
Antiproliferative Water-Soluble Mono- and Binuclear Ruthenium Complexes with Pyridone–Imidazole Ligands

 ,
,  , ,
, ,  , , ,
, , ,  ,
, 

Abstract

1. Introduction
2. Results and Discussion
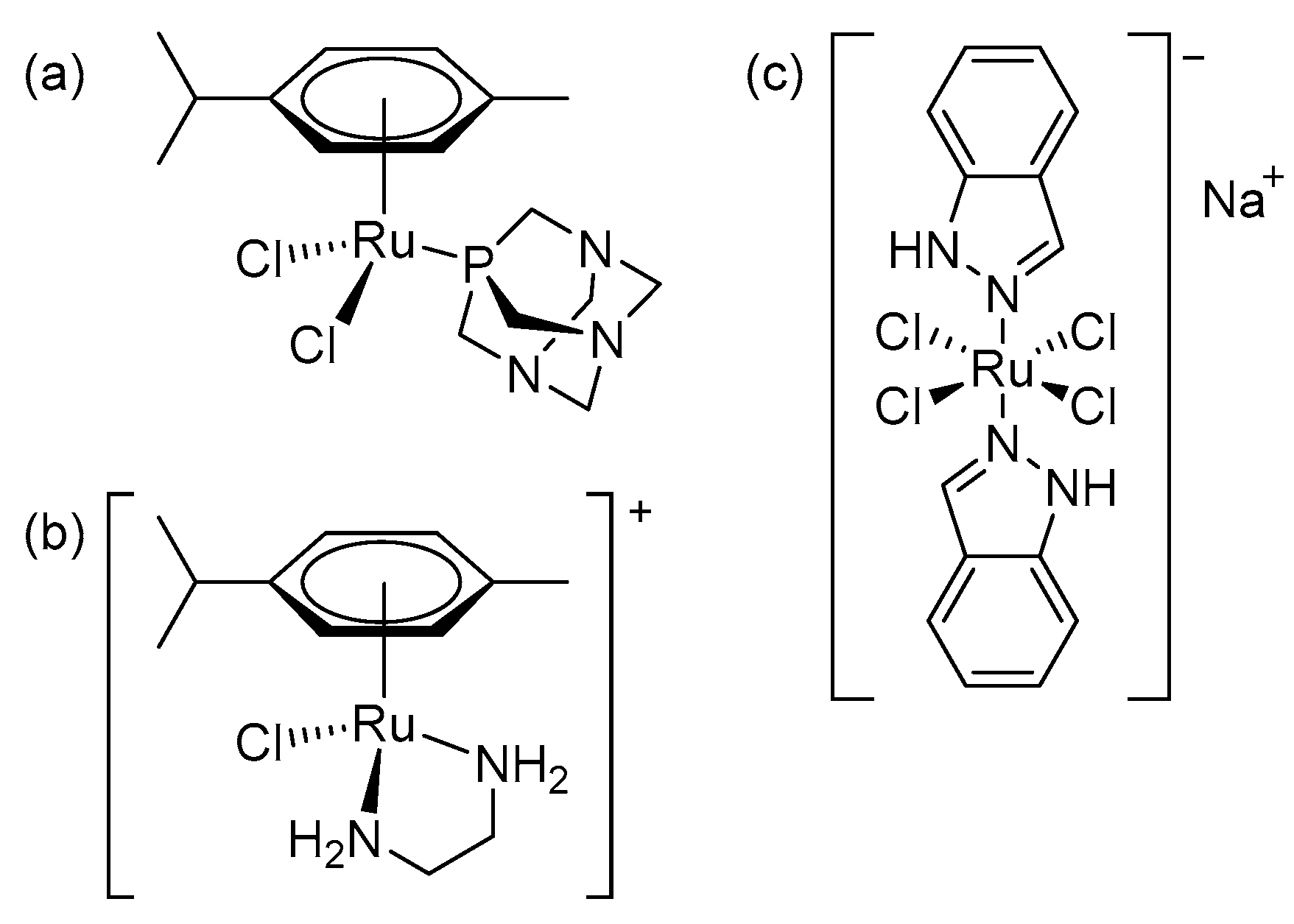
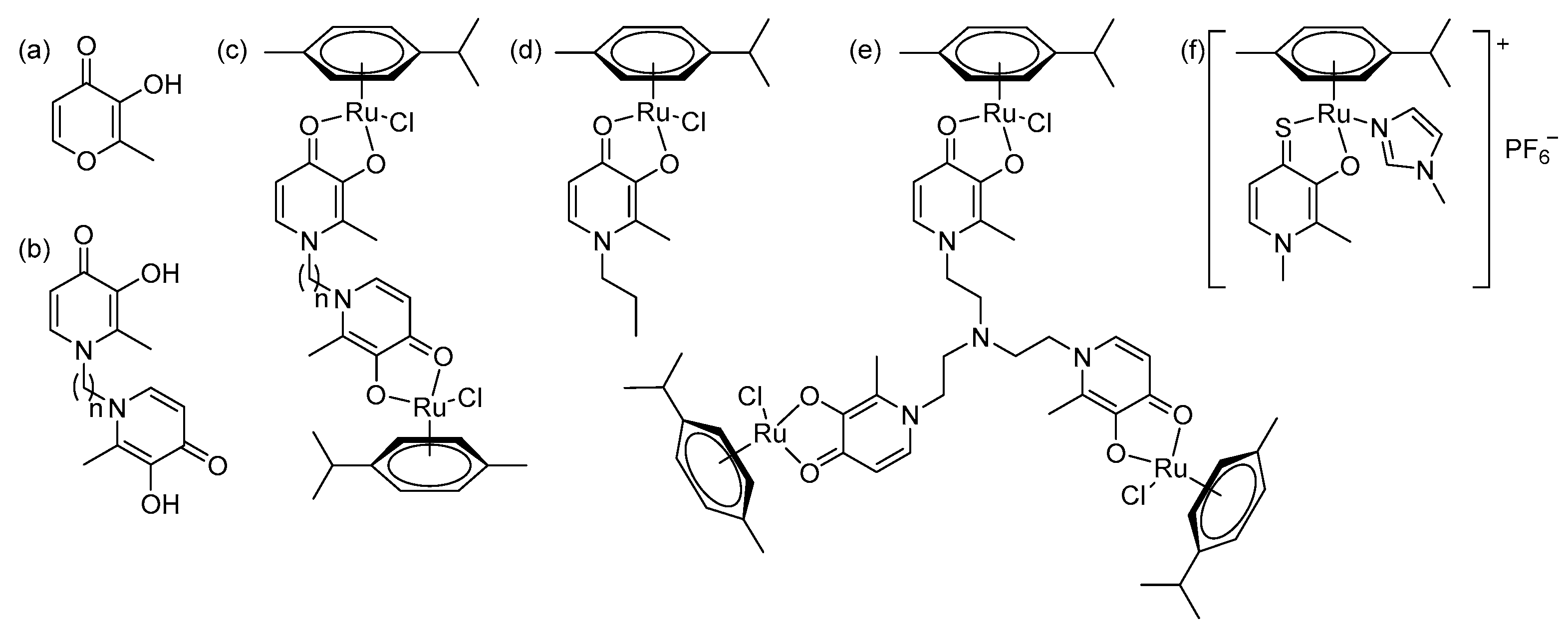
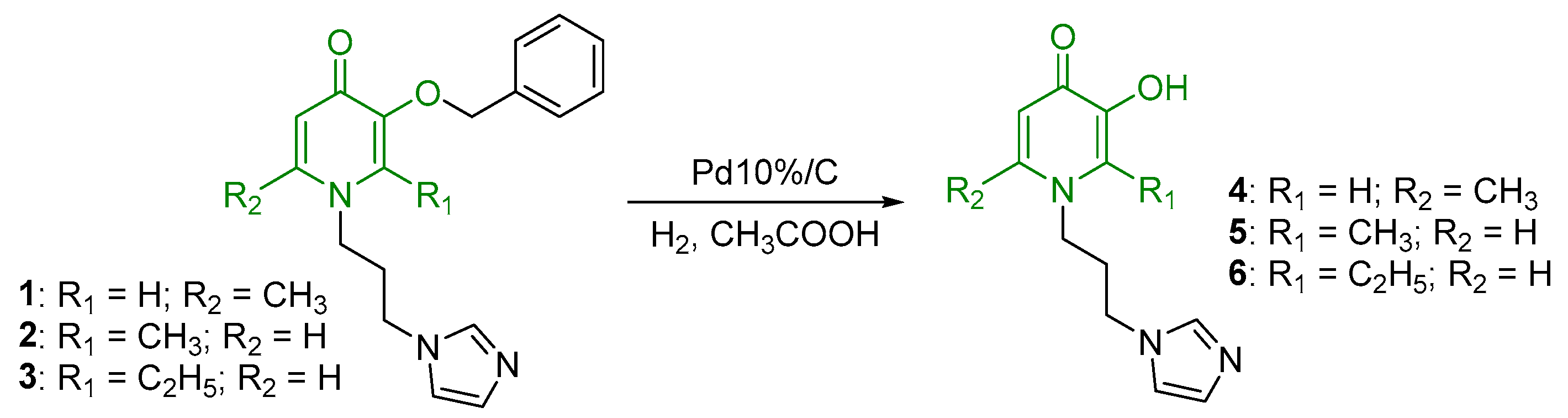
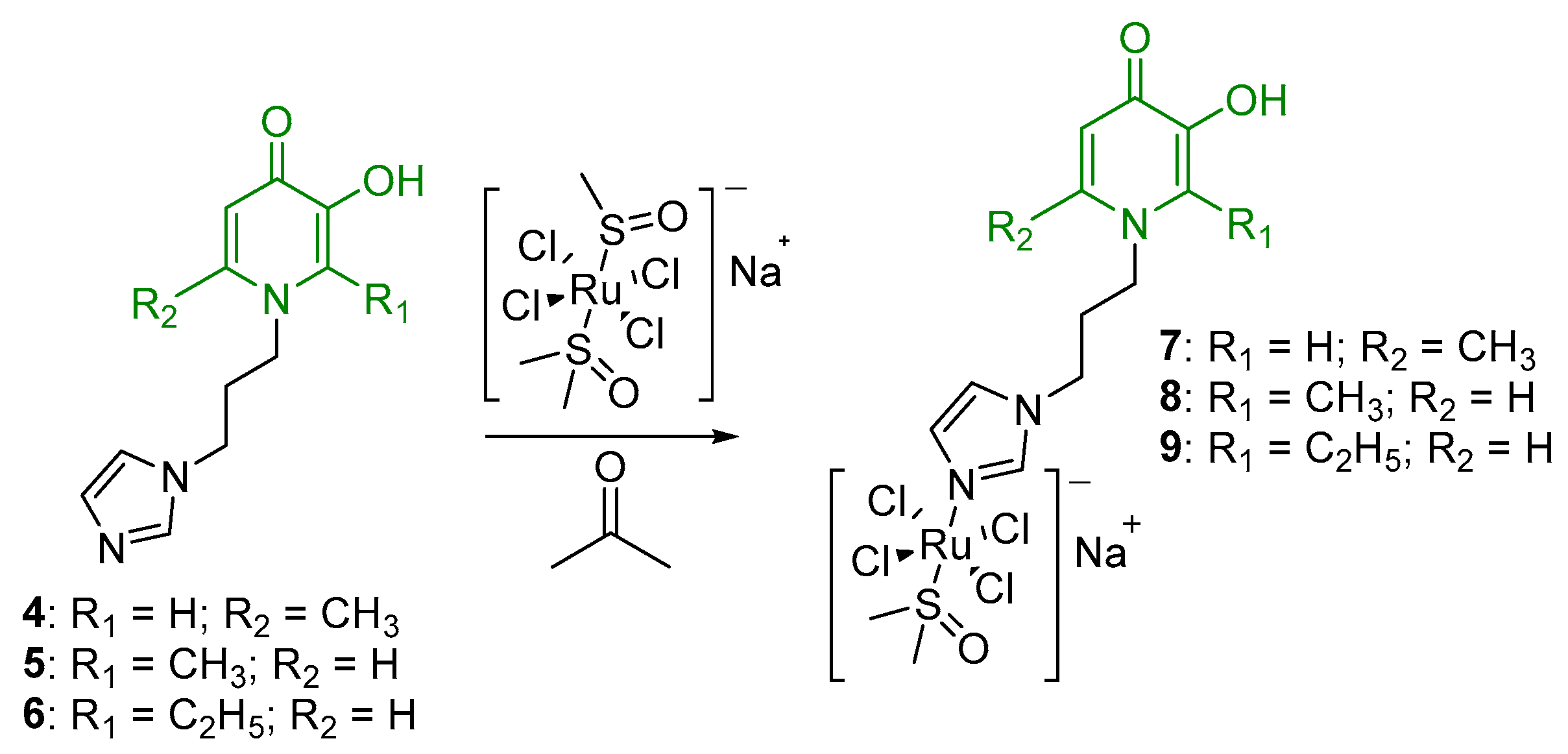
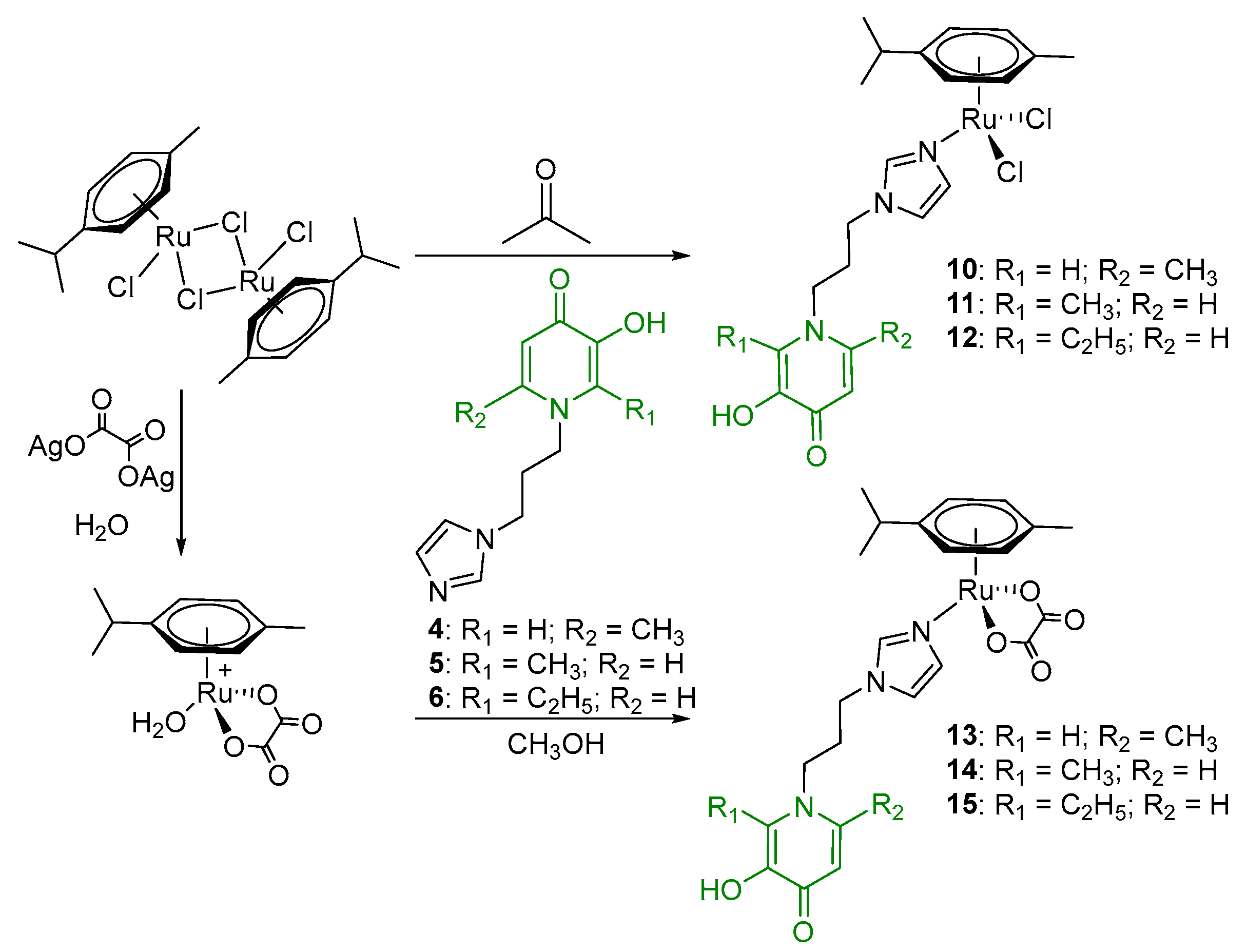
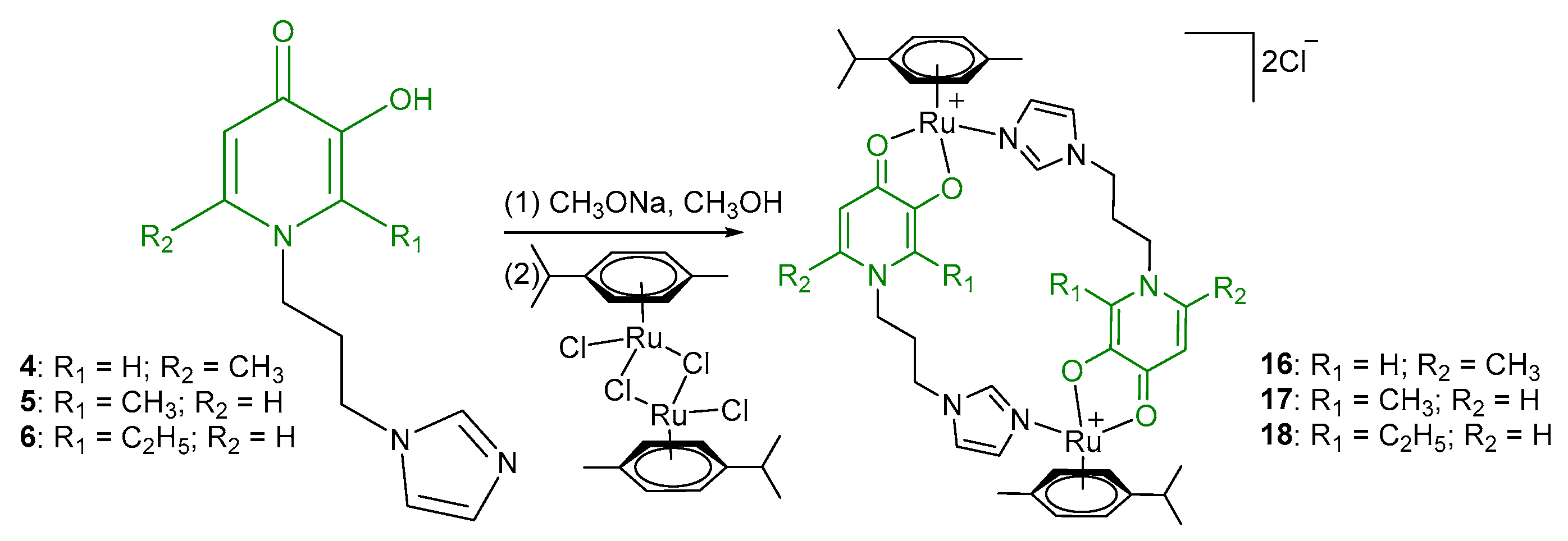
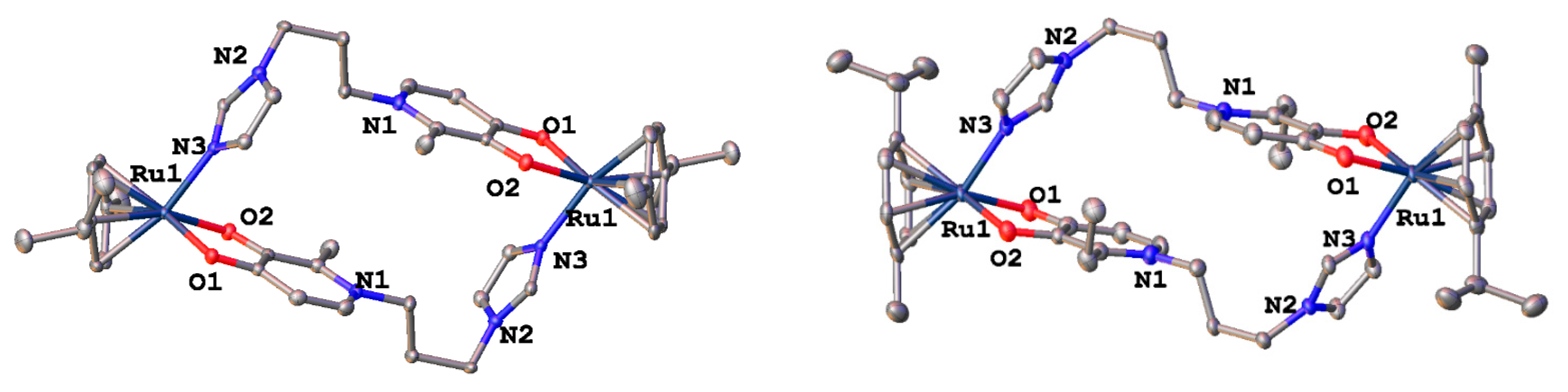
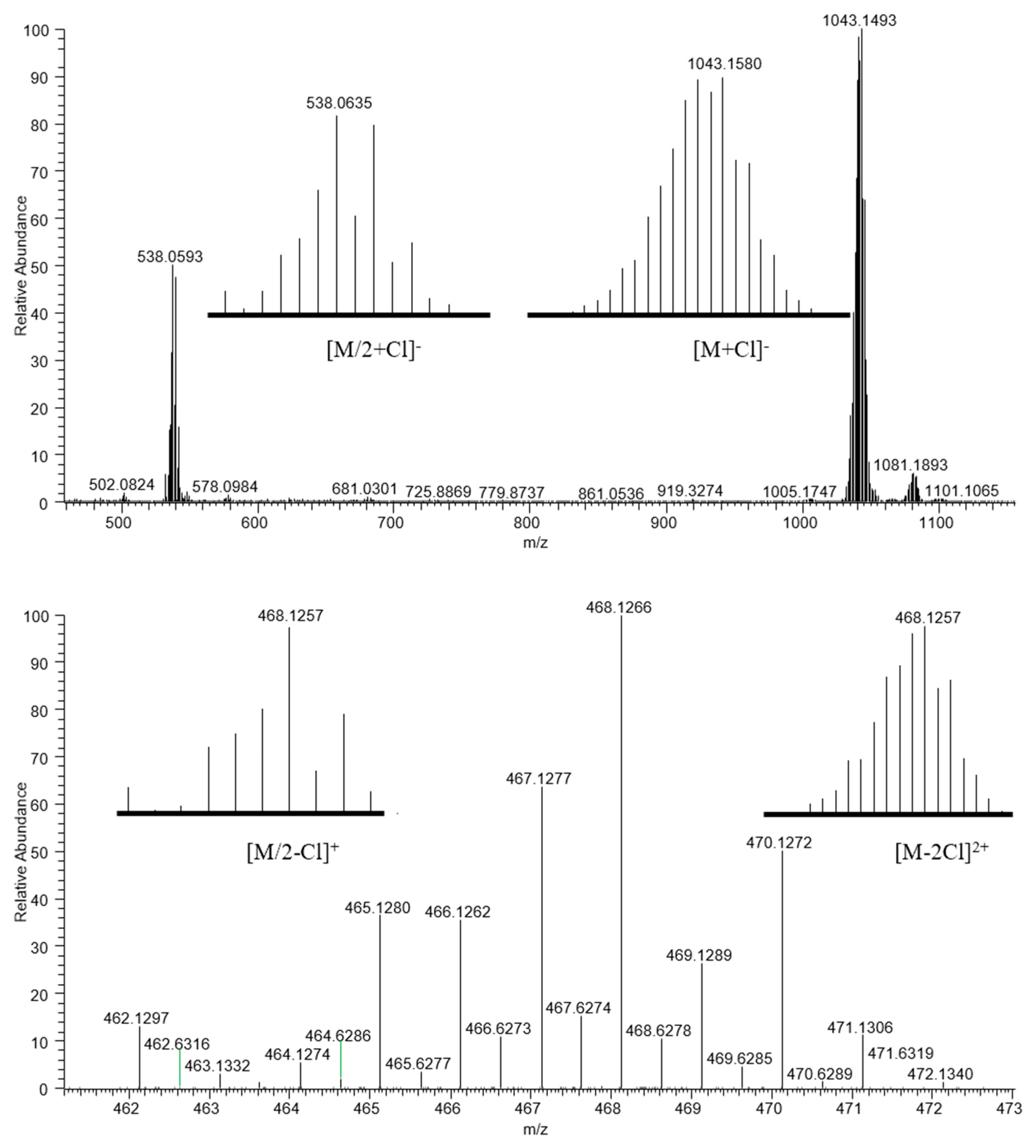
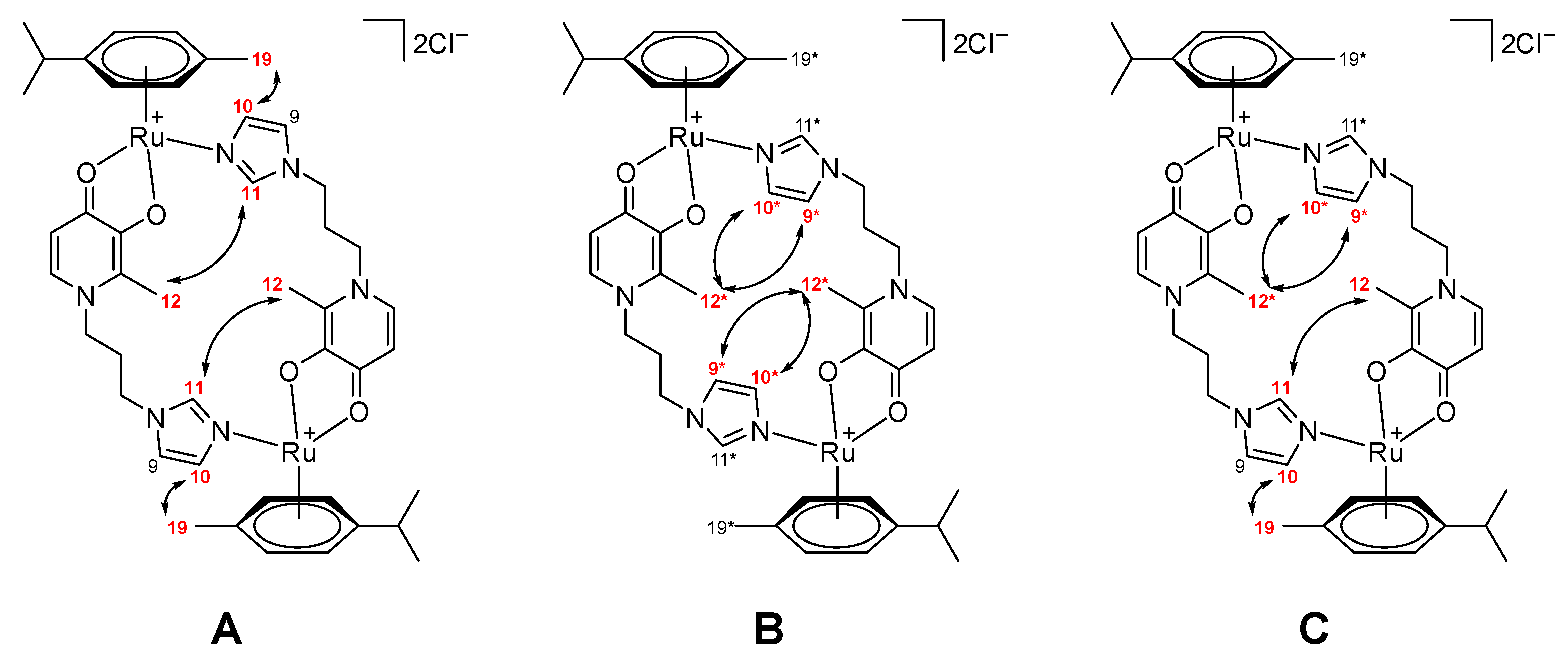
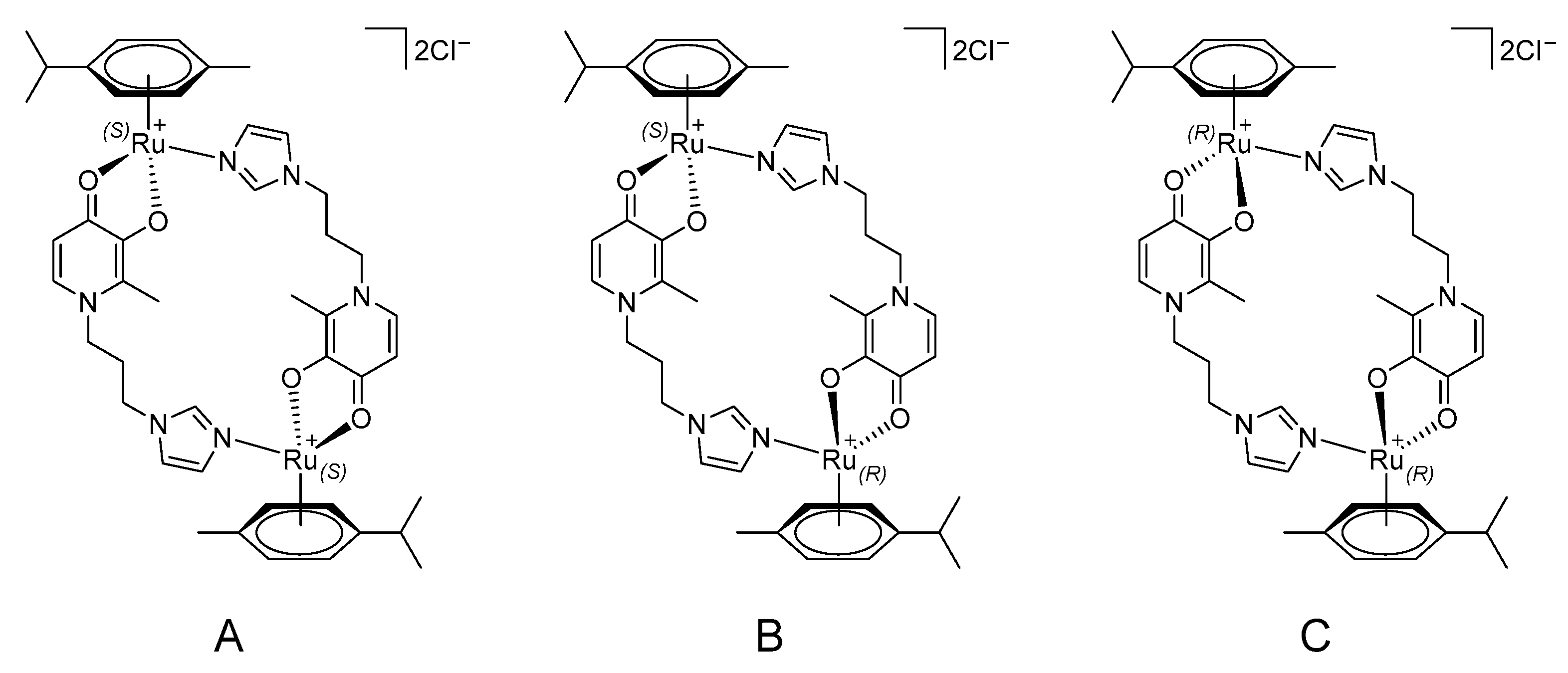
2.1. Synthesis and Characterization
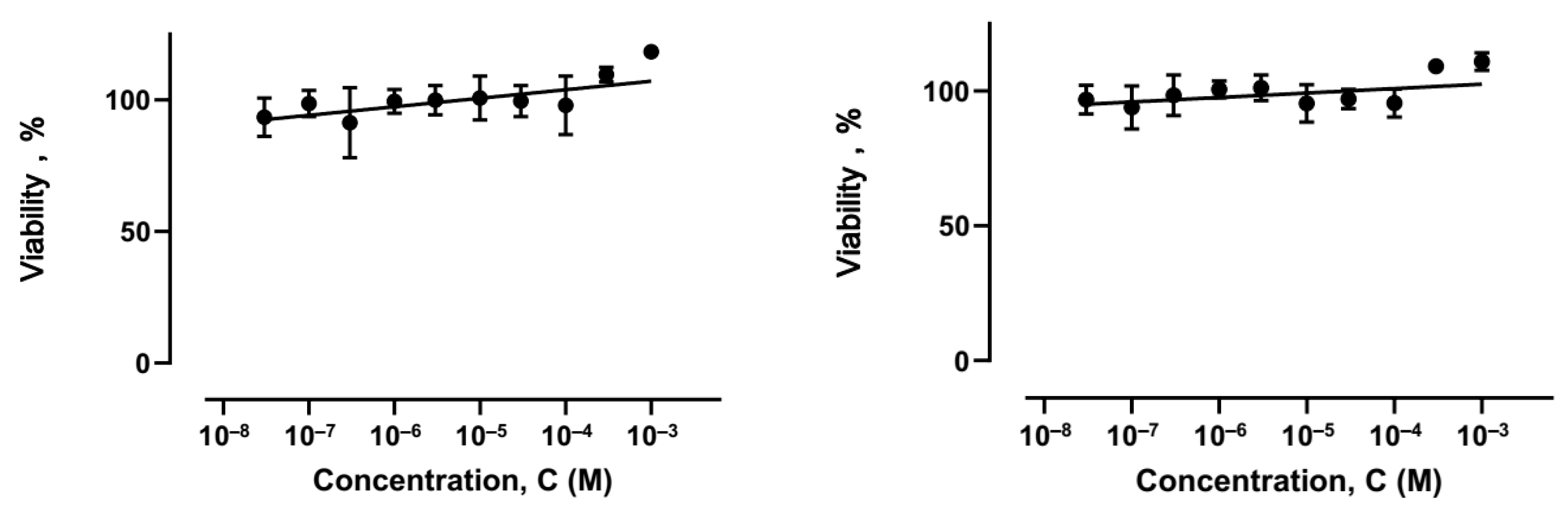
2.2. Antiproliferative Activity Study
3. Materials and Methods
3.1. General
3.2. Synthesis
- N-1-(3-aminopropyl)imidazole-5-hydroxy-2-methyl-4-pyridone (4)

- N-1-(3-aminopropyl)imidazole-3-hydroxy-2-methyl-4-pyridone (5)
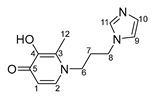
- N-1-(3-aminopropyl)imidazole-3-hydroxy-2-ethyl-4-pyridone (6)
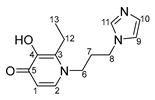
- (η6-p-cymene)(N-1-(3-aminopropyl)imidazole-5-hydroxy-2-methyl-4-pyridone)oxalatoruthenium (II) (13)
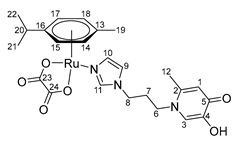
- (η6-p-cymene)(N-1-(3-aminopropyl)imidazole-3-hydroxy-2-methyl-4-pyridone)oxalatoruthenium (II) (14)
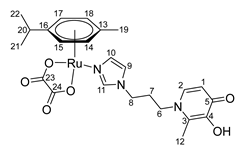
- (η6-p-cymene)(N-1-(3-aminopropyl)imidazole-3-hydroxy-2-ethyl-4-pyridone)oxalatoruthenium (II) (15)

- Di-µ-(1-(3-(1H-imidazol-1-yl)propyl)-6-methyl-4-oxo-1,4-dihydropyridin-3-olate)-bis[(η6-p-cymene)ruthenium (II)] chloride (16)
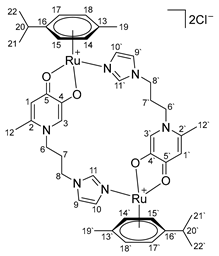
- Di-µ-(1-(3-(1H-imidazol-1-yl)propyl)-2-methyl-4-oxo-1,4-dihydropyridin-3-olate)-bis[(η6-p-cymene)ruthenium(II)] chloride (17)
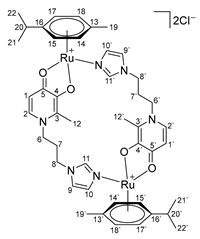
- Di-µ-(1-(3-(1H-imidazol-1-yl)propyl)-2-ethyl-4-oxo-1,4-dihydropyridin-3-olate)-bis[(η6-p-cymene)ruthenium(II)] chloride (18)
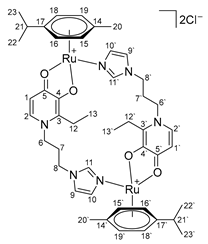
3.3. Water Solubility
3.4. Stability
3.5. Cell Death Assays
3.6. Quantum Chemical Calculations (DFT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA Grants Bold Therapeutics’ BOLD-100 an Orphan Drug Designation (ODD) in the Treatment of Gastric Cancer. Available online: https://www.prnewswire.com/news-releases/fda-grants-bold-therapeutics-bold-100-an-orphan-drug-designation-odd-in-the-treatment-of-gastric-cancer-301288416.html (accessed on 11 May 2021).
- Chen, H.; Parkinson, J.A.; Morris, R.E.; Sadler, P.J. Highly Selective Binding of Organometallic Ruthenium Ethylenediamine Complexes to Nucleic Acids: Novel Recognition Mechanisms. J. Am. Chem. Soc. 2003, 125, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Geldbach, T.J.; Rochat, S.; Dorcier, A.; Gossens, C.; Bergamo, A.; Cocchietto, M.; Tavernelli, I.; Sava, G.; Rothlisberger, U.; et al. Influence of Hydrogen-Bonding Substituents on the Cytotoxicity of RAPTA Compounds. Organometallics 2006, 25, 756–765. [Google Scholar] [CrossRef]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Tremlett, W.D.J.; Goodman, D.M.; Steel, T.R.; Kumar, S.; Wieczorek-Błauż, A.; Walsh, F.P.; Sullivan, M.P.; Hanif, M.; Hartinger, C.G. Design concepts of half-sandwich organoruthenium anticancer agents based on bidentate bioactive ligands. Coord. Chem. Rev. 2021, 445, 213950. [Google Scholar] [CrossRef]
- Fateeva, A.A.; Shutkov, I.A.; Mazur, D.M.; Kovaleva, O.N.; Milaeva, E.R.; Nazarov, A.A. RuII and RuIII complexes with imidazole ligands containing (benzyloxy)pyridinone moiety. Mendeleev Commun. 2022, 32, 186–188. [Google Scholar] [CrossRef]
- Shutkov, I.A.; Okulova, Y.N.; Mazur, D.M.; Melnichuk, N.A.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A.; Milaeva, E.R.; Nazarov, A.A. New Organometallic Ru(II) Compounds with Lonidamine Motif as Antitumor Agents. Pharmaceutics 2023, 15, 1366. [Google Scholar] [CrossRef]
- Shutkov, I.A.; Melnichuk, N.A.; Lyssenko, K.A.; Borisova, N.E.; Kovaleva, O.N.; Nazarov, A.A. Di-µ-(1-(3-(1H-imidazol-1-yl)propyl)-2-methyl-4-oxo-1,4-dihydropyridin-3-olate)-bis[(η5-pentamethylcyclopentadienyl)iridium(III)] Chloride. Molbank 2024, 2024, M1816. [Google Scholar] [CrossRef]
- Peacock, A.F.A.; Melchart, M.; Deeth, R.J.; Habtemariam, A.; Parsons, S.; Sadler, P.J. Osmium(II) and Ruthenium(II) Arene Maltolato Complexes: Rapid Hydrolysis and Nucleobase Binding. Chem. Eur. J. 2007, 13, 2601–2613. [Google Scholar] [CrossRef]
- Kandioller, W.; Hartinger, C.G.; Nazarov, A.A.; Kuznetsov, M.L.; John, R.O.; Bartel, C.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. From Pyrone to Thiopyrone Ligands−Rendering Maltol-Derived Ruthenium(II)−Arene Complexes That Are Anticancer Active in Vitro. Organometallics 2009, 28, 4249–4251. [Google Scholar] [CrossRef]
- Hanif, M.; Schaaf, P.; Kandioller, W.; Hejl, M.; Jakupec, M.A.; Roller, A.; Keppler, B.K.; Hartinger, C.G. Influence of the Arene Ligand and the Leaving Group on the Anticancer Activity of (Thio)maltol Ruthenium(II)-(η6-Arene) Complexes. Aust. J. Chem. 2010, 63, 1521–1528. [Google Scholar] [CrossRef]
- Kandioller, W.; Kurzwernhart, A.; Hanif, M.; Meier, S.M.; Henke, H.; Keppler, B.K.; Hartinger, C.G. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Organomet. Chem. 2011, 696, 999–1010. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Bognár, G.M.; Kiss, T.; Hanif, M.; Hartinger, C.G. Solution equilibrium studies on anticancer ruthenium(II)–η6-p-cymene complexes of 3-hydroxy-2(1H)-pyridones. J. Organomet. Chem. 2013, 734, 38–44. [Google Scholar] [CrossRef]
- Mendoza-Ferri, M.G.; Hartinger, C.G.; Mendoza, M.A.; Groessl, M.; Egger, A.E.; Eichinger, R.E.; Mangrum, J.B.; Farrell, N.P.; Maruszak, M.; Bednarski, P.J.; et al. Transferring the Concept of Multinuclearity to Ruthenium Complexes for Improvement of Anticancer Activity. J. Med. Chem. 2009, 52, 916–925. [Google Scholar] [CrossRef]
- Hackl, C.M.; Legina, M.S.; Pichler, V.; Schmidlehner, M.; Roller, A.; Dömötör, O.; Enyedy, E.A.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Thiomaltol-Based Organometallic Complexes with 1-Methylimidazole as Leaving Group: Synthesis, Stability, and Biological Behavior. Chem. Eur. J. 2016, 22, 17269–17281. [Google Scholar] [CrossRef]
- Parveen, S.; Hanif, M.; Movassaghi, S.; Sullivan, M.P.; Kubanik, M.; Shaheen, M.A.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Cationic Ru(η6-p-cymene) Complexes of 3-Hydroxy-4-pyr(id)ones—Lipophilic Triphenylphosphine as Co-Ligand Is Key to Highly Stable and Cytotoxic Anticancer Agents. Eur. J. Inorg. Chem. 2017, 2017, 1721–1727. [Google Scholar] [CrossRef]
- Fry, A.M.; Chresta, C.M.; Davies, S.M.; Walker, M.C.; Harris, A.L.; Hartley, J.A.; Masters, J.R.; Hickson, I.D. Relationship between topoisomerase II level and chemosensitivity in human tumor cell lines. Cancer Res. 1991, 51, 6592–6595. [Google Scholar]
- Koshiyama, M.; Fujii, H.; Kinezaki, M.; Yoshida, M. Correlation between Topo II alpha expression and chemosensitivity testing for Topo II-targeting drugs in gynaecological carcinomas. Anticancer Res. 2001, 21, 905–910. [Google Scholar]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef]
- Chen, C.-F.; He, X.; Beck, W.T. Abstract 4683: Inducible knockdown of DNA topoisomerase IIα affects drug sensitivity and cell growth. Cancer Res. 2011, 71, 4683. [Google Scholar] [CrossRef]
- Schmidlehner, M.; Pichler, V.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K. Organometallic complexes of (thio)allomaltol-based Mannich-products: Synthesis, stability and preliminary biological investigations. J. Organomet. Chem. 2015, 782, 69–76. [Google Scholar] [CrossRef]
- Legina, M.S.; Nogueira, J.J.; Kandioller, W.; Jakupec, M.A.; González, L.; Keppler, B.K. Biological evaluation of novel thiomaltol-based organometallic complexes as topoisomerase IIα inhibitors. JBIC J. Biol. Inorg. Chem. 2020, 25, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Polborn, K.; Severin, T.; Severin, K. Halfsandwich complexes of ruthenium(II), rhodium(III) and iridium(III) with N-substituted 3-hydroxy-2-methyl-4-pyridone ligands. Inorg. Chim. Acta 1999, 294, 62–67. [Google Scholar] [CrossRef]
- Habereder, T.; Warchhold, M.; Nöth, H.; Severin, K. Chemically Triggered Assembly of Chiral Triangular Metallomacrocycles. Angew. Chem. Int. Ed. 1999, 38, 3225–3228. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Oxford, UK, 2003; p. 608. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theoret. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solubility in Water, g/100 mL |
|---|---|
| 16 | 18.4 ± 0.4 |
| 17 | >20 |
| 18 | 14.3 ± 0.2 |
| Compound | IC50, μM | |||
|---|---|---|---|---|
| A549 | HCT116 | MCF7 | SW480 | |
| cisplatin | 20 ± 2 | 10 ± 4 | 15 ± 5 | 18 ± 5 |
| 4 | >200 | >200 | >200 | nd |
| 5 | 120 ± 13 | 131 ± 6 | 153 ± 24 | nd |
| 6 | 97 ± 14 | 91 ± 11 | 118 ± 18 | nd |
| 13 | >200 | >200 | >200 | nd |
| 14 | 118 ± 15 | 133 ± 17 | 124 ± 20 | nd |
| 15 | 104 ± 18 | 114 ± 17 | 113 ± 20 | nd |
| 16 | >200 | >200 | >200 | >200 |
| 17 | 50 ± 8 | 34 ± 5 | 45 ± 5 | 25 ± 4 |
| 18 | 28 ± 4 | 33 ± 5 | 34 ± 5 | 26 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shutkov, I.A.; Melnichuk, N.A.; Ovakimyan, S.A.; Mazur, D.M.; Borisova, N.E.; Kuznetsov, M.L.; Godovikov, I.A.; Lyssenko, K.A.; Yakovlev, D.S.; Spasov, A.A.; et al. Antiproliferative Water-Soluble Mono- and Binuclear Ruthenium Complexes with Pyridone–Imidazole Ligands. Int. J. Mol. Sci. 2025, 26, 5214. https://doi.org/10.3390/ijms26115214
Shutkov IA, Melnichuk NA, Ovakimyan SA, Mazur DM, Borisova NE, Kuznetsov ML, Godovikov IA, Lyssenko KA, Yakovlev DS, Spasov AA, et al. Antiproliferative Water-Soluble Mono- and Binuclear Ruthenium Complexes with Pyridone–Imidazole Ligands. International Journal of Molecular Sciences. 2025; 26(11):5214. https://doi.org/10.3390/ijms26115214
Chicago/Turabian StyleShutkov, Ilya A., Nikolai A. Melnichuk, Sofya A. Ovakimyan, Dmitrii M. Mazur, Nataliya E. Borisova, Maxim L. Kuznetsov, Ivan A. Godovikov, Konstantin A. Lyssenko, Dmitrii S. Yakovlev, Alexander A. Spasov, and et al. 2025. "Antiproliferative Water-Soluble Mono- and Binuclear Ruthenium Complexes with Pyridone–Imidazole Ligands" International Journal of Molecular Sciences 26, no. 11: 5214. https://doi.org/10.3390/ijms26115214
APA StyleShutkov, I. A., Melnichuk, N. A., Ovakimyan, S. A., Mazur, D. M., Borisova, N. E., Kuznetsov, M. L., Godovikov, I. A., Lyssenko, K. A., Yakovlev, D. S., Spasov, A. A., Milaeva, E. R., & Nazarov, A. A. (2025). Antiproliferative Water-Soluble Mono- and Binuclear Ruthenium Complexes with Pyridone–Imidazole Ligands. International Journal of Molecular Sciences, 26(11), 5214. https://doi.org/10.3390/ijms26115214