases AF inducibility |
| Ramos et al. (2023) [104] | lncRNAs: UCA1, SARRAH, LIPCAR | Not specified (correlated with electrophysiologic conduction features) | Human right atrial appendage and serum samples (AF vs. sinus rhythm); epicardial mapping during surgery | UCA1 levels inversely correlate with conduction velocity and positively with conduction block/delay; SARRAH and LIPCAR downregulated in RAA | Tissue expression reflects AF-associated electropathology; circulating levels elevated in AF | Potential biomarkers of electropathology severity; UCA1 may serve as a bioelectrical fingerprint for AF staging |
| Wang et al. (2020) [105] | lncRNA LIPCAR | TGF-β1/Smad2/3 signaling | Human atrial tissue (AF vs. SR); Ang II-treated human atrial fibroblasts; siRNA knockdown and overexpression | LIPCAR upregulation enhances Ang II-induced TGF-β1 expression and Smad2/3 phosphorylation; silencing LIPCAR reverses effects | lncRNA LIPCAR modulates fibrotic signaling via interaction with TGF-β/Smad pathway | Promotes fibroblast proliferation, increases expression of α-SMA, Collagen I/III; contributes to atrial fibrosis in AF |
| Ruan et al. (2015) [106] | lncRNAs (e.g., uc001eqh.1, ENST00000575612, TCONS_00006371, etc.) | Not specified | Human left atrial appendage tissue from AF vs. non-AF patients; lncRNA microarray; qRT-PCR validation | Differential expression analysis and co-expression networks suggest lncRNA involvement in electrical and structural remodeling | Putative regulatory roles inferred from GO and KEGG enrichment; associated with calcium signaling, RAS, NF-κB pathways | Identified 50 highly conserved lncRNAs potentially modulating pathways linked to fibrosis, APD shortening, and AF maintenance |
| Xu et al. (2016) [107] | lncRNAs: NONHSAT040387, NONHSAG007503, etc. | Not directly specified; inferred via co-expression with TFs like GATA1, TAF7, and EBF1 | Peripheral blood samples from AF patients vs. controls; Agilent human lncRNA microarray; qRT-PCR validation | 177 differentially expressed lncRNAs (≥2-fold); co-expression network implicates TFs in regulation of lncRNA expression | LncRNA-associated transcriptional regulation and potential miRNA sponge activity | Aberrant lncRNA expression reflects structural remodeling in AF and identifies potential serum biomarkers |
| Mei et al. (2018) [108] | lncRNAs: SNORD115-22, BC041938, uc010vaf.1, etc. | Not directly specified; inferred via co-expression with ECM-related and immune genes | Right atrial tissue from RHD patients with permanent AF vs. NSR; lncRNA microarray; RT-qPCR validation | 182 differentially expressed lncRNAs (fold change > 1.5); co-expression with mRNAs involved in ECM organization and inflammation | Putative regulatory roles through transcriptional modulation and lncRNA-mRNA networks | Dysregulated lncRNAs may contribute to atrial remodeling, immune responses, and fibrotic processes in AF |
| Su et al. (2018) [109] | lncRNAs: ENST00000559960, uc004aef.3 | Putative: KCNA5 (based on co-expression network) | Human leukocyte samples (PAF vs. control); microarray; qRT-PCR validation; CNC network analysis | ENST00000559960 negatively correlates and uc004aef.3 positively correlates with KCNA5; altered expression associated with electrical remodeling | Transcriptional modulation via co-expression with ion channel genes; mechanism remains putative | Potential regulators of KCNA5 and atrial electrical remodeling; ENST00000559960 upregulated and uc004aef.3 downregulated in PAF |
| Chen et al. (2016) [110] | lncRNA: AK055347 | MSS51, Cyp450, ATP synthase | Human LA-PV vs. LAA tissue (AF patients); H9C2 cardiomyocytes (siRNA knockdown); microarray; Western blot; immunofluorescence | AK055347 knockdown inhibits MSS51, Cyp450, and ATP synthase expression; involved in mitochondrial energy production | Transcriptional regulation of mitochondrial genes and metabolic enzymes | AK055347 promotes cardiomyocyte viability and mitochondrial function; contributes to AF-associated metabolic remodeling |
| Qian et al. (2019) [111] | lncRNAs GAS5, HOTAIRM1, RP11-296O14.3 | Multiple mRNAs via ceRNA lncRNA interactions | Human atrial tissue (AF vs. SR); microarray probe reannotation; bioinformatic network and clustering analysis | Eight lncRNAs (e.g., GAS5, HOTAIRM1, RP11-296O14.3) identified in dysregulated lncRNA-mRNA network using ceRNA theory | lncRNAs regulate mRNA targets by sponging miRNAs; ceRNA pairs shift between disease and normal states | lncRNAs influence fibrosis, calcium handling, and metabolism; strong diagnostic value for AF (AUC 0.99) |
| Zhao et al. (2020) [112] | lncRNAs: ENST00000477757, ENST00000477227, ENST00000479930, etc. | Putative targets include PDLIM1, NOS3, TTC3, CTSB, KCNA4 | Human EAT tissue from patients with persistent AF vs. SR (n = 6 each); RNA-sequencing; CNC network; qRT-PCR validation | Differential expression and co-expression network suggest lncRNA regulation of inflammation, fibrosis, ion channel expression | LncRNA-mediated transcriptional and post-transcriptional regulation in EAT impacting atrial remodeling | Altered adipocytokine signaling, fibrosis, and ion channel remodeling; lncRNAs may mediate EAT–myocardium crosstalk in AF |
| Lu et al. (2019) [113] | lncRNA: GAS5 | ALK5 | Human RAA tissues (AF vs. SR); AC16 cardiomyocytes (GAS5 knockdown/overexpression); qRT-PCR; colony assay | GAS5 negatively regulates ALK5; overexpression suppresses, knockdown increases ALK5 expression | Transcriptional regulation of ALK5 by lncRNA GAS5 | GAS5 inhibits fibroblast proliferation and attenuates fibrotic remodeling in AF |
| Sun et al. (2019) [114] | lncRNA: NRON | NFATc3/IL-12 | Mouse atrial myocytes; RAW264.7 macrophages; mouse atrial fibroblasts; Ang II treatment; gene transfection | NRON inhibits NFATc3 nuclear transport, suppressing IL-12 transcription; this limits M1 macrophage polarization and fibroblast activation | Transcriptional repression of IL-12 via NFATc3 inhibition; intercellular signaling modulation through conditioned media | Reduced M1 macrophage activation and inflammatory cytokine secretion; attenuated atrial fibroblast-mediated collagen production and fibrosis |
| Yu et al. (2017) [115] | lncRNAs: TCONS_00076385, TCONS_00194688, TCONS_00024161 | Co-expressed with IFI27, IFIT2, IFI6, IDH1, LAMP3, SAMD9L | Human lymphocytes from pmAF patients vs. controls; RNA-seq; qRT-PCR validation; co-expression network | Upregulated lncRNAs co-express with genes involved in IFN signaling, oxidative stress, and autophagy | lncRNA-associated transcriptional modulation of inflammatory and stress-response pathways | May contribute to inflammatory and oxidative stress mechanisms underlying permanent AF pathophysiology |
| Shen et al. (2018) [116] | lncRNA KCNQ1OT1 | CACNA1C | Ang-II-induced AF mouse model; primary atrial cardiomyocytes; HEK293T cells; ChIP and luciferase assays | lncRNA KCNQ1OT1 sponges miR-384, relieving suppression of CACNA1C; YY1 transcription factor upregulates both KCNQ1OT1 and CACNA1C | ceRNA regulation by KCNQ1OT1; YY1-induced transcriptional upregulation | Increased CACNA1C expression enhances ICaL; shortened ERP, increased AF incidence and duration in Ang-II model |
| Wen et al. (2023) [117] | lncRNA XIST | CADM1 | Peripheral blood mononuclear cells from AF vs. healthy controls; microarray; qRT-PCR; ceRNA network; correlation analysis | lncRNA XIST and circRNA_2773 act as ceRNAs that sponge miR-486-5p, relieving suppression of CADM1 | Post-transcriptional derepression via competitive endogenous RNA (ceRNA) mechanism | Upregulation of CADM1 promotes PI3K/AKT pathway activation; implicated in AF-related fibrosis and inflammation |
| Author (Year) | circRNA | Target(s) | Experimental Model | Mechanism | Epigenetic Impact | Functional Role in AF Pathogenesis |
|---|---|---|---|---|---|---|
| Liu et al. (2023) [121] | hsa_circ_0004214, hsa_circ_0000615, hsa_circ_0003862, hsa_circ_0002202, hsa_circ_0000745, hsa_circ_0008326 | miR-103a, miR-107, miR-320d, miR-1468-3p, miR-6736-3p, miR-3194-3p, miR-5580-5p, miR-4518, miR-16-5p | Peripheral blood of PAF patients with and without LRAF (n = 6); circRNA-seq; qRT-PCR validation; circRNA-miRNA interaction network prediction | Differentially expressed circRNAs act as miRNA sponges (ceRNAs), modulating transcription factors and fibrotic/inflammatory pathways | Post-transcriptional modulation of gene expression via circRNA-miRNA interactions | May influence atrial remodeling and fibrosis; potential circulating biomarkers or therapeutic targets for LRAF |
| Xue et al. (2023) [120] | hsa_circ_0043278, hsa_circ_0000511, hsa_circ_0006220, hsa_circ_0001666 | miR-1207, miR-3192, miR-3200, miR-432, miR-187, miR-548, miR-4254, miR-345 | Serum samples of CABG patients with and without AF; GSE129409 and GSE97455; qRT-PCR validation; bioinformatics prediction | DEcircRNAs act as miRNA sponges (ceRNAs), regulating mRNAs involved in fibrosis and inflammation; ceRNA network confirmed with miRNA and mRNA databases | Post-transcriptional regulation via circRNA–miRNA–mRNA interactions | Increased circRNA expression linked to AF occurrence and recurrence post-CABG; potential diagnostic biomarkers |
| Hu et al. (2019) [122] | circRNA_20118, circRNA_17558, circRNA_16688, circRNA_11058, circRNA_11017, circRNA_11109, circRNA_19591, circRNA_19596, circRNA_16175 | miR-29b-1-5p, miR-29b-2-5p (and others in circRNA–miRNA co-expression network) | Human left atrial appendage tissue (persistent AF with RHD vs. healthy donor hearts); RNA-seq; qRT-PCR validation; GO and KEGG pathway analyses; circRNA–miRNA network construction | Differentially expressed circRNAs act as miRNA sponges; predicted by miRanda and visualized using Cytoscape | Post-transcriptional repression via circRNA–miRNA interactions | Dysregulated circRNAs may influence fibrosis, inflammation, and cardiomyopathic pathways in AF with RHD; identified as potential regulatory hubs |
| Costa et al. (2019) [123] | hsa_circ_0025470, hsa_circ_0035132, hsa_circ_0035148, hsa_circ_0057344, hsa_circ_0085900, hsa_circ_0105720, hsa_circ_0112651, hsa_circ_0112664, hsa_circ_0112682 | miR-181d-5p, miR-3180-3p, miR-6868-3p, miR-2277-5p (among others) | Left atrial biopsies from patients with permanent AF, paroxysmal AF, and sinus rhythm; RNA-seq; circRNA-miRNA network analysis using miRanda and NAViGaTOR | circRNAs exclusively expressed in permanent AF sponge specific miRNAs, contributing to their downregulation during disease progression | Post-transcriptional repression through circRNA-miRNA interactions | CircRNA-mediated miRNA downregulation contributes to progression from paroxysmal to permanent AF via regulatory ncRNA crosstalk |
| Ruan et al. (2020) [124] | circRNA_7571, circRNA_4648, circRNA_4631, circRNA_2875 | hsa-miR-328 (primary predicted interaction) | Peripheral blood monocytes from AF patients vs. healthy controls (n = 4 per group); circRNA microarray; qRT-PCR validation; circRNA–miRNA network using Cytoscape | Differentially expressed circRNAs act as miRNA sponges; circRNA–miRNA co-expression network identified major hubs | Post-transcriptional regulation of miRNA availability; potential modulation of fibrosis- and inflammation-related targets | Key circRNAs may contribute to AF pathogenesis via miR-328 sequestration; may serve as novel biomarkers or therapeutic targets |
| Zhu et al. (2020) [125] | circ_255-ITGA7, circ_418-KCNN2, circ_13913-MIB1, circ_44782-LAMA2, circ_81906-RYR2, circ_3136-TNNI3K | miR-302a-3p, miR-302b-3p, miR-302c-3p, miR-302d-3p (RELA); miR-7-5p (CALM2, CALM3) | LAA tissues from VHD patients with vs. without PeAF (n = 10 + 18); RNA-seq; qRT-PCR; Sanger sequencing; circRNA-miRNA-mRNA network prediction | circRNAs function as miRNA sponges; circ_418-KCNN2 regulates RELA via miR-302 family; circ_81906-RYR2 modulates CALM2/3 via miR-7-5p | Post-transcriptional regulation via circRNA-mediated miRNA sequestration | Involvement in calcium handling and inflammatory pathways (e.g., cAMP, Wnt, Rap1); contribute to atrial fibrosis and AF in VHD |
| Du et al. (2025) [126] | circNAB1 | EGR1, Runx1, Gadd45b | Human atrial tissue (AF vs. SR); circNAB1-transgenic and knockout mice; HL-1, AC16, and HCF cell lines; TAC model; LKB1 knockout mice | circNAB1 encodes novel peptide NAB1-356 that interacts with EGR1 and represses transcription of Runx1 and Gadd45b, reducing cytokine expression and fibrosis | Translation of circRNA-derived peptide modulating transcription factor binding and gene expression | Reduces atrial fibrosis and inflammation; decreases AF incidence and duration; potential therapeutic target in AF with pressure overload or genetic predisposition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakasis, P.; Theofilis, P.; Milaras, N.; Vlachakis, P.K.; Patoulias, D.; Karamitsos, T.; Antoniadis, A.P.; Fragakis, N. Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2025, 26, 5253. https://doi.org/10.3390/ijms26115253
Karakasis P, Theofilis P, Milaras N, Vlachakis PK, Patoulias D, Karamitsos T, Antoniadis AP, Fragakis N. Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets. International Journal of Molecular Sciences. 2025; 26(11):5253. https://doi.org/10.3390/ijms26115253
Chicago/Turabian StyleKarakasis, Paschalis, Panagiotis Theofilis, Nikias Milaras, Panayotis K. Vlachakis, Dimitrios Patoulias, Theodoros Karamitsos, Antonios P. Antoniadis, and Nikolaos Fragakis. 2025. "Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets" International Journal of Molecular Sciences 26, no. 11: 5253. https://doi.org/10.3390/ijms26115253
APA StyleKarakasis, P., Theofilis, P., Milaras, N., Vlachakis, P. K., Patoulias, D., Karamitsos, T., Antoniadis, A. P., & Fragakis, N. (2025). Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets. International Journal of Molecular Sciences, 26(11), 5253. https://doi.org/10.3390/ijms26115253