
Correction of a Traffic-Defective Missense ABCB11 Variant Responsible for Progressive Familial Intrahepatic Cholestasis Type 2 †

 , , , , ,
, , , , ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
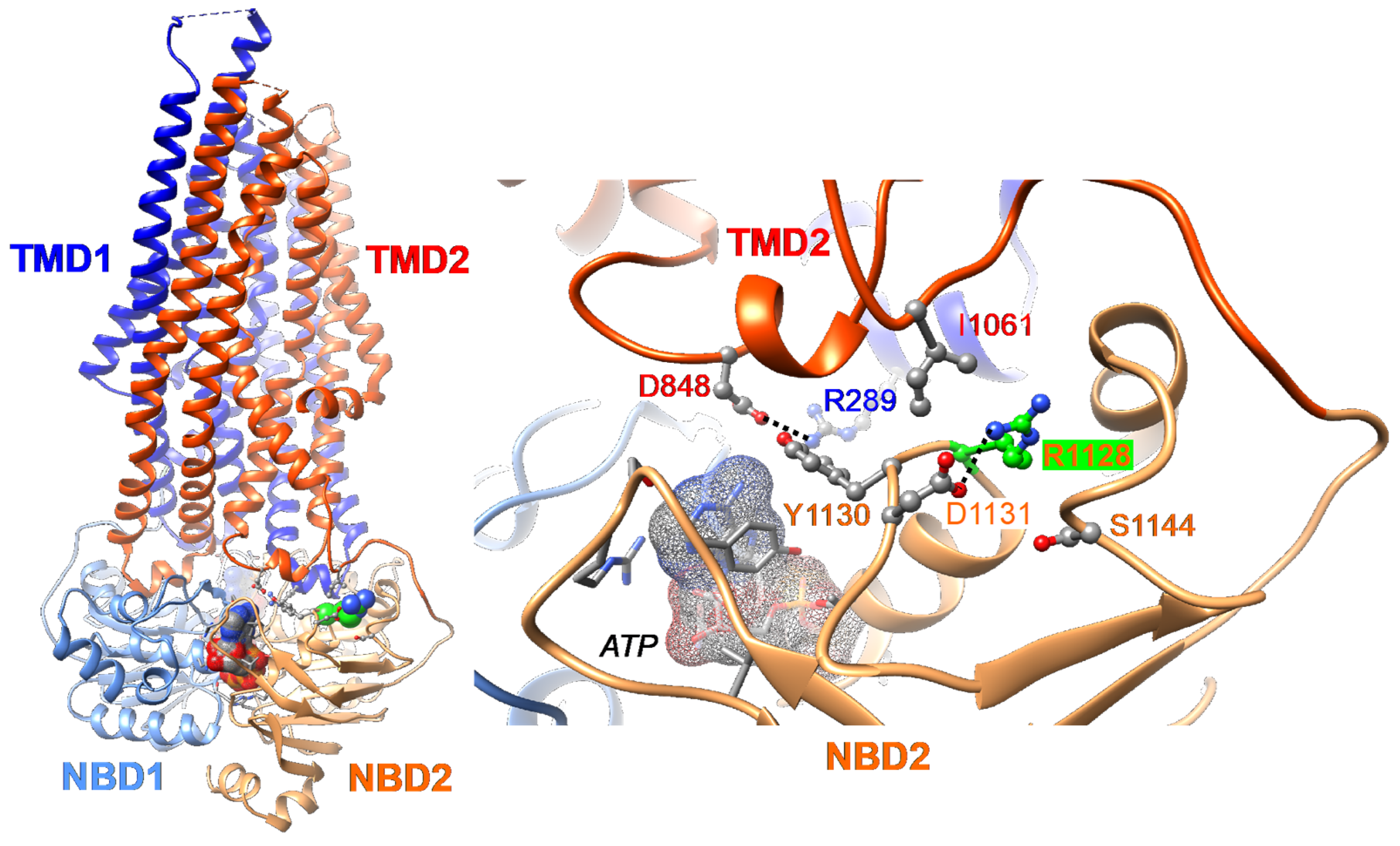
2.1. Three-Dimensional Structure Analysis Predicts a Folding Defect of the Abcb11R1128C Variant
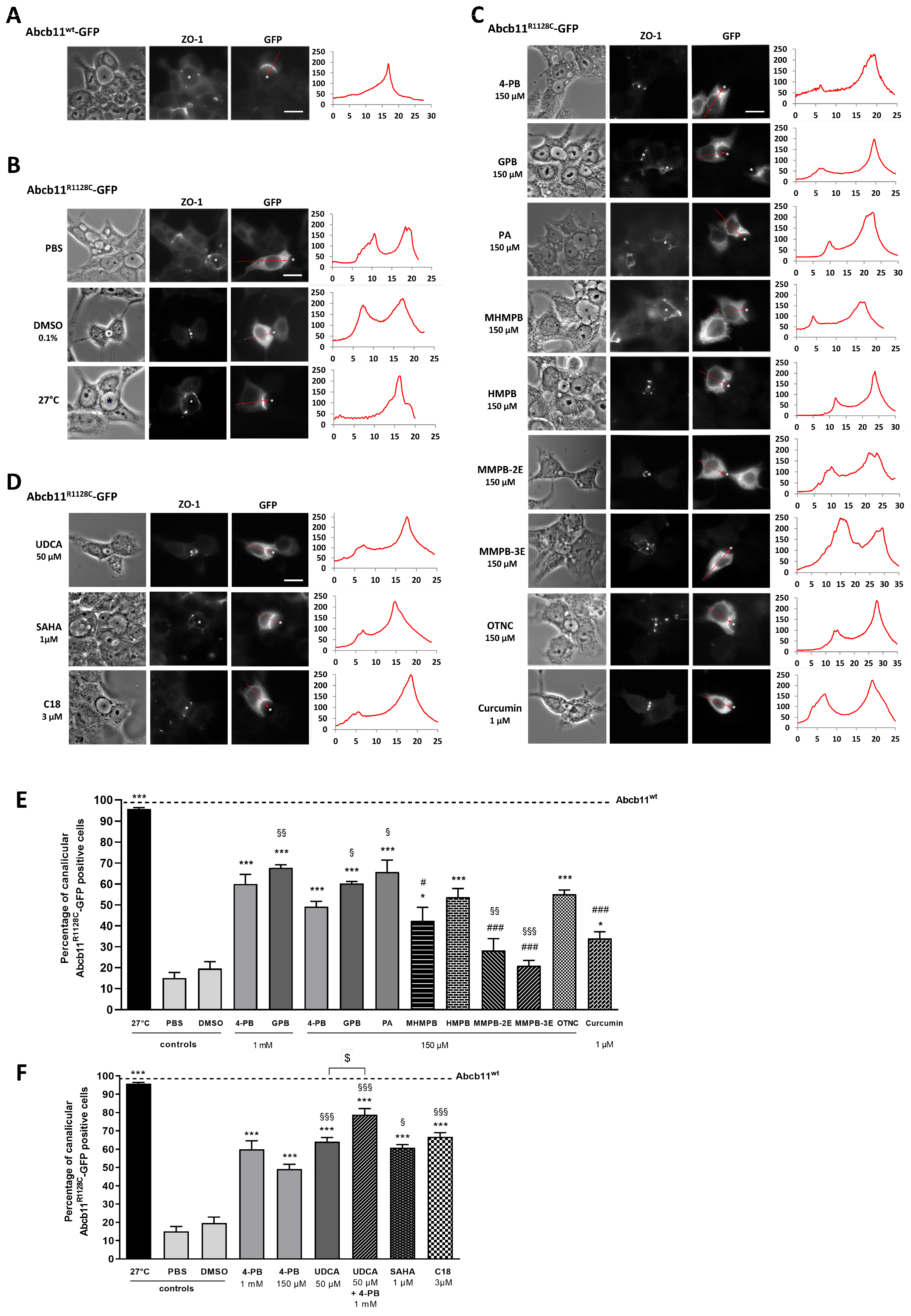
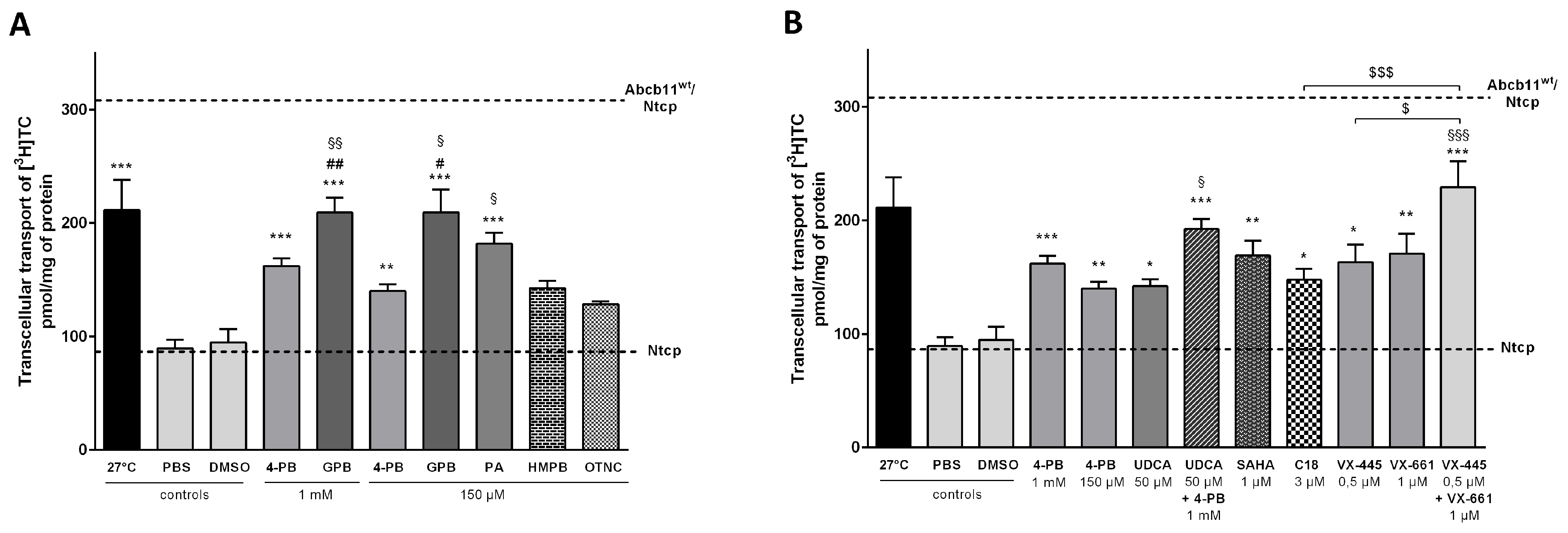
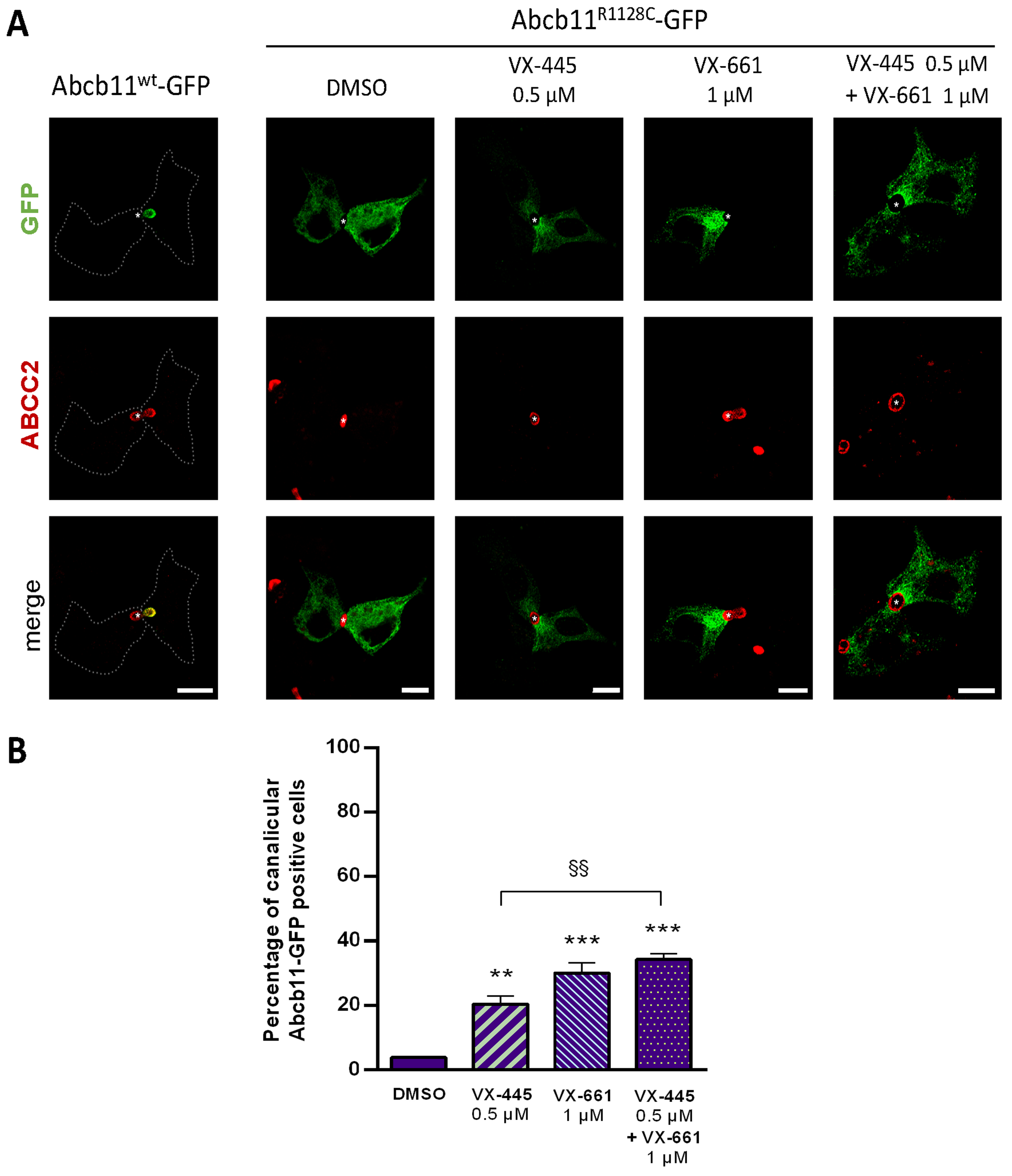
2.2. Effect of 4-Phenylbutyrate, Glycerol Phenylbutyrate and Phenylacetate on the Localization and the Function of the Abcb11R1128C Variant
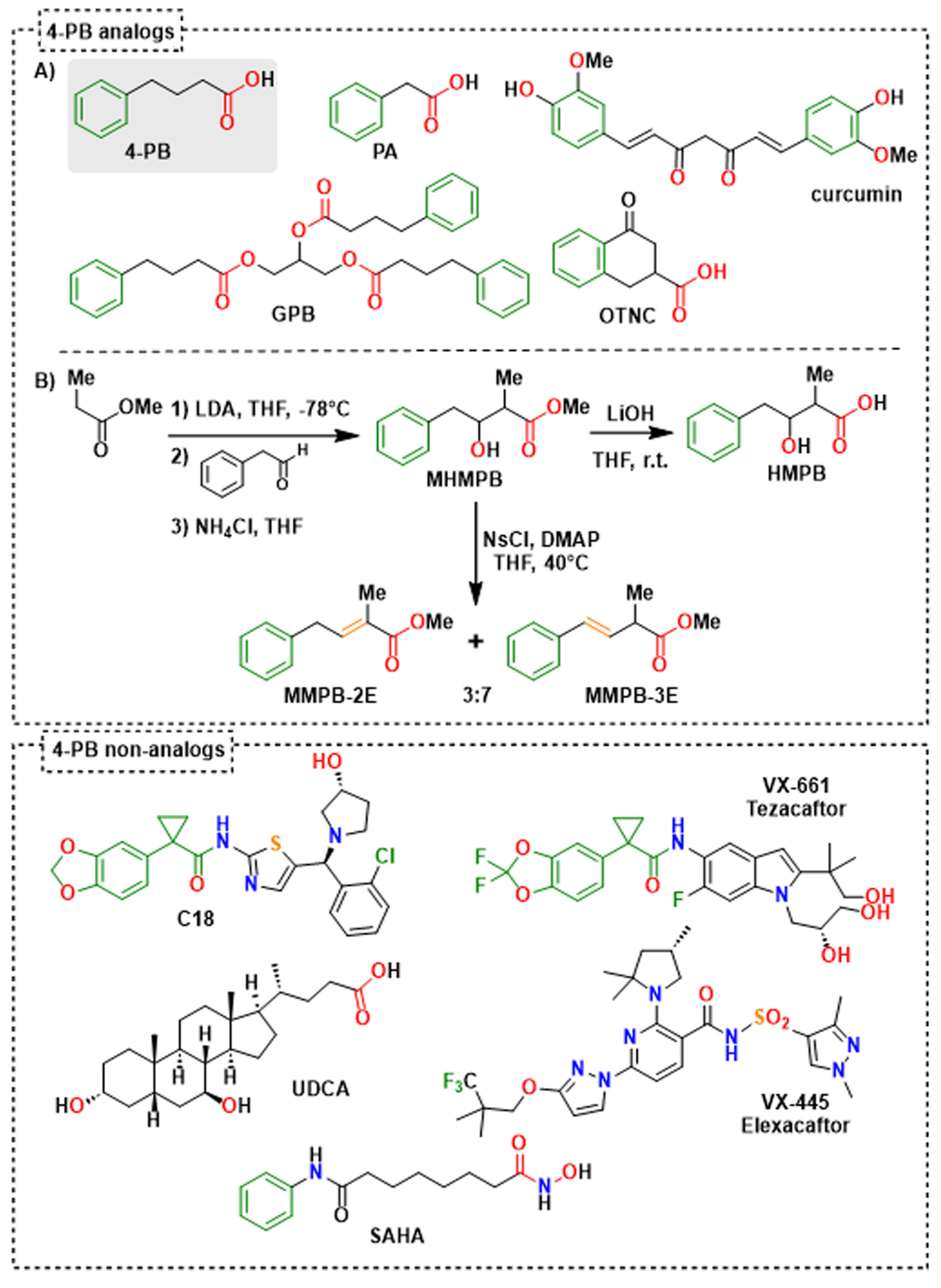
2.3. Identification of 4-Phenylbutyrate-Analogs or Homologs
2.4. Effect of the 4-Phenylbutyrate-Analogs or Homologs on the Localization and the Function of the Abcb11R1128C Variant
2.5. Effect of UDCA and of Other Non-4-PB Related Chemical Correctors on the Localization and the Function of the Abcb11R1128C Variant
3. Discussion
4. Materials and Methods
4.1. Chemistry Materials
4.2. Chemical Synthetic Procedures
4.2.1. The Methyl 3-Hydroxy-2-methyl-4-phenyl Butyric Ester (MHMPB)
4.2.2. The 3-Hydroxy-2-methyl-4-phenylbutyrate (HMPB)
4.2.3. General Procedure for Methyl (E)-2-Methyl-4-phenylbut-3-enoate (MMPB-3E) and Methyl (E)-2-Methyl-4-phenylbut-2-enoate (MMPB-2E)
4.3. Three-Dimensional (3D) Structure Analysis
4.4. DNA Constructs and Mutagenesis
4.5. Transfection, Treatment and Quantification of Cells with Abcb11 Localized at the Canalicular Membrane
4.6. Generation of MDCK Clones Stably Expressing Abcb11 and Ntcp
4.7. Taurocholate Transport Assay
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BA | Bile acid |
| BSEP | Bile salt export pump |
| CF | Cystic fibrosis |
| ER | Endoplasmic reticulum |
| GPB | Glycerol phenylbutyrate |
| HMPB | 3-Hydroxy-2-methyl-4-phenylbutyrate |
| [3H]-TC | 3H-Taurocholate |
| MHMPB | Methyl 3-hydroxy-2-methyl-4-phenyl butyric ester |
| MMPB-2E | Methyl (E)-2-methyl-4-phenylbut-2-enoic ester |
| MMPB-3E | Methyl (E)-2-methyl-4-phenylbut-3-enoic ester |
| OTNC | 4-Oxo-1,2,3,4-tetrahydro-naphthalene-2-carboxylate |
| PA | Phenylacetate |
| 4-PB | 4-phenylbutyrate |
| PFIC2 | Progressive Familial Intrahepatic Cholestasis Type 2 |
| SAHA | Suberoylanilide hydroxamic acid |
| UDCA | Ursodeoxycholic acid |
| wt | Wild type |
References
- Davit-Spraul, A.; Fabre, M.; Branchereau, S.; Baussan, C.; Gonzales, E.; Stieger, B.; Bernard, O.; Jacquemin, E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010, 51, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerova, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134, 1203–1214. [Google Scholar] [CrossRef]
- Amzal, R.; Thebaut, A.; Lapalus, M.; Almes, M.; Grosse, B.; Mareux, E.; Collado-Hilly, M.; Davit-Spraul, A.; Bidou, L.; Namy, O.; et al. Pharmacological Premature Termination Codon Readthrough of ABCB11 in Bile Salt Export Pump Deficiency: An In Vitro Study. Hepatology 2021, 73, 1449–1463. [Google Scholar] [CrossRef]
- Marahatta, A.; Bhandary, B.; Lee, M.R.; Kim, D.S.; Lee, Y.C.; Kim, S.R.; Kim, H.R.; Chae, H.J. Determination of phenylbutyric acid and its metabolite phenylacetic acid in different tissues of mouse by liquid chromatography with tandem mass spectrometry and its application in drug tissue distribution. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 903, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet J. Rare Dis. 2009, 4, 1. [Google Scholar] [CrossRef]
- Knisely, A.S.; Strautnieks, S.S.; Meier, Y.; Stieger, B.; Byrne, J.A.; Portmann, B.C.; Bull, L.N.; Pawlikowska, L.; Bilezikci, B.; Ozcay, F.; et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006, 44, 478–486. [Google Scholar] [CrossRef]
- Thompson, R.J.; Arnell, H.; Artan, R.; Baumann, U.; Calvo, P.L.; Czubkowski, P.; Dalgic, B.; D’Antiga, L.; Durmaz, O.; Fischler, B.; et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: A randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 2022, 7, 830–842. [Google Scholar] [CrossRef]
- Hayashi, H.; Takada, T.; Suzuki, H.; Akita, H.; Sugiyama, Y. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology 2005, 41, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Grosse, B.; Cassio, D.; Davit-Spraul, A.; Fabre, M.; Jacquemin, E. Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. J. Hepatol. 2012, 57, 695–698. [Google Scholar] [CrossRef]
- Sohail, M.I.; Donmez-Cakil, Y.; Szollosi, D.; Stockner, T.; Chiba, P. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. Int. J. Mol. Sci. 2021, 22, 784. [Google Scholar] [CrossRef]
- Gonzales, E.; Grosse, B.; Schuller, B.; Davit-Spraul, A.; Conti, F.; Guettier, C.; Cassio, D.; Jacquemin, E. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 2015, 62, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, V.; Housset, C.; Falguieres, T. Targeted pharmacotherapies for defective ABC transporters. Biochem. Pharmacol. 2017, 136, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sugiyama, Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology 2007, 45, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Naoi, S.; Hirose, Y.; Matsuzaka, Y.; Tanikawa, K.; Igarashi, K.; Nagasaka, H.; Kage, M.; Inui, A.; Kusuhara, H. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol. Res. 2016, 46, 192–200. [Google Scholar] [CrossRef]
- Naoi, S.; Hayashi, H.; Inoue, T.; Tanikawa, K.; Igarashi, K.; Nagasaka, H.; Kage, M.; Takikawa, H.; Sugiyama, Y.; Inui, A.; et al. Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. J. Pediatr. 2014, 164, 1219–1227.e3. [Google Scholar] [CrossRef]
- Malatack, J.J.; Doyle, D. A Drug Regimen for Progressive Familial Cholestasis Type 2. Pediatrics 2018, 141, e20163877. [Google Scholar] [CrossRef]
- Carducci, M.A.; Gilbert, J.; Bowling, M.K.; Noe, D.; Eisenberger, M.A.; Sinibaldi, V.; Zabelina, Y.; Chen, T.L.; Grochow, L.B.; Donehower, R.C. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin. Cancer Res. 2001, 7, 3047–3055. [Google Scholar]
- Gore, S.D.; Weng, L.J.; Figg, W.D.; Zhai, S.; Donehower, R.C.; Dover, G.; Grever, M.R.; Griffin, C.; Grochow, L.B.; Hawkins, A.; et al. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 963–970. [Google Scholar]
- Monteleone, J.P.; Mokhtarani, M.; Diaz, G.A.; Rhead, W.; Lichter-Konecki, U.; Berry, S.A.; Lemons, C.; Dickinson, K.; Coakley, D.; Lee, B.; et al. Population pharmacokinetic modeling and dosing simulations of nitrogen-scavenging compounds: Disposition of glycerol phenylbutyrate and sodium phenylbutyrate in adult and pediatric patients with urea cycle disorders. J. Clin. Pharmacol. 2013, 53, 699–710. [Google Scholar] [CrossRef]
- Almes, M.; Jobert, A.; Lapalus, M.; Mareux, E.; Gonzales, E.; Jacquemin, E. Glycerol Phenylbutyrate Therapy in Progressive Familial Intrahepatic Cholestasis Type 2. J. Pediatr. Gastroenterol. Nutr. 2020, 70, e139–e140. [Google Scholar] [CrossRef]
- Kasumov, T.; Brunengraber, L.L.; Comte, B.; Puchowicz, M.A.; Jobbins, K.; Thomas, K.; David, F.; Kinman, R.; Wehrli, S.; Dahms, W.; et al. New secondary metabolites of phenylbutyrate in humans and rats. Drug Metab. Dispos. 2004, 32, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Revencu, N.; Stephenne, X.; Scheers, I.; Smets, F.; Beleza-Meireles, A.; Reding, R.; Roskams, T.; Sokal, E.M. Retargeting of bile salt export pump and favorable outcome in children with progressive familial intrahepatic cholestasis type 2. Hepatology 2015, 62, 198–206. [Google Scholar] [CrossRef]
- Liu, H.; Irobalieva, R.N.; Kowal, J.; Ni, D.; Nosol, K.; Bang-Sorensen, R.; Lancien, L.; Stahlberg, H.; Stieger, B.; Locher, K.P. Structural basis of bile salt extrusion and small-molecule inhibition in human BSEP. Nat. Commun. 2023, 14, 7296. [Google Scholar] [CrossRef]
- Delaunay, J.L.; Durand-Schneider, A.M.; Delautier, D.; Rada, A.; Gautherot, J.; Jacquemin, E.; Ait-Slimane, T.; Maurice, M. A missense mutation in ABCB4 gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by low temperature. Hepatology 2009, 49, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Yang, C.C.; Chan, D.C.; Wu, C.T.; Chen, L.P.; Huang, J.W.; Hung, K.Y.; Chiang, C.K. Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef]
- Upagupta, C.; Carlisle, R.E.; Dickhout, J.G. Analysis of the potency of various low molecular weight chemical chaperones to prevent protein aggregation. Biochem. Biophys. Res. Commun. 2017, 486, 163–170. [Google Scholar] [CrossRef]
- White, J.D.; Amedio, J.C., Jr.; Gut, S.; Ohira, S.; Jayasinghe, L.R. Stereoselective synthesis of the pyrrolizidine alkaloids (-)-integerrimine and (+)-usaramine. J. Org. Chem. 1992, 57, 2270–2284. [Google Scholar] [CrossRef]
- Kang, J.H.; Benzaria, S.; Sigano, D.M.; Lewin, N.E.; Pu, Y.; Peach, M.L.; Blumberg, P.M.; Marquez, V.E. Conformationally constrained analogues of diacylglycerol. 26. Exploring the chemical space surrounding the C1 domain of protein kinase C with DAG-lactones containing aryl groups at the sn-1 and sn-2 positions. J. Med. Chem. 2006, 49, 3185–3203. [Google Scholar] [CrossRef]
- Holder, G.M.; Plummer, J.L.; Ryan, A.J. The metabolism and excretion of curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) in the rat. Xenobiotica 1978, 8, 761–768. [Google Scholar] [CrossRef]
- Lakli, M.; Onnee, M.; Carrez, T.; Becq, F.; Falguieres, T.; Fanen, P. ABC transporters involved in respiratory and cholestatic diseases: From rare to very rare monogenic diseases. Biochem. Pharmacol. 2024, 229, 116468. [Google Scholar] [CrossRef] [PubMed]
- Mareux, E.; Lapalus, M.; Saad, A.B.; Zelli, R.; Lakli, M.; Riahi, Y.; Almes, M.; Banet, M.; Callebaut, I.; Decout, J.L.; et al. In Vitro Rescue of the Bile Acid Transport Function of ABCB11 Variants by CFTR Potentiators. Int. J. Mol. Sci. 2022, 23, 10758. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, H.; Soroka, C.J.; Wei, N.; Boyer, J.L.; Hochstrasser, M. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 2008, 48, 1558–1569. [Google Scholar] [CrossRef]
- McGuire, B.M.; Zupanets, I.A.; Lowe, M.E.; Xiao, X.; Syplyviy, V.A.; Monteleone, J.; Gargosky, S.; Dickinson, K.; Martinez, A.; Mokhtarani, M.; et al. Pharmacology and safety of glycerol phenylbutyrate in healthy adults and adults with cirrhosis. Hepatology 2010, 51, 2077–2085. [Google Scholar] [CrossRef]
- Mulliner, D.; Wondrousch, D.; Schuurmann, G. Predicting Michael-acceptor reactivity and toxicity through quantum chemical transition-state calculations. Org. Biomol. Chem. 2011, 9, 8400–8412. [Google Scholar] [CrossRef]
- Kagawa, T.; Watanabe, N.; Mochizuki, K.; Numari, A.; Ikeno, Y.; Itoh, J.; Tanaka, H.; Arias, I.M.; Mine, T. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G58–G67. [Google Scholar] [CrossRef]
- Kagawa, T.; Orii, R.; Hirose, S.; Arase, Y.; Shiraishi, K.; Mizutani, A.; Tsukamoto, H.; Mine, T. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membrane in MDCK II cells. J. Gastroenterol. 2014, 49, 890–899. [Google Scholar] [CrossRef]
- van der Woerd, W.L.; Wichers, C.G.; Vestergaard, A.L.; Andersen, J.P.; Paulusma, C.C.; Houwen, R.H.; van de Graaf, S.F. Rescue of defective ATP8B1 trafficking by CFTR correctors as a therapeutic strategy for familial intrahepatic cholestasis. J. Hepatol. 2016, 64, 1339–1347. [Google Scholar] [CrossRef]
- Bouchecareilh, M.; Hutt, D.M.; Szajner, P.; Flotte, T.R.; Balch, W.E. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency. J. Biol. Chem. 2012, 287, 38265–38278. [Google Scholar] [CrossRef]
- Bodas, M.; Mazur, S.; Min, T.; Vij, N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir. Res. 2018, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Molinski, S.V.; Shahani, V.M.; Subramanian, A.S.; MacKinnon, S.S.; Woollard, G.; Laforet, M.; Laselva, O.; Morayniss, L.D.; Bear, C.E.; Windemuth, A. Comprehensive mapping of cystic fibrosis mutations to CFTR protein identifies mutation clusters and molecular docking predicts corrector binding site. Proteins 2018, 86, 833–843. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Elbahnsi, A.; Dudas, B.; Callebaut, I.; Hinzpeter, A.; Miteva, M.A. ATP-Binding Cassette and Solute Carrier Transporters: Understanding Their Mechanisms and Drug Modulation Through Structural and Modeling Approaches. Pharmaceuticals 2024, 17, 1602. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Baatallah, N.; Elbahnsi, A.; Mornon, J.P.; Chevalier, B.; Pranke, I.; Servel, N.; Zelli, R.; Decout, J.L.; Edelman, A.; Sermet-Gaudelus, I.; et al. Pharmacological chaperones improve intra-domain stability and inter-domain assembly via distinct binding sites to rescue misfolded CFTR. Cell Mol. Life Sci. 2021, 78, 7813–7829. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Delta508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef]
- Baatallah, N.; Elbahnsi, A.; Chevalier, B.; Castanier, S.; Mornon, J.P.; Pranke, I.; Edelman, A.; Sermet-Gaudelus, I.; Callebaut, I.; Hinzpeter, A. Acting on the CFTR Membrane-Spanning Domains Interface Rescues Some Misfolded Mutants. Int. J. Mol. Sci. 2022, 23, 16225. [Google Scholar] [CrossRef]
- Ferreira, F.C.; Buarque, C.D.; Lopes-Pacheco, M. Organic Synthesis and Current Understanding of the Mechanisms of CFTR Modulator Drugs Ivacaftor, Tezacaftor, and Elexacaftor. Molecules 2024, 29, 821. [Google Scholar] [CrossRef]
- Mareux, E.; Lapalus, M.; Amzal, R.; Almes, M.; Ait-Slimane, T.; Delaunay, J.L.; Adnot, P.; Collado-Hilly, M.; Davit-Spraul, A.; Falguieres, T.; et al. Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. 2020, 40, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Caro, Y.; Masaguer, C.F.; Raviña, E. Preparation of (R)-(−)- and (S)-(+)-3-hydroxymethyl-1-tetralone tosylates, key intermediates in the synthesis of new CNS drugs, via resolution of precursors. Tetrahedron Asymmetry 2003, 14, 381–387. [Google Scholar] [CrossRef]
- Veen, R.H.V.D.; Geenevasen, J.A.J.; Cerfontain, H. Reactions of α-aryl carbonyl compounds with lithium ester enolates. Can. J. Chem. 1984, 62, 2202–2205. [Google Scholar] [CrossRef]
- Tran, L.; Deshmukh, R.A.; Biehl, E. Facile Synthesis of (E)-4-Aryl-2-Methyl-3-Butenoic Acids and Their Methyl Esters by the Condensation of Tiglic Acid Dianion with Arynes. Synth. Commun. 1996, 26, 963–971. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lapalus, M.; Mareux, E.; Amzal, R. Trafficking rescue of a missense ABCB11 mutation responsible for progressive familial intrahepatic cholestasis type 2. In Proceedings of the XXVII International Bile acid Meeting: Bile Acids in Health and Disease 2024—Falk Symposium 237, Edinburgh, UK, 5–6 July 2024. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapalus, M.; Mareux, E.; Amzal, R.; Drège, E.; Riahi, Y.; Petit, S.; Banet, M.; Falguières, T.; Callebaut, I.; Figadère, B.; et al. Correction of a Traffic-Defective Missense ABCB11 Variant Responsible for Progressive Familial Intrahepatic Cholestasis Type 2. Int. J. Mol. Sci. 2025, 26, 5232. https://doi.org/10.3390/ijms26115232
Lapalus M, Mareux E, Amzal R, Drège E, Riahi Y, Petit S, Banet M, Falguières T, Callebaut I, Figadère B, et al. Correction of a Traffic-Defective Missense ABCB11 Variant Responsible for Progressive Familial Intrahepatic Cholestasis Type 2. International Journal of Molecular Sciences. 2025; 26(11):5232. https://doi.org/10.3390/ijms26115232
Chicago/Turabian StyleLapalus, Martine, Elodie Mareux, Rachida Amzal, Emmanuelle Drège, Yosra Riahi, Sylvain Petit, Manon Banet, Thomas Falguières, Isabelle Callebaut, Bruno Figadère, and et al. 2025. "Correction of a Traffic-Defective Missense ABCB11 Variant Responsible for Progressive Familial Intrahepatic Cholestasis Type 2" International Journal of Molecular Sciences 26, no. 11: 5232. https://doi.org/10.3390/ijms26115232
APA StyleLapalus, M., Mareux, E., Amzal, R., Drège, E., Riahi, Y., Petit, S., Banet, M., Falguières, T., Callebaut, I., Figadère, B., Joseph, D., Gonzales, E., & Jacquemin, E. (2025). Correction of a Traffic-Defective Missense ABCB11 Variant Responsible for Progressive Familial Intrahepatic Cholestasis Type 2. International Journal of Molecular Sciences, 26(11), 5232. https://doi.org/10.3390/ijms26115232