
Serotonergic Regulation in Alzheimer’s Disease

 , and
, and
Abstract
1. Introduction
2. Stress, Depression, and the Development of AD
3. AβO, 5-HT, and Mitochondrial Dysfunction
4. Metabolism and Functions of Serotonin in the CNS
5. Serotonin Receptors

{kind=link}
{kind=link}
{kind=link}
| Receptor Kd Subtype | Signaling | Agonists | Antagonists | Localization | AD Related [References] | ||
|---|---|---|---|---|---|---|---|
| CNS | CVS | Other | |||||
| 5-HT1A 2.65 nM Metabotropic | Gi/Go—AC—PKA Gi/Go—PI3K—PKB—eNOS Gi/Go—P38MAPK—HMGB1 Gi/Go—NOX1/4—ROS—ROCK—ERK1/2 | Xaliprofen Ipsapirone BP 554 8-OH-DPAT U92016A | WAY100635 NAN190 | + | + | ✓ [11] | |
| 5-HT1B 16.01 nM Metabotropic | Gi/Go—AC—PKA | CGS 12066B CP-93129 Ergotamine Eltoprazine Triptans (Zolmitriptan Sumatriptan, Eletriptan) Nonyloxytryptamine | GR-127935 GR55562 Isamoltane SB236057 NAS-181 | + | + | ✓ [156] | |
| 5-HT1D 10.05 nM Metabotropic | Gi/Go—AC—PKA | PNU-109291 L-703,664 GR 46611 Ergotamine Alniditan Triptans (Zolmitriptan, Sumatriptan, Eletriptan, Frovatriptan, Naratriptan, Almotriptan) | SB272183 LY310762 BRL15572 Cyanopindolol | + | + | ✓ [157] | |
| 5-HT1E 7.0 nM Metabotropic | Gi/Go—AC—PKA | 5-CT BRL54443 | + | + | |||
| 5-HT1F 67.60 nM Metabotropic | Gq/11—PLC—PIP2—IP3—Ca2+—DAG Gq/11—NOX—ROS—PI3K—PKB—mTOR—p70S6K1 | LY334370 BRL54443 LY344864 | BRL-54443 Lasmiditan | + | |||
| 5-HT1P Metabotropic | 5-Hydroxy-indalpine | 5-HTP-DP | GI Tract | ||||
| 5-HT2A 970.80 nM Metabotropic | Gq/11—PLC—PIP2—IP3—Ca2+—DAG Gq/11—ERK1/2—eNOS | DOB DOI α-Methyl-5HT TCB-2 | R-95544 Volinanserin Sarpogrelate | + | + | GI Tract, Platelets, PNS, SMC | ✓ [11] |
| 5-HT2B 11.35 nM Metabotropic | Gq/11—PLC—PIP2—IP3—Ca2+—DAG | BW723C86 DOB DOI | LY272015 RS127445 | + | + | GI Tract, Platelets, PNS, SMC | ✓ [158] |
| 5-HT2C 35.58 nM Metabotropic | Gq/11—PLC—PIP2—IP3—Ca2+—DAG | WAY 163909 MK212 1-Methyl-psilocin DOB DOI | RS102221 SB242084 | + | + | GI Tract, Platelets, PNS, SMC | ✓ [159] |
| 5-HT3 190.33 nM Ionotropic | SR57227 2-Methyl-5HT Phenylbiguanide | MDL 72222 Tropisetron Ondansetron Granisetron | + | GI Tract, PNS | ✓ [160] | ||
| 5-HT4 117.0 nM Metabotropic | Gs—AC—PKA | RS67506 BIMU1 BIMU8 RS67333 Zacopride | GR113808 RS100235 SB204070 | + | GI Tract, PNS | ✓ [161] | |
| 5-HT5A Metabotropic | Gi/Go—AC—PKA | SB 699551 | + | ✓ [162] | |||
| 5-HT5B Metabotropic | Rodent CNS | ||||||
| 5-HT6 116.53 нM Metabotropic | Gs—AC-PKA Fyn—ras-MEK-ERK1/2 | WAY 181,187 EMD 386088 | Ro 04-6790 SB 399885 SB271046 | + | ✓ [163] | ||
| 5-HT7(a-d) 3.65 нM Metabotropic | Gs—AC—PKA | LP12 LP44 AS-19 5-CT LP211 | SB-269970 SB-258719 | + | + | GI Tract | ✓ [164] |
5.1. 5-HT1 Receptors
5.2. 5-HT2 Receptors
5.3. 5-HT3 Receptors
5.4. 5-HT4 Receptors
5.5. 5-HT5 Receptors
5.6. 5-HT6 Receptor
5.7. 5-HT7 Receptors
6. Serotonin Level, Inflammation, and Immunity
7. Conclusions: Serotonin Is a Factor of Healthy Longevity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5HT | Serotonin |
| 5-HT system | Serotonergic system |
| AD | Alzheimer’s disease |
| Aβ | Amyloid β |
| AβO | Amyloid β oligomers |
| AC | Adenylate cyclase |
| ACC | Anterior cingulate cortex |
| AChE | Acetylcholinesterase |
| APP | Amyloid precursor protein |
| sAPPα | Secreted form of APP |
| AQP4 | Aquaporin-4 |
| BChE | Butyrylcholinesterase |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CVS | Cardiovascular system |
| DRN | Dorsal raphe nucleus |
| ETC | Electron transport chain |
| FGF | Fibroblast growth factor |
| GABA | γ-aminobutyric acid |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| mAHP | Medium-duration afterhyperpolarization |
| MDD | Major depressive disorder |
| mPTP | Mitochondrial permeability transition pore |
| MRN | Median raphe nucleus |
| NMDAR | NMDA receptor |
| Nrf2 | Nuclear respiratory factor 2 |
| PANoptosis | Pyroptosis, apoptosis, and necroptosis |
| PFC | Prefrontal cortex |
| PNS | Peripheral nervous system |
| PrPc | Cellular prion protein |
| pTau | Tau protein |
| ROS | Reactive oxygen species |
| SCN | Suprachiasmatic nucleus |
| SERT | Serotonin transporter |
| SSRIs | Selective serotonin reuptake inhibitors |
| TIM | Translocase of the inner membrane |
| TOM | Translocase of the outer membrane |
Appendix A. Chemical Names for the Research Compounds
- 5-HTP-DP—N-acetyl-5-hydroxytrytophyl-5-hydroxytryptophan amide.
- 8-OH-DPAT—7-(Dipropylamino)-5,6,7,8-tetrahydronaphthalen-1-ol.
- AS-19—(2S)-5-(1,3,5-Trimethylpyrazol-4-yl)-2-(dimethylamino)tetralin.
- BIMU1—33-ethyl-N-[(5S)-8-methyl-8-azabicyclo [3.2.1]octan-3-yl]-2-oxobenzimidazole-1-carboxamide.
- BIMU8—N-[(1R,5S)-8-methyl-8-azabicyclo [3.2.1]oct-3-yl]-2-oxo-3-(propan-2-yl)-2,3-dihydro-1H-benzimidazole-1-carboxamide hydrochloride.
- BP 554—1-[3-(3,4-methylenedioxyphenoxy)propyl]-4-phenyl-piperazine maleate.
- BRL15572—3-(4-(3-chlorophenyl)piperazin-1-yl)-1,1-diphenyl-2-propanol.
- BRL54443—5-Hydroxy-3-(1-methylpiperidin-4-yl)-1H-indole.
- BW723C86—α-Methyl-5-(2-thienylmethoxy)-1H-indole-3-ethanamine hydrochloride.
- CGS 12066B—7-Trifluoromethyl-4-(4-methyl-1-piperazinyl)pyrrolo [1,2-a]-quinoxalinedimaleate.
- CP-93129—1,4-Dihydro-3-(1,2,3,6-tetrahydro-4-pyridinyl)-5H-pyrrol [3,2-b]pyridin-5-one dihydrochloride.
- DOI—2,5-Dimethoxy-4-iodoamphetamine.
- EMD386088—5-Chloro-2-methyl-3-(1,2,3,6-tetrahydro-4-pyridinyl)-1H-indole.
- GR113808—[1-[2-[(Methylsulfonyl)amino]ethyl]-4-piperidinyl]methyl 1-methyl-1H-indole-3-carboxylate.
- GR-127935—N-[4-methoxy-3-(4-methyl-1-piperazinyl)phenyl]-2′-methyl-4′-(5-methyl-1,2,4-oxadiazol-3-yl)-1-1′-biphenyl-4-carboxamide.
- GR 4661—3-[3-(2-Dimethylaminoethyl)-1H-indol-5-yl]-N-(4-methoxybenzyl)acrylamid.
- GR55562—3-[3-(Dimethylamino)propyl]-4-hydroxy-N-[4-(4-pyridinyl)phenyl]benzamide dihydrochloride.
- L-703,664—2-((3-(2-(dimethylamino)ethyl)-1H-indol-5-yl)methyl)-5-methyl-1,2,5-thiadiazolidine 1,1-dioxide succinate.
- LP12—4-(1,1′-Biphenyl)-2-yl-N-(1,2,3,4-tetrahydro-1-naphthalenyl)-1-piperazinehexanamide hydrochloride.
- LP44—6-[4-(2-methylsulfanylphenyl)piperazin-1-yl]-N-(1,2,3,4-tetrahydronaphthalen-1-yl)hexanamide hydrochloride.
- LP211—4-[1,1′-Biphenyl]-2-yl-N-[(4-cyanophenyl)methyl]-1-piperazinehexanamide.
- LY272015—1-[(3,4-dimethoxyphenyl)methyl]-6-methyl-2,3,4,9-tetrahydro-1H-pyrido [3,4-b]indole hydrochloride.
- LY310762—1-[2-[4-(4-fluorobenzoyl)piperidin-1-yl]ethyl]-3,3-dimethylindol-2-one hydrochloride.
- LY334370—4-fluoro-N-[3-(1-methylpiperidin-4-yl)-1H-indol-5-yl]benzamide.
- LY344864—N-[(6R)-6-(dimethylamino)-6,7,8,9-tetrahydro-5H-carbazol-3-yl]-4-fluorobenzamide.
- MDL72222—3-tropanyl-3,5-dichlorobenzoat.
- MK212—2-chloro-6-piperazin-1-ylpyrazine hydrochloride.
- NAN190—1-(2-Methoxyphenyl)-4-(4-phthalimidobutyl)piperazine hydrobromide.
- NAS-181—(2R)-2-[[[3-(4-morpholinylmethyl)-2H-1-benzopyran-8-yl]oxy]methyl]-morpholine bimethanesulfonate.
- PNU-109291—(S)-3,4-Dihydro-1-[2-[4-(4-methoxyphenyl)-1-piperazinyl]ethyl]-N-methyl-1H-2-benzopyran-6-carboxamide.
- R-96544—(2R,4R)-5-[2-[2-[2-(3-Methoxyphenyl)ethyl]phenoxy]ethyl]-1-methyl-3-pyrrolidinol hydrochloride.
- Ro 04-6790—4-amino-N-[2,6-bis(methylamino)pyrimidin-4-yl]benzenesulfonamide.
- RS100235—1-(5-amino-6-chloro-2,3-dihydro-1,4-benzodioxin-8-yl)-3-[1-[3-(3,4-dimethoxyphenyl)propyl]piperidin-4-yl]propan-1-one.
- RS102221—N-[5-[5-(2,4-dioxo-1,3,8-triazaspiro [4.5]decan-8-yl)pentanoyl]-2,4-dimethoxyphenyl]-4-(trifluoromethyl)benzenesulfonamide.
- RS127445—4-(4-fluoronaphthalen-1-yl)-6-propan-2-ylpyrimidin-2-amine.
- RS67333—1-(4-amino-5-chloro-2-methoxyphenyl)-3-(1-butylpiperidin-4-yl)propan-1-one.
- RS67506—N-[2-[4-[3-(4-amino-5-chloro-2-methoxyphenyl)-3-oxopropyl]piperidin-1-yl]ethyl]methanesulfonamide hydrochloride.
- SB204070—(1-butylpiperidin-4-yl)methyl 5-amino-6-chloro-2,3-dihydro-1,4-benzodioxine-8-carboxylate.
- SB236057—(1′-ethylspiro [6,7-dihydro-2H-furo [2,3-f]indole-3,4′-piperidine]-5-yl)-[4-[2-methyl-4-(5-methyl-1,3,4-oxadiazol-2-yl)phenyl]phenyl]methanone.
- SB242084—6-chloro-5-methyl-N-[6-(2-methylpyridin-3-yl)oxypyridin-3-yl]-2,3-dihydroindole-1-carboxamide.
- SB258719—(1R)-3,N-dimethyl-N-[1-methyl-3-(4-methylpiperidin-1-yl)propyl]benzenesulfonamide.
- SB269970—3-[(2R)-2-[2-(4-methylpiperidin-1-yl)ethyl]pyrrolidin-1-yl]sulfonylphenol.
- SB271046—5-chloro-N-(4-methoxy-3-piperazin-1-ylphenyl)-3-methyl-1-benzothiophene-2-sulfonamide.
- SB272183—5-chloro-6-(4-methylpiperazin-1-yl)-N-(4-pyridin-4-ylnaphthalen-1-yl)-2,3-dihydroindole-1-carboxamide.
- SB399885—N-(3,5-dichloro-2-methoxyphenyl)-4-methoxy-3-piperazin-1-ylbenzenesulfonamide.
- SB699551—3-cyclopentyl-N-[2-(dimethylamino)ethyl]-N-[[4-[4-[(2-phenylethylamino)methyl]phenyl]phenyl]methyl]propanamide;dihydrochloride.
- SR57227A—1-(6-chloropyridin-2-yl)piperidin-4-amine.
- U92016A—(8R)-8-(dipropylamino)-6,7,8,9-tetrahydro-3H-benzo[e]indole-2-carbonitrile
- WAY 100635—N-[2-[4-(2-Methoxyphenyl)-1-piperazinyl]ethyl]-N-(2-pyridyl)cyclohexanecarboxamide.
- WAY 163909—(11R,15R)-7,10-diazatetracyclo [8.5.1.05,16.011,15]hexadeca-1,3,5(16)-triene.
- WAY 181187—2-[1-(6-chloroimidazo [2,1-b][1,3]thiazol-5-yl)sulfonylindol-3-yl]ethanamine.
References
- Rodríguez, J.J.; Noristani, H.N.; Verkhratsky, A. The Serotonergic System in Ageing and Alzheimer’s Disease. Prog. Neurobiol. 2012, 99, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Eremin, D.V.; Kondaurova, E.M.; Rodnyy, A.Y.; Molobekova, C.A.; Kudlay, D.A.; Naumenko, V.S. Serotonin Receptors as a Potential Target in the Treatment of Alzheimer’s Disease. Biochem. Biokhimiia 2023, 88, 2023–2042. [Google Scholar] [CrossRef] [PubMed]
- Butzlaff, M.; Ponimaskin, E. The Role of Serotonin Receptors in Alzheimer’s Disease. Opera Medica Physiol. 2016, 2, 91–100. [Google Scholar] [CrossRef]
- Päivärinta, M.A.; Marttila, R.J.; Lönnberg, P.; Rinne, U.K. Decreased Raphe Serotonin in Rabbits with Experimental Herpes Simplex Encephalitis. Neurosci. Lett. 1993, 156, 1–4. [Google Scholar] [CrossRef]
- Melnikov, M.; Sviridova, A.; Rogovskii, V.; Oleskin, A.; Boziki, M.; Bakirtzis, C.; Kesidou, E.; Grigoriadis, N.; Boyko, A. Serotoninergic System Targeting in Multiple Sclerosis: The Prospective for Pathogenetic Therapy. Mult. Scler. Relat. Disord. 2021, 51, 102888. [Google Scholar] [CrossRef]
- Carneiro, I.B.C.; Toscano, A.E.; Da Cunha, M.D.S.B.; Lacerda, D.C.; Pontes, P.B.; De Castro, R.M.; De Jesus Deiró, T.C.B.; Medeiros, J.M.B. Serotonergic Mechanisms Associated with Experimental Models of Hypoxia: A Systematic Review. Int. J. Dev. Neurosci. 2022, 82, 667–679. [Google Scholar] [CrossRef]
- Tian, J.; Stucky, C.S.; Wang, T.; Muma, N.A.; Johnson, M.; Du, H. Mitochondrial Dysfunction Links to Impaired Hippocampal Serotonin Release in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. JAD 2023, 93, 605–619. [Google Scholar] [CrossRef]
- Cardon, I.; Grobecker, S.; Jenne, F.; Jahner, T.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. Serotonin Effects on Human iPSC-Derived Neural Cell Functions: From Mitochondria to Depression. Mol. Psychiatry 2024, 29, 2689–2700. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Burstein, E.S. Relevance of 5-HT2A Receptor Modulation of Pyramidal Cell Excitability for Dementia-Related Psychosis: Implications for Pharmacotherapy. CNS Drugs 2021, 35, 727–741. [Google Scholar] [CrossRef]
- Afshar, S.; Shahidi, S.; Rohani, A.H.; Soleimani Asl, S.; Komaki, A. Protective Effects of 5-HT1A Receptor Antagonist and 5-HT2A Receptor Agonist on the Biochemical and Histological Features in a Rat Model of Alzheimer’s Disease. J. Chem. Neuroanat. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Bernedo, V.; Insua, D.; Suárez, M.-L.; Santamarina, G.; Sarasa, M.; Pesini, P. Beta-Amyloid Cortical Deposits Are Accompanied by the Loss of Serotonergic Neurons in the Dog. J. Comp. Neurol. 2009, 513, 417–429. [Google Scholar] [CrossRef]
- Song, N.-N.; Huang, Y.; Yu, X.; Lang, B.; Ding, Y.-Q.; Zhang, L. Divergent Roles of Central Serotonin in Adult Hippocampal Neurogenesis. Front. Cell. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative Efficacy and Acceptability of 21 Antidepressant Drugs for the Acute Treatment of Adults with Major Depressive Disorder: A Systematic Review and Network Meta-Analysis. Lancet Lond. Engl. 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Funk, K.A.; Bostwick, J.R. A Comparison of the Risk of QT Prolongation among SSRIs. Ann. Pharmacother. 2013, 47, 1330–1341. [Google Scholar] [CrossRef]
- Noristani, H.N.; Verkhratsky, A.; Rodríguez, J.J. High Tryptophan Diet Reduces CA1 Intraneuronal β-Amyloid in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Aging Cell 2012, 11, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s Disease: Strategies for Disease Modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Olajide, O.J.; Suvanto, M.E.; Chapman, C.A. Molecular Mechanisms of Neurodegeneration in the Entorhinal Cortex That Underlie Its Selective Vulnerability during the Pathogenesis of Alzheimer’s Disease. Biol. Open 2021, 10, bio056796. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia Prevent Beta-Amyloid Plaque Formation in the Early Stage of an Alzheimer’s Disease Mouse Model with Suppression of Glymphatic Clearance. Alzheimers Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Role of Oxygen Radicals in Alzheimer’s Disease: Focus on Tau Protein. Oxygen 2021, 1, 96–120. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-Beta and Tau Synergistically Impair the Oxidative Phosphorylation System in Triple Transgenic Alzheimer’s Disease Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Spuch, C.; Ortolano, S.; Navarro, C. New Insights in the Amyloid-Beta Interaction with Mitochondria. J. Aging Res. 2012, 2012, 324968. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.-C.; Wang, Z.; Luo, Y.; Zhang, X.; Liu, X.-P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau Accumulation Impairs Mitophagy via Increasing Mitochondrial Membrane Potential and Reducing Mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yan, S.S. Mitochondrial Permeability Transition Pore in Alzheimer’s Disease: Cyclophilin D and Amyloid Beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid Precursor Protein Trafficking, Processing, and Function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Haass, C.; Willem, M. Secreted APP Modulates Synaptic Activity: A Novel Target for Therapeutic Intervention? Neuron 2019, 101, 557–559. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid Precursor Protein Processing and Bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Gakhar-Koppole, N.; Hundeshagen, P.; Mandl, C.; Weyer, S.W.; Allinquant, B.; Müller, U.; Ciccolini, F. Activity Requires Soluble Amyloid Precursor Protein Alpha to Promote Neurite Outgrowth in Neural Stem Cell-Derived Neurons via Activation of the MAPK Pathway. Eur. J. Neurosci. 2008, 28, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, Y.; Gao, Z.; Li, J.; Zhang, L.; Shi, H.; Dong, J.; Song, S.; Qian, C. sAPPα Peptide Promotes Damaged Microglia to Clear Alzheimer’s Amyloid-β via Restoring Mitochondrial Function. Chem. Weinh. Bergstr. Ger. 2024, 30, e202400870. [Google Scholar] [CrossRef]
- Demars, M.P.; Hollands, C.; Zhao, K.D.T.; Lazarov, O. Soluble Amyloid Precursor Protein-α Rescues Age-Linked Decline in Neural Progenitor Cell Proliferation. Neurobiol. Aging 2013, 34, 2431–2440. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of Secreted Oligomers of Amyloid Β-protein on Hippocampal Synaptic Plasticity: A Potent Role for Trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Clausen, D.; Pérez-González, R.; Peng, H.; Meszaros, A.; Eckman, C.B.; Levy, E.; Eckman, E.A. Intracellular Metalloprotease Activity Controls Intraneuronal Aβ Aggregation and Limits Secretion of Aβ via Exosomes. FASEB J. 2019, 33, 3758–3771. [Google Scholar] [CrossRef]
- Brewer, G.J.; Herrera, R.A.; Philipp, S.; Sosna, J.; Reyes-Ruiz, J.M.; Glabe, C.G. Age-Related Intraneuronal Aggregation of Amyloid-β in Endosomes, Mitochondria, Autophagosomes, and Lysosomes. J. Alzheimers Dis. 2020, 73, 229–246. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-Protein Oligomers and Alzheimer’s Disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef]
- Bode, D.C.; Freeley, M.; Nield, J.; Palma, M.; Viles, J.H. Amyloid-β Oligomers Have a Profound Detergent-like Effect on Lipid Membrane Bilayers, Imaged by Atomic Force and Electron Microscopy. J. Biol. Chem. 2019, 294, 7566–7572. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High Molecular Weight Amyloid β 1-42 Oligomers Induce Neurotoxicity via Plasma Membrane Damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-Beta Dynamics Are Regulated by Orexin and the Sleep-Wake Cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Boespflug, E.L.; Iliff, J.J. The Emerging Relationship Between Interstitial Fluid-Cerebrospinal Fluid Exchange, Amyloid-β, and Sleep. Biol. Psychiatry 2018, 83, 328–336. [Google Scholar] [CrossRef]
- Quentin, E.; Belmer, A.; Maroteaux, L. Somato-Dendritic Regulation of Raphe Serotonin Neurons; A Key to Antidepressant Action. Front. Neurosci. 2018, 12, 982. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Cupples, L.A.; Kurz, A.; Auerbach, S.; Go, R.; Sadovnick, D.; Duara, R.; Kukull, W.A.; Chui, H.; Edeki, T.; et al. Depression as a Risk Factor for Alzheimer Disease: The MIRAGE Study. Arch. Neurol. 2003, 60, 753–759. [Google Scholar] [CrossRef]
- Swartz, J.R.; Miller, B.L.; Lesser, I.M.; Booth, R.; Darby, A.; Wohl, M.; Benson, D.F. Behavioral Phenomenology in Alzheimer’s Disease, Frontotemporal Dementia, and Late-Life Depression: A Retrospective Analysis. J. Geriatr. Psychiatry Neurol. 1997, 10, 67–74. [Google Scholar] [CrossRef]
- Mitolo, M.; Tonon, C.; La Morgia, C.; Testa, C.; Carelli, V.; Lodi, R. Effects of Light Treatment on Sleep, Cognition, Mood, and Behavior in Alzheimer’s Disease: A Systematic Review. Dement. Geriatr. Cogn. Disord. 2018, 46, 371–384. [Google Scholar] [CrossRef]
- Padovani, A.; Antonini, A.; Barone, P.; Bellelli, G.; Fagiolini, A.; Ferini Strambi, L.; Sorbi, S.; Stocchi, F. Exploring Depression in Alzheimer’s Disease: An Italian Delphi Consensus on Phenomenology, Diagnosis, and Management. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2023, 44, 4323–4332. [Google Scholar] [CrossRef]
- Rapp, M.A.; Schnaider-Beeri, M.; Purohit, D.P.; Perl, D.P.; Haroutunian, V.; Sano, M. Increased Neurofibrillary Tangles in Patients with Alzheimer Disease with Comorbid Depression. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2008, 16, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. History and Evolution of the Monoamine Hypothesis of Depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6. [Google Scholar] [PubMed]
- Cowen, P.J. Serotonin and Depression: Pathophysiological Mechanism or Marketing Myth? Trends Pharmacol. Sci. 2008, 29, 433–436. [Google Scholar] [CrossRef]
- Albert, P.R.; Benkelfat, C.; Descarries, L. The Neurobiology of Depression--Revisiting the Serotonin Hypothesis. I. Cellular and Molecular Mechanisms. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012, 367, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Leyton, M.; Young, S.N.; Benkelfat, C. Relapse of Depression after Rapid Depletion of Tryptophan. Lancet Lond. Engl. 1997, 349, 1840–1841. [Google Scholar] [CrossRef]
- Delgado, P.L. Monoamine Depletion Studies: Implications for Antidepressant Discontinuation Syndrome. J. Clin. Psychiatry 2006, 67 (Suppl. 4), 22–26. [Google Scholar]
- Ślifirski, G.; Król, M.; Turło, J. 5-HT Receptors and the Development of New Antidepressants. Int. J. Mol. Sci. 2021, 22, 9015. [Google Scholar] [CrossRef]
- Orrico-Sanchez, A.; Chausset-Boissarie, L.; Alves De Sousa, R.; Coutens, B.; Rezai Amin, S.; Vialou, V.; Louis, F.; Hessani, A.; Dansette, P.M.; Zornoza, T.; et al. Antidepressant Efficacy of a Selective Organic Cation Transporter Blocker in a Mouse Model of Depression. Mol. Psychiatry 2020, 25, 1245–1259. [Google Scholar] [CrossRef]
- Spencer, R.L.; Chun, L.E.; Hartsock, M.J.; Woodruff, E.R. Glucocorticoid Hormones Are Both a Major Circadian Signal and Major Stress Signal: How This Shared Signal Contributes to a Dynamic Relationship between the Circadian and Stress Systems. Front. Neuroendocrinol. 2018, 49, 52–71. [Google Scholar] [CrossRef]
- Bunney, B.G.; Li, J.Z.; Walsh, D.M.; Stein, R.; Vawter, M.P.; Cartagena, P.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Watson, S.J.; et al. Circadian Dysregulation of Clock Genes: Clues to Rapid Treatments in Major Depressive Disorder. Mol. Psychiatry 2015, 20, 48–55. [Google Scholar] [CrossRef]
- Kent, J.; Meredith, A.L. BK Channels Regulate Spontaneous Action Potential Rhythmicity in the Suprachiasmatic Nucleus. PLoS ONE 2008, 3, e3884. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, D.E.; Griffin, P.; Sheehan, P.W.; Kim, D.-H.; Musiek, E.S.; Yoon, S.-Y. Inhibition of REV-ERBs Stimulates Microglial Amyloid-Beta Clearance and Reduces Amyloid Plaque Deposition in the 5XFAD Mouse Model of Alzheimer’s Disease. Aging Cell 2020, 19, e13078. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhan, G.; Fenik, P.; Brandes, M.; Bell, P.; Francois, N.; Shulman, K.; Veasey, S. Chronic Sleep Disruption Advances the Temporal Progression of Tauopathy in P301S Mutant Mice. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 10255–10270. [Google Scholar] [CrossRef] [PubMed]
- Daut, R.A.; Fonken, L.K. Circadian Regulation of Depression: A Role for Serotonin. Front. Neuroendocrinol. 2019, 54, 100746. [Google Scholar] [CrossRef]
- Kiss, J.; Léránth, C.; Halász, B. Serotoninergic Endings on VIP-Neurons in the Suprachiasmatic Nucleus and on ACTH-Neurons in the Arcuate Nucleus of the Rat Hypothalamus. A Combination of High Resolution Autoradiography and Electron Microscopic Immunocytochemistry. Neurosci. Lett. 1984, 44, 119–124. [Google Scholar] [CrossRef]
- Bosler, O.; Beaudet, A. VIP Neurons as Prime Synaptic Targets for Serotonin Afferents in Rat Suprachiasmatic Nucleus: A Combined Radioautographic and Immunocytochemical Study. J. Neurocytol. 1985, 14, 749–763. [Google Scholar] [CrossRef]
- Manrique, C.; Segu, L.; Héry, F.; Héry, M.; Faudon, M.; François-Bellan, A.M. Increase of Central 5-HT1B Binding Sites Following 5,7-Dihydroxytryptamine Axotomy in the Adult Rat. Brain Res. 1993, 623, 345–348. [Google Scholar] [CrossRef]
- Amir, S.; Robinson, B.; Ratovitski, T.; Rea, M.A.; Stewart, J.; Simantov, R. A Role for Serotonin in the Circadian System Revealed by the Distribution of Serotonin Transporter and Light-Induced Fos Immunoreactivity in the Suprachiasmatic Nucleus and Intergeniculate Leaflet. Neuroscience 1998, 84, 1059–1073. [Google Scholar] [CrossRef]
- Tao, L.; Jiang, R.; Zhang, K.; Qian, Z.; Chen, P.; Lv, Y.; Yao, Y. Light Therapy in Non-Seasonal Depression: An Update Meta-Analysis. Psychiatry Res. 2020, 291, 113247. [Google Scholar] [CrossRef]
- Richardson, C.R.; Faulkner, G.; McDevitt, J.; Skrinar, G.S.; Hutchinson, D.S.; Piette, J.D. Integrating Physical Activity into Mental Health Services for Persons with Serious Mental Illness. Psychiatr. Serv. Wash. DC 2005, 56, 324–331. [Google Scholar] [CrossRef]
- Hu, D.; Mao, Y.; Xu, G.; Liao, W.; Ren, J.; Yang, H.; Yang, J.; Sun, L.; Chen, H.; Wang, W.; et al. Time-Restricted Feeding Causes Irreversible Metabolic Disorders and Gut Microbiota Shift in Pediatric Mice. Pediatr. Res. 2019, 85, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Anjum, B.; Godbole, N.M.; Rajak, S.; Shukla, P.; Tiwari, S.; Sinha, R.A.; Godbole, M.M. Time-Restricted Feeding Reduces High-Fat Diet Associated Placental Inflammation and Limits Adverse Effects on Fetal Organ Development. Biochem. Biophys. Res. Commun. 2019, 514, 415–421. [Google Scholar] [CrossRef]
- Yan, T.; Qiu, Y.; Yu, X.; Yang, L. Glymphatic Dysfunction: A Bridge Between Sleep Disturbance and Mood Disorders. Front. Psychiatry 2021, 12, 658340. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.C.; Karimy, J.K.; Kundishora, A.J.; Mestre, H.; Cerci, H.M.; Matouk, C.; Alper, S.L.; Lundgaard, I.; Nedergaard, M.; Kahle, K.T. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 2020, 26, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Shokri-Kojori, E.; Wang, G.-J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid Accumulation in the Human Brain after One Night of Sleep Deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef]
- López-Doménech, G.; Kittler, J.T. Mitochondrial Regulation of Local Supply of Energy in Neurons. Curr. Opin. Neurobiol. 2023, 81, 102747. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease, and Parkinson’s Disease, Huntington’s Disease and Amyotrophic Lateral Sclerosis—An Updated Review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Huang, N.; Sheng, Z.-H. Programming Axonal Mitochondrial Maintenance and Bioenergetics in Neurodegeneration and Regeneration. Neuron 2022, 110, 1899–1923. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, Nonfibrillar Ligands Derived from Abeta1-42 Are Potent Central Nervous System Neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. JAD 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-Β42 Oligomers but Not Amyloid-Β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef]
- Arispe, N. Architecture of the Alzheimer’s A Beta P Ion Channel Pore. J. Membr. Biol. 2004, 197, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.; Ramamoorthy, A. Two-Step Mechanism of Membrane Disruption by Aβ through Membrane Fragmentation and Pore Formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-Protein-Interacting Amyloid-β Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na, K-ATPase A3 Is a Death Target of Alzheimer Patient Amyloid-β Assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer Amyloid-β Oligomer Bound to Postsynaptic Prion Protein Activates Fyn to Impair Neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yang, D.-S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty Autolysosome Acidification in Alzheimer’s Disease Mouse Models Induces Autophagic Build-up of Aβ in Neurons, Yielding Senile Plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Schützmann, M.P.; Hasecke, F.; Bachmann, S.; Zielinski, M.; Hänsch, S.; Schröder, G.F.; Zempel, H.; Hoyer, W. Endo-Lysosomal Aβ Concentration and pH Trigger Formation of Aβ Oligomers That Potently Induce Tau Missorting. Nat. Commun. 2021, 12, 4634. [Google Scholar] [CrossRef]
- Sebollela, A.; Cline, E.N.; Popova, I.; Luo, K.; Sun, X.; Ahn, J.; Barcelos, M.A.; Bezerra, V.N.; Lyra E Silva, N.M.; Patel, J.; et al. A Human scFv Antibody That Targets and Neutralizes High Molecular Weight Pathogenic Amyloid-β Oligomers. J. Neurochem. 2017, 142, 934–947. [Google Scholar] [CrossRef]
- Viola, K.L.; Bicca, M.A.; Bebenek, A.M.; Kranz, D.L.; Nandwana, V.; Waters, E.A.; Haney, C.R.; Lee, M.; Gupta, A.; Brahmbhatt, Z.; et al. The Therapeutic and Diagnostic Potential of Amyloid β Oligomers Selective Antibodies to Treat Alzheimer’s Disease. Front. Neurosci. 2021, 15, 768646. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Mitochondrial Alterations in Alzheimer’s Disease. J. Alzheimers Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Ratnaike, T.E.; De Gruyter, H.L.M.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA Deletions Cause the Biochemical Defect Observed in Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef]
- McKenna, M.C.; Stridh, M.H.; McNair, L.F.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Glutamate Oxidation in Astrocytes: Roles of Glutamate Dehydrogenase and Aminotransferases. J. Neurosci. Res. 2016, 94, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Arrázola, M.S.; Ramos-Fernández, E.; Cisternas, P.; Ordenes, D.; Inestrosa, N.C. Wnt Signaling Prevents the Aβ Oligomer-Induced Mitochondrial Permeability Transition Pore Opening Preserving Mitochondrial Structure in Hippocampal Neurons. PLoS ONE 2017, 12, e0168840. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing Mitochondrial Proteostasis Reduces Amyloid-β Proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial Dysfunction in Alzheimer’s Disease: Role in Pathogenesis and Novel Therapeutic Opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased Mitochondrial Calcium Levels Associated with Neuronal Death in a Mouse Model of Alzheimer’s Disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Sayyed, U.M.H.; Mahalakshmi, R. Mitochondrial Protein Translocation Machinery: From TOM Structural Biogenesis to Functional Regulation. J. Biol. Chem. 2022, 298, 101870. [Google Scholar] [CrossRef]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic Exposure to Sub-Lethal Beta-Amyloid (Abeta) Inhibits the Import of Nuclear-Encoded Proteins to Mitochondria in Differentiated PC12 Cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Meng, X.; Song, Q.; Liu, Z.; Liu, X.; Wang, Y.; Liu, J. Neurotoxic β-Amyloid Oligomers Cause Mitochondrial Dysfunction-the Trigger for PANoptosis in Neurons. Front. Aging Neurosci. 2024, 16, 1400544. [Google Scholar] [CrossRef]
- Sahay, A.; Hen, R. Adult Hippocampal Neurogenesis in Depression. Nat. Neurosci. 2007, 10, 1110–1115. [Google Scholar] [CrossRef]
- Vaidya, V.A.; Fernandes, K.; Jha, S. Regulation of Adult Hippocampal Neurogenesis: Relevance to Depression. Expert Rev. Neurother. 2007, 7, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Diksic, M. Labelled Alpha-Methyl-L-Tryptophan as a Tracer for the Study of the Brain Serotonergic System. J. Psychiatry Neurosci. JPN 2001, 26, 293–303. [Google Scholar] [PubMed]
- Albrecht, J.; Zielińska, M. Exchange-Mode Glutamine Transport across CNS Cell Membranes. Neuropharmacology 2019, 161, 107560. [Google Scholar] [CrossRef] [PubMed]
- Abela, A.R.; Browne, C.J.; Sargin, D.; Prevot, T.D.; Ji, X.D.; Li, Z.; Lambe, E.K.; Fletcher, P.J. Median Raphe Serotonin Neurons Promote Anxiety-like Behavior via Inputs to the Dorsal Hippocampus. Neuropharmacology 2020, 168, 107985. [Google Scholar] [CrossRef]
- Zhang, X.; Beaulieu, J.-M.; Sotnikova, T.D.; Gainetdinov, R.R.; Caron, M.G. Tryptophan Hydroxylase-2 Controls Brain Serotonin Synthesis. Science 2004, 305, 217. [Google Scholar] [CrossRef]
- Galano, A.; Castañeda-Arriaga, R.; Pérez-González, A.; Tan, D.-X.; Reiter, R.J. Phenolic Melatonin-Related Compounds: Their Role as Chemical Protectors against Oxidative Stress. Molecules 2016, 21, 1442. [Google Scholar] [CrossRef]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin Regulates Mitochondrial Biogenesis and Function in Rodent Cortical Neurons via the 5-HT 2A Receptor and SIRT1–PGC-1α Axis. Proc. Natl. Acad. Sci. USA 2019, 116, 11028–11037. [Google Scholar] [CrossRef]
- Azmitia, E.C. Serotonin and Brain: Evolution, Neuroplasticity, and Homeostasis. Int. Rev. Neurobiol. 2007, 77, 31–56. [Google Scholar] [CrossRef]
- Velasquez, J.C.; Goeden, N.; Bonnin, A. Placental Serotonin: Implications for the Developmental Effects of SSRIs and Maternal Depression. Front. Cell. Neurosci. 2013, 7, 47. [Google Scholar] [CrossRef]
- Chaouloff, F.; Berton, O.; Mormède, P. Serotonin and Stress. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 21, 28S–32S. [Google Scholar] [CrossRef]
- Dalva, M.B.; McClelland, A.C.; Kayser, M.S. Cell Adhesion Molecules: Signalling Functions at the Synapse. Nat. Rev. Neurosci. 2007, 8, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Daubert, E.A.; Condron, B.G. Serotonin: A Regulator of Neuronal Morphology and Circuitry. Trends Neurosci. 2010, 33, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.; Cases, O.; Maroteaux, L. The Developmental Role of Serotonin: News from Mouse Molecular Genetics. Nat. Rev. Neurosci. 2003, 4, 1002–1012. [Google Scholar] [CrossRef]
- Jin, Y.; Dougherty, S.E.; Wood, K.; Sun, L.; Cudmore, R.H.; Abdalla, A.; Kannan, G.; Pletnikov, M.; Hashemi, P.; Linden, D.J. Regrowth of Serotonin Axons in the Adult Mouse Brain Following Injury. Neuron 2016, 91, 748–762. [Google Scholar] [CrossRef]
- Kajstura, T.J.; Dougherty, S.E.; Linden, D.J. Serotonin Axons in the Neocortex of the Adult Female Mouse Regrow after Traumatic Brain Injury. J. Neurosci. Res. 2018, 96, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Zahrai, A.; Vahid-Ansari, F.; Daigle, M.; Albert, P.R. Fluoxetine-Induced Recovery of Serotonin and Norepinephrine Projections in a Mouse Model of Post-Stroke Depression. Transl. Psychiatry 2020, 10, 334. [Google Scholar] [CrossRef]
- Turlejski, K. Evolutionary Ancient Roles of Serotonin: Long-Lasting Regulation of Activity and Development. Acta Neurobiol. Exp. 1996, 56, 619–636. [Google Scholar] [CrossRef]
- Lesch, K.-P.; Waider, J. Serotonin in the Modulation of Neural Plasticity and Networks: Implications for Neurodevelopmental Disorders. Neuron 2012, 76, 175–191. [Google Scholar] [CrossRef]
- Brummelte, S.; Mc Glanaghy, E.; Bonnin, A.; Oberlander, T.F. Developmental Changes in Serotonin Signaling: Implications for Early Brain Function, Behavior and Adaptation. Neuroscience 2017, 342, 212–231. [Google Scholar] [CrossRef]
- Lupien, S.J. Brains under Stress. Can. J. Psychiatry Rev. Can. Psychiatr. 2009, 54, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Alzari, P.M. Structural Insights into Sialic Acid Enzymology. Curr. Opin. Chem. Biol. 2008, 12, 565–572. [Google Scholar] [CrossRef]
- Cummings, K.J.; Leiter, J.C. Take a Deep Breath and Wake up: The Protean Role of Serotonin Preventing Sudden Death in Infancy. Exp. Neurol. 2020, 326, 113165. [Google Scholar] [CrossRef] [PubMed]
- Paulus, E.V.; Mintz, E.M. Circadian Rhythms of Clock Gene Expression in the Cerebellum of Serotonin-Deficient Pet-1 Knockout Mice. Brain Res. 2016, 1630, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Olivier, J.D.A.; Vinkers, C.H.; Olivier, B. The Role of the Serotonergic and GABA System in Translational Approaches in Drug Discovery for Anxiety Disorders. Front. Pharmacol. 2013, 4, 74. [Google Scholar] [CrossRef]
- De Deurwaerdère, P.; Di Giovanni, G. Serotonin in Health and Disease. Int. J. Mol. Sci. 2020, 21, 3500. [Google Scholar] [CrossRef]
- De Deurwaerdère, P.; Chagraoui, A.; Di Giovanni, G. Serotonin/Dopamine Interaction: Electrophysiological and Neurochemical Evidence. Prog. Brain Res. 2021, 261, 161–264. [Google Scholar] [CrossRef]
- Iovino, L.; Mutolo, D.; Cinelli, E.; Contini, M.; Pantaleo, T.; Bongianni, F. Breathing Stimulation Mediated by 5-HT1A and 5-HT3 Receptors within the preBötzinger Complex of the Adult Rabbit. Brain Res. 2019, 1704, 26–39. [Google Scholar] [CrossRef]
- Murphy, D.L.; Lerner, A.; Rudnick, G.; Lesch, K.-P. Serotonin Transporter: Gene, Genetic Disorders, and Pharmacogenetics. Mol. Interv. 2004, 4, 109–123. [Google Scholar] [CrossRef]
- Lin, Z.; Madras, B.K. Human Genetics and Pharmacology of Neurotransmitter Transporters. Handb. Exp. Pharmacol. 2006, 175, 327–371. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jørgensen, T.N.; Sørensen, L.; Eriksen, J.; Loland, C.J.; Strømgaard, K.; Gether, U. SLC6 Neurotransmitter Transporters: Structure, Function, and Regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef]
- Duan, H.; Wang, J. Selective Transport of Monoamine Neurotransmitters by Human Plasma Membrane Monoamine Transporter and Organic Cation Transporter 3. J. Pharmacol. Exp. Ther. 2010, 335, 743–753. [Google Scholar] [CrossRef]
- Bockaert, J.; Claeysen, S.; Bécamel, C.; Dumuis, A.; Marin, P. Neuronal 5-HT Metabotropic Receptors: Fine-Tuning of Their Structure, Signaling, and Roles in Synaptic Modulation. Cell Tissue Res. 2006, 326, 553–572. [Google Scholar] [CrossRef]
- Serotonin Receptors in Neurobiology; Chattopadhyay, A., Ed.; Frontiers in Neuroscience; CRC Press: Boca Raton, FL, USA, 2007; ISBN 978-0-8493-3977-6. [Google Scholar]
- Nadeev, A.D.; Zharkikh, I.L.; Avdonin, P.V.; Goncharov, N.V. Serotonin and its receptors in the cardiovascular system. Eksp. Klin. Farmakol. 2014, 77, 32–37. [Google Scholar] [PubMed]
- Wang, Y.; Tu, D.; Du, J.; Han, X.; Sun, Y.; Xu, Q.; Zhai, G.; Zhou, Y. Classification of Subcortical Vascular Cognitive Impairment Using Single MRI Sequence and Deep Learning Convolutional Neural Networks. Front. Neurosci. 2019, 13, 627. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Higashida, H.; Aoki, S.; Wada, K. Multiple Signal Transduction Pathways Mediated by 5-HT Receptors. Mol. Neurobiol. 2004, 29, 31–40. [Google Scholar] [CrossRef]
- Asada, M.; Ebihara, S.; Yamanda, S.; Niu, K.; Okazaki, T.; Sora, I.; Arai, H. Depletion of Serotonin and Selective Inhibition of 2B Receptor Suppressed Tumor Angiogenesis by Inhibiting Endothelial Nitric Oxide Synthase and Extracellular Signal-Regulated Kinase 1/2 Phosphorylation. Neoplasia 2009, 11, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Charnay, Y.; Léger, L. Brain Serotonergic Circuitries. Dialogues Clin. Neurosci. 2010, 12, 471–487. [Google Scholar] [CrossRef]
- Davis, R.P.; Pattison, J.; Thompson, J.M.; Tiniakov, R.; Scrogin, K.E.; Watts, S.W. 5-Hydroxytryptamine (5-HT) Reduces Total Peripheral Resistance during Chronic Infusion: Direct Arterial Mesenteric Relaxation Is Not Involved. BMC Pharmacol. 2012, 12, 4. [Google Scholar] [CrossRef]
- Fujita, M.; Minamino, T.; Sanada, S.; Asanuma, H.; Hirata, A.; Ogita, H.; Okada, K.; Tsukamoto, O.; Takashima, S.; Tomoike, H.; et al. Selective Blockade of Serotonin 5-HT2A Receptor Increases Coronary Blood Flow via Augmented Cardiac Nitric Oxide Release through 5-HT1B Receptor in Hypoperfused Canine Hearts. J. Mol. Cell. Cardiol. 2004, 37, 1219–1223. [Google Scholar] [CrossRef]
- Liu, Y.; Fanburg, B.L. Serotonin-Induced Growth of Pulmonary Artery Smooth Muscle Requires Activation of Phosphatidylinositol 3-Kinase/Serine-Threonine Protein Kinase B/Mammalian Target of Rapamycin/P70 Ribosomal S6 Kinase 1. Am. J. Respir. Cell Mol. Biol. 2006, 34, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Wang, W.W.; Fanburg, B.L. Superoxide as an Intermediate Signal for Serotonin-Induced Mitogenesis. Free Radic. Biol. Med. 1998, 24, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFalpha-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Linder, A.E.; Gaskell, G.L.; Szasz, T.; Thompson, J.M.; Watts, S.W. Serotonin Receptors in Rat Jugular Vein: Presence and Involvement in the Contraction. J. Pharmacol. Exp. Ther. 2010, 334, 116–123. [Google Scholar] [CrossRef]
- Lu, R.; Alioua, A.; Kumar, Y.; Kundu, P.; Eghbali, M.; Weisstaub, N.V.; Gingrich, J.A.; Stefani, E.; Toro, L. C-Src Tyrosine Kinase, a Critical Component for 5-HT2A Receptor-Mediated Contraction in Rat Aorta. J. Physiol. 2008, 586, 3855–3869. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.M.R. The Serotonin Hypothesis in Pulmonary Hypertension Revisited: Targets for Novel Therapies (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018759125. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, Y.V.; Garnovskaya, M.N.; Collinsworth, G.; Grewal, J.S.; Pendergrass, D.; Nagai, T.; Pinckney, S.; Greene, E.L.; Raymond, J.R. 5-Hydroxytryptamine1A Receptor/Gibetagamma Stimulates Mitogen-Activated Protein Kinase via NAD(P)H Oxidase and Reactive Oxygen Species Upstream of Src in Chinese Hamster Ovary Fibroblasts. Biochem. J. 2000, 347 Pt 1, 61–67. [Google Scholar] [CrossRef]
- Nelson, D.L. 5-HT5 Receptors. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 53–58. [Google Scholar] [CrossRef]
- Nichols, D.E.; Nichols, C.D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef]
- Ogden, K.; Thompson, J.M.; Hickner, Z.; Huang, T.; Tang, D.D.; Watts, S.W. A New Signaling Paradigm for Serotonin: Use of Crk-Associated Substrate in Arterial Contraction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2857–H2863. [Google Scholar] [CrossRef] [PubMed]
- Ullmer, C.; Boddeke, H.G.; Schmuck, K.; Lübbert, H. 5-HT2B Receptor-Mediated Calcium Release from Ryanodine-Sensitive Intracellular Stores in Human Pulmonary Artery Endothelial Cells. Br. J. Pharmacol. 1996, 117, 1081–1088. [Google Scholar] [CrossRef]
- Villalón, C.M.; Centurión, D. Cardiovascular Responses Produced by 5-Hydroxytriptamine:A Pharmacological Update on the Receptors/Mechanisms Involved and Therapeutic Implications. Naunyn. Schmiedebergs Arch. Pharmacol. 2007, 376, 45–63. [Google Scholar] [CrossRef]
- Watts, S.W. 5-HT in Systemic Hypertension: Foe, Friend or Fantasy? Clin. Sci. Lond. Engl. 1979 2005, 108, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, Y.; Kaneto, H.; Fanburg, B.L. JNK Regulates Serotonin-Mediated Proliferation and Migration of Pulmonary Artery Smooth Muscle Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2010, 298, L863–L869. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Yu, J.; Ma, Z.; Li, M.; Wang, J.; Hu, P.; Zou, J.; Liu, X.; Liu, Y.; et al. A Novel 5-HT1B Receptor Agonist of Herbal Compounds and One of the Therapeutic Uses for Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 735876. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Xie, H.; Laudon, M.; Zhou, S.; Tian, S.; You, Y. Piromelatine Ameliorates Memory Deficits Associated with Chronic Mild Stress-Induced Anhedonia in Rats. Psychopharmacology 2016, 233, 2229–2239. [Google Scholar] [CrossRef]
- Acquarone, E.; Argyrousi, E.K.; Arancio, O.; Watterson, D.M.; Roy, S.M. The 5HT2b Receptor in Alzheimer’s Disease: Increased Levels in Patient Brains and Antagonist Attenuation of Amyloid and Tau Induced Dysfunction. J. Alzheimers Dis. JAD 2024, 98, 1349–1360. [Google Scholar] [CrossRef]
- Liu, H.; He, Y.; Liu, H.; Brouwers, B.; Yin, N.; Lawler, K.; Keogh, J.M.; Henning, E.; Lee, D.-K.; Yu, M.; et al. Neural Circuits Expressing the Serotonin 2C Receptor Regulate Memory in Mice and Humans. Sci. Adv. 2024, 10, eadl2675. [Google Scholar] [CrossRef]
- Tian, Z.R.; Sharma, A.; Muresanu, D.F.; Sharma, S.; Feng, L.; Zhang, Z.; Li, C.; Buzoianu, A.D.; Lafuente, J.V.; Nozari, A.; et al. Nicotine Neurotoxicity Exacerbation Following Engineered Ag and Cu (50–60 Nm) Nanoparticles Intoxication. Neuroprotection with Nanowired Delivery of Antioxidant Compound H-290/51 Together with Serotonin 5-HT3 Receptor Antagonist Ondansetron. Int. Rev. Neurobiol. 2023, 172, 189–233. [Google Scholar] [CrossRef]
- Jiang, S.; Sydney, E.J.; Runyan, A.M.; Serpe, R.; Srikanth, M.; Figueroa, H.Y.; Yang, M.; Myeku, N. 5-HT4 Receptor Agonists Treatment Reduces Tau Pathology and Behavioral Deficit in the PS19 Mouse Model of Tauopathy. Front. Cell. Neurosci. 2024, 18, 1338502. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Bezprozvanny, I. Evaluation of Dimebon in Cellular Model of Huntington’s Disease. Mol. Neurodegener. 2008, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Chaumont-Dubel, S.; Dupuy, V.; Bockaert, J.; Bécamel, C.; Marin, P. The 5-HT6 Receptor Interactome: New Insight in Receptor Signaling and Its Impact on Brain Physiology and Pathologies. Neuropharmacology 2020, 172, 107839. [Google Scholar] [CrossRef]
- Quintero-Villegas, A.; Valdés-Ferrer, S.I. Central Nervous System Effects of 5-HT7 Receptors: A Potential Target for Neurodegenerative Diseases. Mol. Med. Camb. Mass 2022, 28, 70. [Google Scholar] [CrossRef]
- McDevitt, R.A.; Neumaier, J.F. Regulation of Dorsal Raphe Nucleus Function by Serotonin Autoreceptors: A Behavioral Perspective. J. Chem. Neuroanat. 2011, 41, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Courtney, N.A.; Ford, C.P. Mechanisms of 5-HT1A Receptor-Mediated Transmission in Dorsal Raphe Serotonin Neurons. J. Physiol. 2016, 594, 953–965. [Google Scholar] [CrossRef]
- Llamosas, N.; Bruzos-Cidón, C.; Rodríguez, J.J.; Ugedo, L.; Torrecilla, M. Deletion of GIRK2 Subunit of GIRK Channels Alters the 5-HT 1A Receptor-Mediated Signaling and Results in a Depression-Resistant Behavior. Int. J. Neuropsychopharmacol. 2015, 18, pyv051. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, A.; Corradetti, R.; Mlinar, B. Pharmacological Characterization of 5-HT1A Autoreceptor-Coupled GIRK Channels in Rat Dorsal Raphe 5-HT Neurons. PLoS ONE 2015, 10, e0140369. [Google Scholar] [CrossRef]
- Grunnet, M.; Jespersen, T.; Perrier, J.-F. 5-HT1A Receptors Modulate Small-Conductance Ca2+-Activated K+ Channels. J. Neurosci. Res. 2004, 78, 845–854. [Google Scholar] [CrossRef]
- Kirby, L.G.; Pernar, L.; Valentino, R.J.; Beck, S.G. Distinguishing Characteristics of Serotonin and Non-Serotonin-Containing Cells in the Dorsal Raphe Nucleus: Electrophysiological and Immunohistochemical Studies. Neuroscience 2003, 116, 669–683. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin Stimulates Mitochondrial Transport in Hippocampal Neurons. Mol. Cell. Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef]
- Albert, P.R.; Lemonde, S. 5-HT1A Receptors, Gene Repression, and Depression: Guilt by Association. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2004, 10, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Altieri, S.C.; Garcia-Garcia, A.L.; Leonardo, E.D.; Andrews, A.M. Rethinking 5-HT1A Receptors: Emerging Modes of Inhibitory Feedback of Relevance to Emotion-Related Behavior. ACS Chem. Neurosci. 2013, 4, 72–83. [Google Scholar] [CrossRef]
- Vargas, M.V.; Dunlap, L.E.; Dong, C.; Carter, S.J.; Tombari, R.J.; Jami, S.A.; Cameron, L.P.; Patel, S.D.; Hennessey, J.J.; Saeger, H.N.; et al. Psychedelics Promote Neuroplasticity through the Activation of Intracellular 5-HT2A Receptors. Science 2023, 379, 700–706. [Google Scholar] [CrossRef]
- Hornung, J.-P. The Human Raphe Nuclei and the Serotonergic System. J. Chem. Neuroanat. 2003, 26, 331–343. [Google Scholar] [CrossRef]
- Carhart-Harris, R.L. Serotonin, Psychedelics and Psychiatry. World Psychiatry Off. J. World Psychiatr. Assoc. WPA 2018, 17, 358–359. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Phillips, A.G. Neuroplasticity as a Convergent Mechanism of Ketamine and Classical Psychedelics. Trends Pharmacol. Sci. 2021, 42, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Wallach, J.; Cao, A.B.; Calkins, M.M.; Heim, A.J.; Lanham, J.K.; Bonniwell, E.M.; Hennessey, J.J.; Bock, H.A.; Anderson, E.I.; Sherwood, A.M.; et al. Identification of 5-HT2A Receptor Signaling Pathways Associated with Psychedelic Potential. Nat. Commun. 2023, 14, 8221. [Google Scholar] [CrossRef]
- Cornea-Hébert, V.; Watkins, K.C.; Roth, B.L.; Kroeze, W.K.; Gaudreau, P.; Leclerc, N.; Descarries, L. Similar Ultrastructural Distribution of the 5-HT(2A) Serotonin Receptor and Microtubule-Associated Protein MAP1A in Cortical Dendrites of Adult Rat. Neuroscience 2002, 113, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Béïque, J.-C.; Imad, M.; Mladenovic, L.; Gingrich, J.A.; Andrade, R. Mechanism of the 5-Hydroxytryptamine 2A Receptor-Mediated Facilitation of Synaptic Activity in Prefrontal Cortex. Proc. Natl. Acad. Sci. USA 2007, 104, 9870–9875. [Google Scholar] [CrossRef]
- Andrade, R. Serotonergic Regulation of Neuronal Excitability in the Prefrontal Cortex. Neuropharmacology 2011, 61, 382–386. [Google Scholar] [CrossRef]
- Aznar, S.; Hervig, M.E.-S. The 5-HT2A Serotonin Receptor in Executive Function: Implications for Neuropsychiatric and Neurodegenerative Diseases. Neurosci. Biobehav. Rev. 2016, 64, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.C.; Olson, D.E.; Preller, K.H.; Roth, B.L. The Neural Basis of Psychedelic Action. Nat. Neurosci. 2022, 25, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Yakel, J.L.; Shao, X.M.; Jackson, M.B. The Selectivity of the Channel Coupled to the 5-HT3 Receptor. Brain Res. 1990, 533, 46–52. [Google Scholar] [CrossRef]
- Malone, H.M.; Peters, J.A.; Lambert, J.J. Physiological and Pharmacological Properties of 5-HT3 Receptors--a Patch Clamp-Study. Neuropeptides 1991, 19, 25–30. [Google Scholar] [CrossRef]
- Maricq, A.V.; Peterson, A.S.; Brake, A.J.; Myers, R.M.; Julius, D. Primary Structure and Functional Expression of the 5HT3 Receptor, a Serotonin-Gated Ion Channel. Science 1991, 254, 432–437. [Google Scholar] [CrossRef]
- Niesler, B.; Frank, B.; Kapeller, J.; Rappold, G.A. Cloning, Physical Mapping and Expression Analysis of the Human 5-HT3 Serotonin Receptor-like Genes HTR3C, HTR3D and HTR3E. Gene 2003, 310, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Blandina, P.; Goldfarb, J.; Craddock-Royal, B.; Green, J.P. Release of Endogenous Dopamine by Stimulation of 5-Hydroxytryptamine3 Receptors in Rat Striatum. J. Pharmacol. Exp. Ther. 1989, 251, 803–809. [Google Scholar] [CrossRef]
- Miquel, M.-C.; Emerit, M.B.; Nosjean, A.; Simon, A.; Rumajogee, P.; Brisorgueil, M.-J.; Doucet, E.; Hamon, M.; Vergé, D. Differential Subcellular Localization of the 5-HT3-As Receptor Subunit in the Rat Central Nervous System. Eur. J. Neurosci. 2002, 15, 449–457. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lummis, S.C.R. 5-HT3 Receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [Google Scholar] [CrossRef]
- Bloom, F.E.; Morales, M. The Central 5-HT3 Receptor in CNS Disorders. Neurochem. Res. 1998, 23, 653–659. [Google Scholar] [CrossRef]
- Morales, M.; Bloom, F.E. The 5-HT3 Receptor Is Present in Different Subpopulations of GABAergic Neurons in the Rat Telencephalon. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 3157–3167. [Google Scholar] [CrossRef]
- Turner, T.J.; Mokler, D.J.; Luebke, J.I. Calcium Influx through Presynaptic 5-HT3 Receptors Facilitates GABA Release in the Hippocampus: In Vitro Slice and Synaptosome Studies. Neuroscience 2004, 129, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Krauthausen, M.; Kummer, M.P.; Zimmermann, J.; Reyes-Irisarri, E.; Terwel, D.; Bulic, B.; Heneka, M.T.; Müller, M. CXCR3 Promotes Plaque Formation and Behavioral Deficits in an Alzheimer’s Disease Model. J. Clin. Investig. 2015, 125, 365–378. [Google Scholar] [CrossRef]
- Liu, L.-F.; Liu, Y.-T.; Wu, D.-D.; Cheng, J.; Li, N.-N.; Zheng, Y.-N.; Huang, L.; Yuan, Q.-L. Inhibiting 5-Hydroxytryptamine Receptor 3 Alleviates Pathological Changes of a Mouse Model of Alzheimer’s Disease. Neural Regen. Res. 2023, 18, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Claeysen, S.; Compan, V.; Dumuis, A. 5-HT(4) Receptors: History, Molecular Pharmacology and Brain Functions. Neuropharmacology 2008, 55, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, P.; Hall, H.; Gommeren, W.; Cras, P.; Langlois, X.; Jurzak, M.; Leysen, J.E. Mapping of Serotonin 5-HT(4) Receptor mRNA and Ligand Binding Sites in the Post-Mortem Human Brain. Synapse 2000, 36, 35–46. [Google Scholar] [CrossRef]
- Karayol, R.; Medrihan, L.; Warner-Schmidt, J.L.; Fait, B.W.; Rao, M.N.; Holzner, E.B.; Greengard, P.; Heintz, N.; Schmidt, E.F. Serotonin Receptor 4 in the Hippocampus Modulates Mood and Anxiety. Mol. Psychiatry 2021, 26, 2334–2349. [Google Scholar] [CrossRef]
- Lucas, G.; Rymar, V.V.; Du, J.; Mnie-Filali, O.; Bisgaard, C.; Manta, S.; Lambas-Senas, L.; Wiborg, O.; Haddjeri, N.; Piñeyro, G.; et al. Serotonin(4) (5-HT(4)) Receptor Agonists Are Putative Antidepressants with a Rapid Onset of Action. Neuron 2007, 55, 712–725. [Google Scholar] [CrossRef]
- Faye, C.; Hen, R.; Guiard, B.P.; Denny, C.A.; Gardier, A.M.; Mendez-David, I.; David, D.J. Rapid Anxiolytic Effects of RS67333, a Serotonin Type 4 Receptor Agonist, and Diazepam, a Benzodiazepine, Are Mediated by Projections From the Prefrontal Cortex to the Dorsal Raphe Nucleus. Biol. Psychiatry 2020, 87, 514–525. [Google Scholar] [CrossRef]
- Peñas-Cazorla, R.; Vilaró, M.T. Serotonin 5-HT4 Receptors and Forebrain Cholinergic System: Receptor Expression in Identified Cell Populations. Brain Struct. Funct. 2015, 220, 3413–3434. [Google Scholar] [CrossRef]
- Köhler-Forsberg, K.; Dam, V.H.; Ozenne, B.; Sankar, A.; Beliveau, V.; Landman, E.B.; Larsen, S.V.; Poulsen, A.S.; Ip, C.-T.; Jørgensen, A.; et al. Serotonin 4 Receptor Brain Binding in Major Depressive Disorder and Association With Memory Dysfunction. JAMA Psychiatry 2023, 80, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.R. 5-ht5A Receptors as a Therapeutic Target. Pharmacol. Ther. 2006, 111, 707–714. [Google Scholar] [CrossRef]
- Vidal-Cantú, G.C.; Jiménez-Hernández, M.; Rocha-González, H.I.; Villalón, C.M.; Granados-Soto, V.; Muñoz-Islas, E. Role of 5-HT5A and 5-HT1B/1D Receptors in the Antinociception Produced by Ergotamine and Valerenic Acid in the Rat Formalin Test. Eur. J. Pharmacol. 2016, 781, 109–116. [Google Scholar] [CrossRef]
- Yun, H.-M.; Kim, S.; Kim, H.-J.; Kostenis, E.; Kim, J.I.; Seong, J.Y.; Baik, J.-H.; Rhim, H. The Novel Cellular Mechanism of Human 5-HT6 Receptor through an Interaction with Fyn. J. Biol. Chem. 2007, 282, 5496–5505. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.-M.; Baik, J.-H.; Kang, I.; Jin, C.; Rhim, H. Physical Interaction of Jab1 with Human Serotonin 6 G-Protein-Coupled Receptor and Their Possible Roles in Cell Survival. J. Biol. Chem. 2010, 285, 10016–10029. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, T.; Bordi, F.; Minetti, P.; Spadoni, G.; Yun, H.-M.; Im, B.-H.; Tarzia, G.; Rhim, H.; Borsini, F. ST1936 Stimulates cAMP, Ca2+, ERK1/2 and Fyn Kinase through a Full Activation of Cloned Human 5-HT6 Receptors. Eur. J. Pharmacol. 2011, 661, 8–14. [Google Scholar] [CrossRef]
- Riccio, O.; Potter, G.; Walzer, C.; Vallet, P.; Szabó, G.; Vutskits, L.; Kiss, J.Z.; Dayer, A.G. Excess of Serotonin Affects Embryonic Interneuron Migration through Activation of the Serotonin Receptor 6. Mol. Psychiatry 2009, 14, 280–290. [Google Scholar] [CrossRef]
- Jacobshagen, M.; Niquille, M.; Chaumont-Dubel, S.; Marin, P.; Dayer, A. The Serotonin 6 Receptor Controls Neuronal Migration during Corticogenesis via a Ligand-Independent Cdk5-Dependent Mechanism. Dev. Camb. Engl. 2014, 141, 3370–3377. [Google Scholar] [CrossRef]
- Guadiana, S.M.; Semple-Rowland, S.; Daroszewski, D.; Madorsky, I.; Breunig, J.J.; Mykytyn, K.; Sarkisian, M.R. Arborization of Dendrites by Developing Neocortical Neurons Is Dependent on Primary Cilia and Type 3 Adenylyl Cyclase. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2626–2638. [Google Scholar] [CrossRef]
- Lesiak, A.J.; Brodsky, M.; Cohenca, N.; Croicu, A.G.; Neumaier, J.F. Restoration of Physiological Expression of 5-HT 6 Receptor into the Primary Cilia of Null Mutant Neurons Lengthens Both Primary Cilia and Dendrites. Mol. Pharmacol. 2018, 94, 731–742. [Google Scholar] [CrossRef]
- Roberts, J.C.; Reavill, C.; East, S.Z.; Harrison, P.J.; Patel, S.; Routledge, C.; Leslie, R.A. The Distribution of 5-HT6 Receptors in Rat Brain: An Autoradiographic Binding Study Using the Radiolabelled 5-HT6 Receptor Antagonist [125I]SB-258585. Brain Res. 2002, 934, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Woolley, M.L.; Marsden, C.A.; Fone, K.C.F. 5-Ht6 Receptors. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Helboe, L.; Egebjerg, J.; de Jong, I.E.M. Distribution of Serotonin Receptor 5-HT6 mRNA in Rat Neuronal Subpopulations: A Double in Situ Hybridization Study. Neuroscience 2015, 310, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, V.; Prieur, M.; Pizzoccaro, A.; Margarido, C.; Valjent, E.; Bockaert, J.; Bouschet, T.; Marin, P.; Chaumont-Dubel, S. Spatiotemporal Dynamics of 5-HT6 Receptor Ciliary Localization during Mouse Brain Development. Neurobiol. Dis. 2023, 176, 105949. [Google Scholar] [CrossRef]
- Meffre, J.; Chaumont-Dubel, S.; Mannoury La Cour, C.; Loiseau, F.; Watson, D.J.G.; Dekeyne, A.; Séveno, M.; Rivet, J.; Gaven, F.; Déléris, P.; et al. 5-HT 6 Receptor Recruitment of mTOR as a Mechanism for Perturbed Cognition in Schizophrenia. EMBO Mol. Med. 2012, 4, 1043–1056. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Hirst, W.D.; Chen, C.P.L.-H.; Lasheras, B.; Francis, P.T.; Ramírez, M.J. Differential Involvement of 5-HT(1B/1D) and 5-HT6 Receptors in Cognitive and Non-Cognitive Symptoms in Alzheimer’s Disease. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 410–416. [Google Scholar] [CrossRef]
- Upton, N.; Chuang, T.T.; Hunter, A.J.; Virley, D.J. 5-HT6 Receptor Antagonists as Novel Cognitive Enhancing Agents for Alzheimer’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 458–469. [Google Scholar] [CrossRef]
- Foley, A.G.; Murphy, K.J.; Hirst, W.D.; Gallagher, H.C.; Hagan, J.J.; Upton, N.; Walsh, F.S.; Regan, C.M. The 5-HT(6) Receptor Antagonist SB-271046 Reverses Scopolamine-Disrupted Consolidation of a Passive Avoidance Task and Ameliorates Spatial Task Deficits in Aged Rats. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 93–100. [Google Scholar] [CrossRef]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6 Receptor Antagonists for the Treatment of Cognitive Deficiency in Alzheimer’s Disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Ivanenkov, Y.A.; Veselov, M.S.; Okun, I.M. AVN-322 Is a Safe Orally Bio-Available Potent and Highly Selective Antagonist of 5-HT6R with Demonstrated Ability to Improve Impaired Memory in Animal Models. Curr. Alzheimer Res. 2017, 14, 268–294. [Google Scholar] [CrossRef]
- Suárez-Santiago, J.E.; Roldán Roldán, G.; Picazo Picazo, O. The 5-HT6R Agonist E-6837 and the Antagonist SB-271046 Reverse the Psychotic-like Behaviors Induced by Ketamine. Behav. Pharmacol. 2022, 33, 249–254. [Google Scholar] [CrossRef]
- Kucwaj-Brysz, K.; Baltrukevich, H.; Czarnota, K.; Handzlik, J. Chemical Update on the Potential for Serotonin 5-HT6 and 5-HT7 Receptor Agents in the Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2021, 49, 128275. [Google Scholar] [CrossRef] [PubMed]
- Heidmann, D.E.; Metcalf, M.A.; Kohen, R.; Hamblin, M.W. Four 5-Hydroxytryptamine7 (5-HT7) Receptor Isoforms in Human and Rat Produced by Alternative Splicing: Species Differences Due to Altered Intron-Exon Organization. J. Neurochem. 1997, 68, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Adham, N.; Zgombick, J.M.; Bard, J.; Branchek, T.A. Functional Characterization of the Recombinant Human 5-Hydroxytryptamine7(a) Receptor Isoform Coupled to Adenylate Cyclase Stimulation. J. Pharmacol. Exp. Ther. 1998, 287, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Guseva, D.; Wirth, A.; Ponimaskin, E. Cellular Mechanisms of the 5-HT7 Receptor-Mediated Signaling. Front. Behav. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Kobe, F.; Guseva, D.; Jensen, T.P.; Wirth, A.; Renner, U.; Hess, D.; Müller, M.; Medrihan, L.; Zhang, W.; Zhang, M.; et al. 5-HT7R/G12 Signaling Regulates Neuronal Morphology and Function in an Age-Dependent Manner. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 2915–2930. [Google Scholar] [CrossRef]
- Speranza, L.; Giuliano, T.; Volpicelli, F.; De Stefano, M.E.; Lombardi, L.; Chambery, A.; Lacivita, E.; Leopoldo, M.; Bellenchi, G.C.; Di Porzio, U.; et al. Activation of 5-HT7 Receptor Stimulates Neurite Elongation through mTOR, Cdc42 and Actin Filaments Dynamics. Front. Behav. Neurosci. 2015, 9, 62. [Google Scholar] [CrossRef]
- Marin, P.; Dityatev, A. 5-HT7 Receptor Shapes Spinogenesis in Cortical and Striatal Neurons: An Editorial Highlight for “Serotonin 5-HT7 Receptor Increases the Density of Dendritic Spines and Facilitates Synaptogenesis in Forebrain Neurons”. J. Neurochem. 2017, 141, 644–646. [Google Scholar] [CrossRef]
- Speranza, L.; Chambery, A.; Di Domenico, M.; Crispino, M.; Severino, V.; Volpicelli, F.; Leopoldo, M.; Bellenchi, G.C.; di Porzio, U.; Perrone-Capano, C. The Serotonin Receptor 7 Promotes Neurite Outgrowth via ERK and Cdk5 Signaling Pathways. Neuropharmacology 2013, 67, 155–167. [Google Scholar] [CrossRef]
- Kvachnina, E.; Liu, G.; Dityatev, A.; Renner, U.; Dumuis, A.; Richter, D.W.; Dityateva, G.; Schachner, M.; Voyno-Yasenetskaya, T.A.; Ponimaskin, E.G. 5-HT7 Receptor Is Coupled to G Alpha Subunits of Heterotrimeric G12-Protein to Regulate Gene Transcription and Neuronal Morphology. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 7821–7830. [Google Scholar] [CrossRef]
- Speranza, L.; Labus, J.; Volpicelli, F.; Guseva, D.; Lacivita, E.; Leopoldo, M.; Bellenchi, G.C.; Di Porzio, U.; Bijata, M.; Perrone-Capano, C.; et al. Serotonin 5- HT 7 Receptor Increases the Density of Dendritic Spines and Facilitates Synaptogenesis in Forebrain Neurons. J. Neurochem. 2017, 141, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Sanger, G.J. 5-Hydroxytryptamine and the Gastrointestinal Tract: Where Next? Trends Pharmacol. Sci. 2008, 29, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Ciranna, L.; Catania, M.V. 5-HT7 Receptors as Modulators of Neuronal Excitability, Synaptic Transmission and Plasticity: Physiological Role and Possible Implications in Autism Spectrum Disorders. Front. Cell. Neurosci. 2014, 8, 250. [Google Scholar] [CrossRef]
- Chang Chien, C.-C.; Hsin, L.-W.; Su, M.-J. Activation of Serotonin 5-HT7 Receptor Induces Coronary Flow Increase in Isolated Rat Heart. Eur. J. Pharmacol. 2015, 748, 68–75. [Google Scholar] [CrossRef]
- Hedlund, P.B.; Huitron-Resendiz, S.; Henriksen, S.J.; Sutcliffe, J.G. 5-HT7 Receptor Inhibition and Inactivation Induce Antidepressantlike Behavior and Sleep Pattern. Biol. Psychiatry 2005, 58, 831–837. [Google Scholar] [CrossRef]
- Blattner, K.M.; Canney, D.J.; Pippin, D.A.; Blass, B.E. Pharmacology and Therapeutic Potential of the 5-HT7 Receptor. ACS Chem. Neurosci. 2019, 10, 89–119. [Google Scholar] [CrossRef]
- Shimizu, M.; Nishida, A.; Zensho, H.; Miyata, M.; Yamawaki, S. Agonist-Induced Desensitization of Adenylyl Cyclase Activity Mediated by 5-Hydroxytryptamine7 Receptors in Rat Frontocortical Astrocytes. Brain Res. 1998, 784, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Mahé, C.; Loetscher, E.; Dev, K.K.; Bobirnac, I.; Otten, U.; Schoeffter, P. Serotonin 5-HT7 Receptors Coupled to Induction of Interleukin-6 in Human Microglial MC-3 Cells. Neuropharmacology 2005, 49, 40–47. [Google Scholar] [CrossRef]
- Hashemi-Firouzi, N.; Komaki, A.; Soleimani Asl, S.; Shahidi, S. The Effects of the 5-HT7 Receptor on Hippocampal Long-Term Potentiation and Apoptosis in a Rat Model of Alzheimer’s Disease. Brain Res. Bull. 2017, 135, 85–91. [Google Scholar] [CrossRef]
- Wan, M.; Ding, L.; Wang, D.; Han, J.; Gao, P. Serotonin: A Potent Immune Cell Modulator in Autoimmune Diseases. Front. Immunol. 2020, 11, 186. [Google Scholar] [CrossRef]
- Leff-Gelman, P.; Mancilla-Herrera, I.; Flores-Ramos, M.; Cruz-Fuentes, C.; Reyes-Grajeda, J.P.; García-Cuétara, M.D.P.; Bugnot-Pérez, M.D.; Pulido-Ascencio, D.E. The Immune System and the Role of Inflammation in Perinatal Depression. Neurosci. Bull. 2016, 32, 398–420. [Google Scholar] [CrossRef] [PubMed]
- Abuelezz, S.A.; Hendawy, N.; Magdy, Y. Targeting Oxidative Stress, Cytokines and Serotonin Interactions Via Indoleamine 2, 3 Dioxygenase by Coenzyme Q10: Role in Suppressing Depressive Like Behavior in Rats. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2017, 12, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Schaulies, J.; Beyersdorf, N. CD4+ Foxp3+ Regulatory T Cell-Mediated Immunomodulation by Anti-Depressants Inhibiting Acid Sphingomyelinase. Biol. Chem. 2018, 399, 1175–1182. [Google Scholar] [CrossRef]
- Stasi, C.; Sadalla, S.; Milani, S. The Relationship Between the Serotonin Metabolism, Gut-Microbiota and the Gut-Brain Axis. Curr. Drug Metab. 2019, 20, 646–655. [Google Scholar] [CrossRef]
- Hersey, M.; Woodruff, J.L.; Maxwell, N.; Sadek, A.T.; Bykalo, M.K.; Bain, I.; Grillo, C.A.; Piroli, G.G.; Hashemi, P.; Reagan, L.P. High-Fat Diet Induces Neuroinflammation and Reduces the Serotonergic Response to Escitalopram in the Hippocampus of Obese Rats. Brain. Behav. Immun. 2021, 96, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, A.; Szklarczyk, J.; Barańska, I.; Majda, A.; Jaworek, J. Association between Levels of Serotonin, Melatonin, Cortisol and the Clinical Condition of Patients with Rheumatoid Arthritis. Rheumatol. Int. 2023, 43, 859–866. [Google Scholar] [CrossRef]
- Han, K.M.; Ham, B.J. How Inflammation Affects the Brain in Depression: A Review of Functional and Structural MRI Studies. J. Clin. Neurol. Seoul Korea 2021, 17, 503–515. [Google Scholar] [CrossRef]
- Haq, S.; Grondin, J.A.; Khan, W.I. Tryptophan-Derived Serotonin-Kynurenine Balance in Immune Activation and Intestinal Inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21888. [Google Scholar] [CrossRef] [PubMed]
- Tchinda Defo, S.H.; Moussa, D.; Bouvourné, P.; Guédang Nyayi, S.D.; Woumitna, G.C.; Kodji, K.; Wado, E.K.; Ngatanko Abaissou, H.H.; Foyet, H.S. Unpredictable Chronic Mild Stress Induced Anxio-Depressive Disorders and Enterobacteria Dysbiosis: Potential Protective Effects of Detariummicrocarpum. J. Ethnopharmacol. 2025, 337, 118940. [Google Scholar] [CrossRef]
- Anderson, G. Depression Pathophysiology: Astrocyte Mitochondrial Melatonergic Pathway as Crucial Hub. Int. J. Mol. Sci. 2022, 24, 350. [Google Scholar] [CrossRef]
- Savonije, K.; Weaver, D.F. The Role of Tryptophan Metabolism in Alzheimer’s Disease. Brain Sci. 2023, 13, 292. [Google Scholar] [CrossRef]
- Sheline, Y.I.; West, T.; Yarasheski, K.; Swarm, R.; Jasielec, M.S.; Fisher, J.R.; Ficker, W.D.; Yan, P.; Xiong, C.; Frederiksen, C.; et al. An Antidepressant Decreases CSF Aβ Production in Healthy Individuals and in Transgenic AD Mice. Sci. Transl. Med. 2014, 6, 236re4. [Google Scholar] [CrossRef]
- Singh, K.; Gupta, J.K.; Sethi, P.; Mathew, S.; Bhatt, A.; Sharma, M.C.; Saha, S.; Shamim; Kumar, S. Recent Advances in the Synthesis of Antioxidant Derivatives: Pharmacological Insights for Neurological Disorders. Curr. Top. Med. Chem. 2024, 24, 1940–1959. [Google Scholar] [CrossRef] [PubMed]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.-J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, Biological Evaluation, and Computational Studies of Tri- and Tetracyclic Nitrogen-Bridgehead Compounds as Potent Dual-Acting AChE Inhibitors and h H3 Receptor Antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Dolles, D.; Nimczick, M.; Scheiner, M.; Ramler, J.; Stadtmüller, P.; Sawatzky, E.; Drakopoulos, A.; Sotriffer, C.; Wittmann, H.-J.; Strasser, A.; et al. Aminobenzimidazoles and Structural Isomers as Templates for Dual-Acting Butyrylcholinesterase Inhibitors and hCB2 R Ligands To Combat Neurodegenerative Disorders. ChemMedChem 2016, 11, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Dolles, D.; Hoffmann, M.; Gunesch, S.; Marinelli, O.; Möller, J.; Santoni, G.; Chatonnet, A.; Lohse, M.J.; Wittmann, H.-J.; Strasser, A.; et al. Structure–Activity Relationships and Computational Investigations into the Development of Potent and Balanced Dual-Acting Butyrylcholinesterase Inhibitors and Human Cannabinoid Receptor 2 Ligands with Pro-Cognitive in Vivo Profiles. J. Med. Chem. 2018, 61, 1646–1663. [Google Scholar] [CrossRef] [PubMed]
- Szałaj, N.; Godyń, J.; Jończyk, J.; Pasieka, A.; Panek, D.; Wichur, T.; Więckowski, K.; Zaręba, P.; Bajda, M.; Pislar, A.; et al. Multidirectional in Vitro and in Cellulo Studies as a Tool for Identification of Multi-Target-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2020, 35, 1944–1952. [Google Scholar] [CrossRef]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities as Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef]
- Dumuis, A.; Sebben, M.; Monferini, E.; Nicola, M.; Turconi, M.; Ladinsky, H.; Bockaert, J. Azabicycloalkyl Benzimidazolone Derivatives as a Novel Class of Potent Agonists at the 5-HT4 Receptor Positively Coupled to Adenylate Cyclase in Brain. Naunyn. Schmiedebergs Arch. Pharmacol. 1991, 343, 245–251. [Google Scholar] [CrossRef]
- Manzke, T.; Guenther, U.; Ponimaskin, E.G.; Haller, M.; Dutschmann, M.; Schwarzacher, S.; Richter, D.W. 5-HT4(a) Receptors Avert Opioid-Induced Breathing Depression without Loss of Analgesia. Science 2003, 301, 226–229. [Google Scholar] [CrossRef]
- Chelusnova, Y.V.; Voronina, P.A.; Belinskaia, D.A.; Goncharov, N.V. Benzimidazole-Carboxamides as Potential Therapeutics for Alzheimer’s Disease: Primary Analysis In Silico and In Vitro. Bull. Exp. Biol. Med. 2023, 175, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Belinskaia, D.A.; Voronina, P.A.; Krivorotov, D.V.; Jenkins, R.O.; Goncharov, N.V. Anticholinesterase and Serotoninergic Evaluation of Benzimidazole-Carboxamides as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease. Pharmaceutics 2023, 15, 2159. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.M.; Suh, G.-H.; Lee, B.C.; Choi, I.-G.; Kim, H.S.; Kim, J.W.; Hwang, J.; Yi, D.; Kim, J.W. High-Intensity Walking in Midlife Is Associated with Improved Memory in Physically Capable Older Adults. Alzheimers Res. Ther. 2023, 15, 143. [Google Scholar] [CrossRef]
- Park, S.-S.; Park, H.-S.; Kim, C.-J.; Baek, S.-S.; Kim, T.-W. Exercise Attenuates Maternal Separation-Induced Mood Disorder-like Behaviors by Enhancing Mitochondrial Functions and Neuroplasticity in the Dorsal Raphe. Behav. Brain Res. 2019, 372, 112049. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, A.; Marques, E.A.; Mota, J.; Carvalho, J. Effects of a Multicomponent Exercise Program in Institutionalized Elders with Alzheimer’s Disease. Dementia 2019, 18, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chu, J.M.T.; Yan, T.; Zhang, Y.; Chen, Y.; Chang, R.C.C.; Wong, G.T.C. Short-Term Resistance Exercise Inhibits Neuroinflammation and Attenuates Neuropathological Changes in 3xTg Alzheimer’s Disease Mice. J. Neuroinflamm. 2020, 17, 4. [Google Scholar] [CrossRef]
- Lv, S.; Wang, Q.; Liu, W.; Zhang, X.; Cui, M.; Li, X.; Xu, Y. Comparison of Various Exercise Interventions on Cognitive Function in Alzheimer’s Patients: A Network Meta-Analysis. Arch. Gerontol. Geriatr. 2023, 115, 105113. [Google Scholar] [CrossRef]
- Adan, R.A.H.; van der Beek, E.M.; Buitelaar, J.K.; Cryan, J.F.; Hebebrand, J.; Higgs, S.; Schellekens, H.; Dickson, S.L. Nutritional Psychiatry: Towards Improving Mental Health by What You Eat. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2019, 29, 1321–1332. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaia, D.A.; Ukolov, A.I.; Jenkins, R.O.; Avdonin, P.V. Organosulfur compounds as nutraceuticals. In Nutraceuticals: Efficacy, Safety, and Toxicity, 2nd ed.; Gupta, R.C., Lall, R., Srivastava, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 911–924. ISBN 978-0-12-821038-3. [Google Scholar] [CrossRef]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a Potential Protective Phytochemical against Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Santana, R.A.; Silva-Islas, C.A.; Chánez-Cárdenas, M.E.; Santamaría, A.; Maldonado, P.D. The Antioxidant Mechanisms Underlying the Aged Garlic Extract- and S-Allylcysteine-Induced Protection. Oxid. Med. Cell. Longev. 2012, 2012, 907162. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, L.; Chu, Y.; Feng, F.; Tang, W.; Chen, C.; Qiu, Y.; Hu, Z.; Diao, H.; Tang, Z. Dietary Taurine Improves Growth Performance and Intestine Health via the GSH/GSSG Antioxidant System and Nrf2/ARE Signaling Pathway in Weaned Piglets. Antioxidants 2023, 12, 1852. [Google Scholar] [CrossRef] [PubMed]
- Seol, S.-I.; Kang, I.S.; Lee, J.S.; Lee, J.-K.; Kim, C. Taurine Chloramine-Mediated Nrf2 Activation and HO-1 Induction Confer Protective Effects in Astrocytes. Antioxidants 2024, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Bellavite, P. Neuroprotective Potentials of Flavonoids: Experimental Studies and Mechanisms of Action. Antioxidants 2023, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, X.; Wu, W.; Wang, J.; Xie, H.; Wu, Z. Regeneration of Glutathione by α-Lipoic Acid via Nrf2/ARE Signaling Pathway Alleviates Cadmium-Induced HepG2 Cell Toxicity. Environ. Toxicol. Pharmacol. 2017, 51, 30–37. [Google Scholar] [CrossRef]
- Toohey, J.I.; Cooper, A.J.L. Thiosulfoxide (Sulfane) Sulfur: New Chemistry and New Regulatory Roles in Biology. Molecules 2014, 19, 12789–12813. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Reprint of: Nrf2/ARE-Mediated Antioxidant Actions of pro-Electrophilic Drugs. Free Radic. Biol. Med. 2014, 66, 45–57. [Google Scholar] [CrossRef]
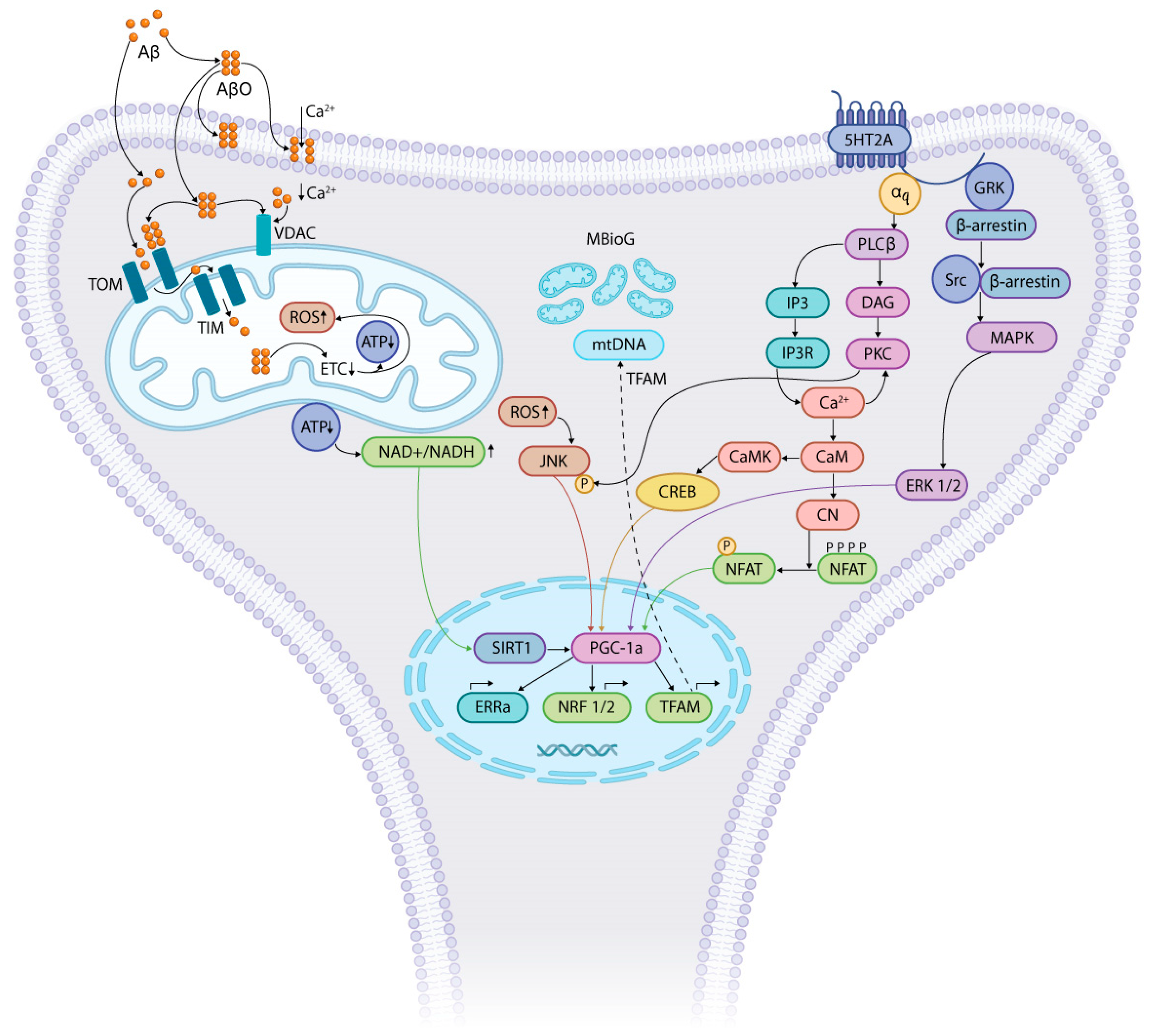
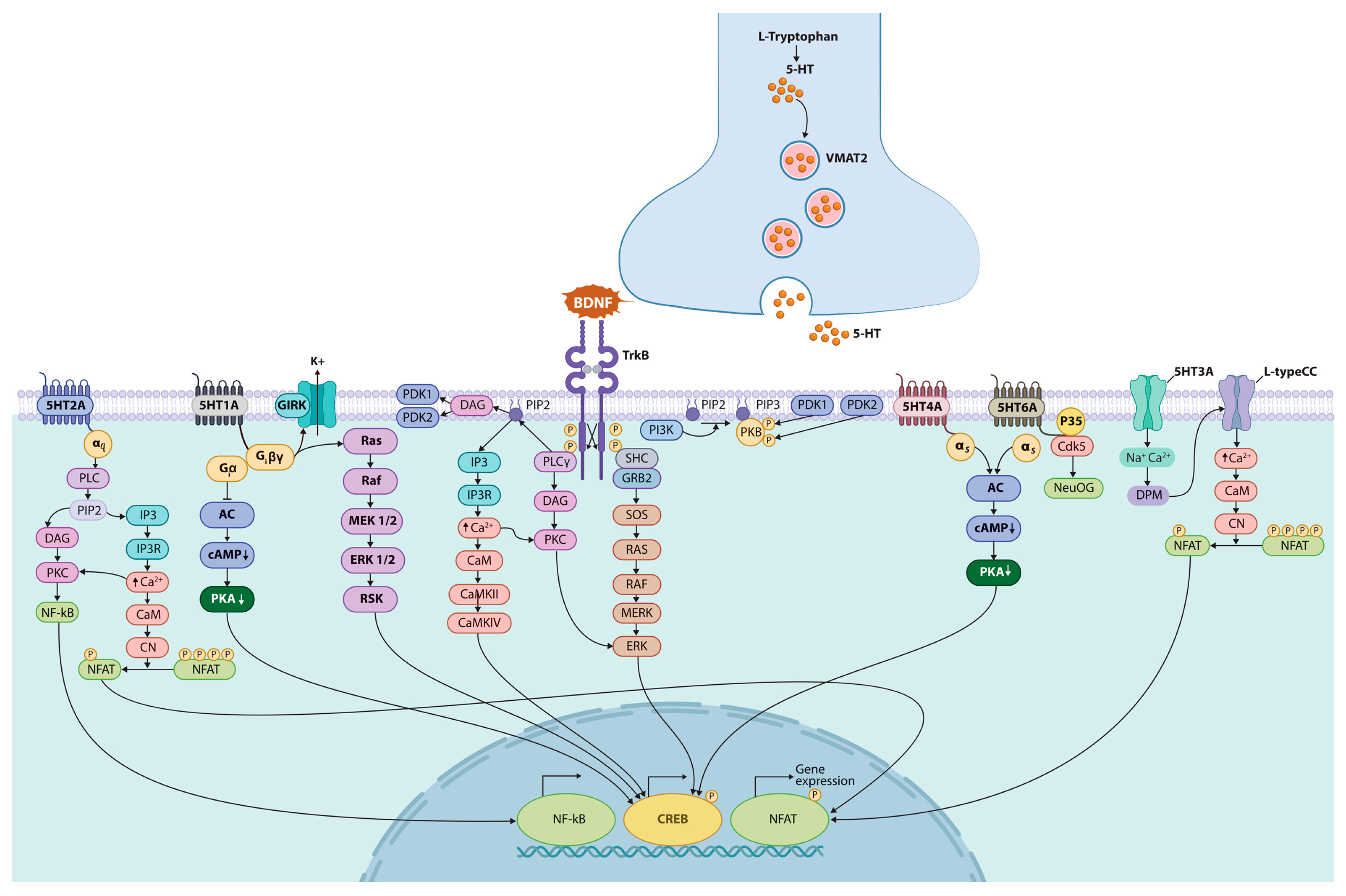
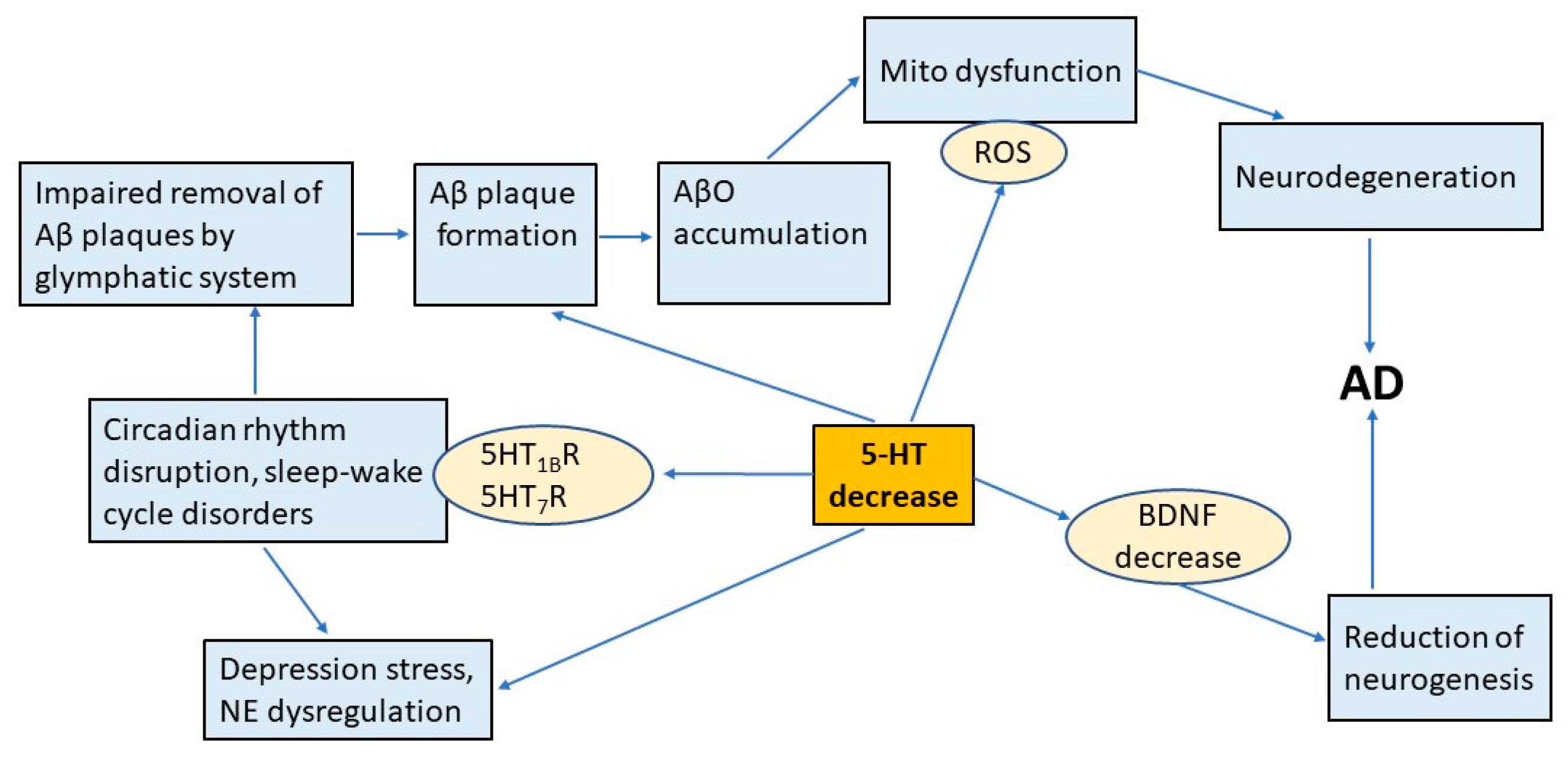
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolgacheva, L.P.; Zinchenko, V.P.; Nadeev, A.D.; Goncharov, N.V. Serotonergic Regulation in Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 5218. https://doi.org/10.3390/ijms26115218
Dolgacheva LP, Zinchenko VP, Nadeev AD, Goncharov NV. Serotonergic Regulation in Alzheimer’s Disease. International Journal of Molecular Sciences. 2025; 26(11):5218. https://doi.org/10.3390/ijms26115218
Chicago/Turabian StyleDolgacheva, Lyudmila P., Valery P. Zinchenko, Alexander D. Nadeev, and Nikolay V. Goncharov. 2025. "Serotonergic Regulation in Alzheimer’s Disease" International Journal of Molecular Sciences 26, no. 11: 5218. https://doi.org/10.3390/ijms26115218
APA StyleDolgacheva, L. P., Zinchenko, V. P., Nadeev, A. D., & Goncharov, N. V. (2025). Serotonergic Regulation in Alzheimer’s Disease. International Journal of Molecular Sciences, 26(11), 5218. https://doi.org/10.3390/ijms26115218