
Alterations in the Expression of a Set of miRNAs in Endometrial Cancer and Their Correlation with Clinical Variables and the p53 Signaling Pathway

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Differentially Expressed miRNAs in Endometrial Cancer
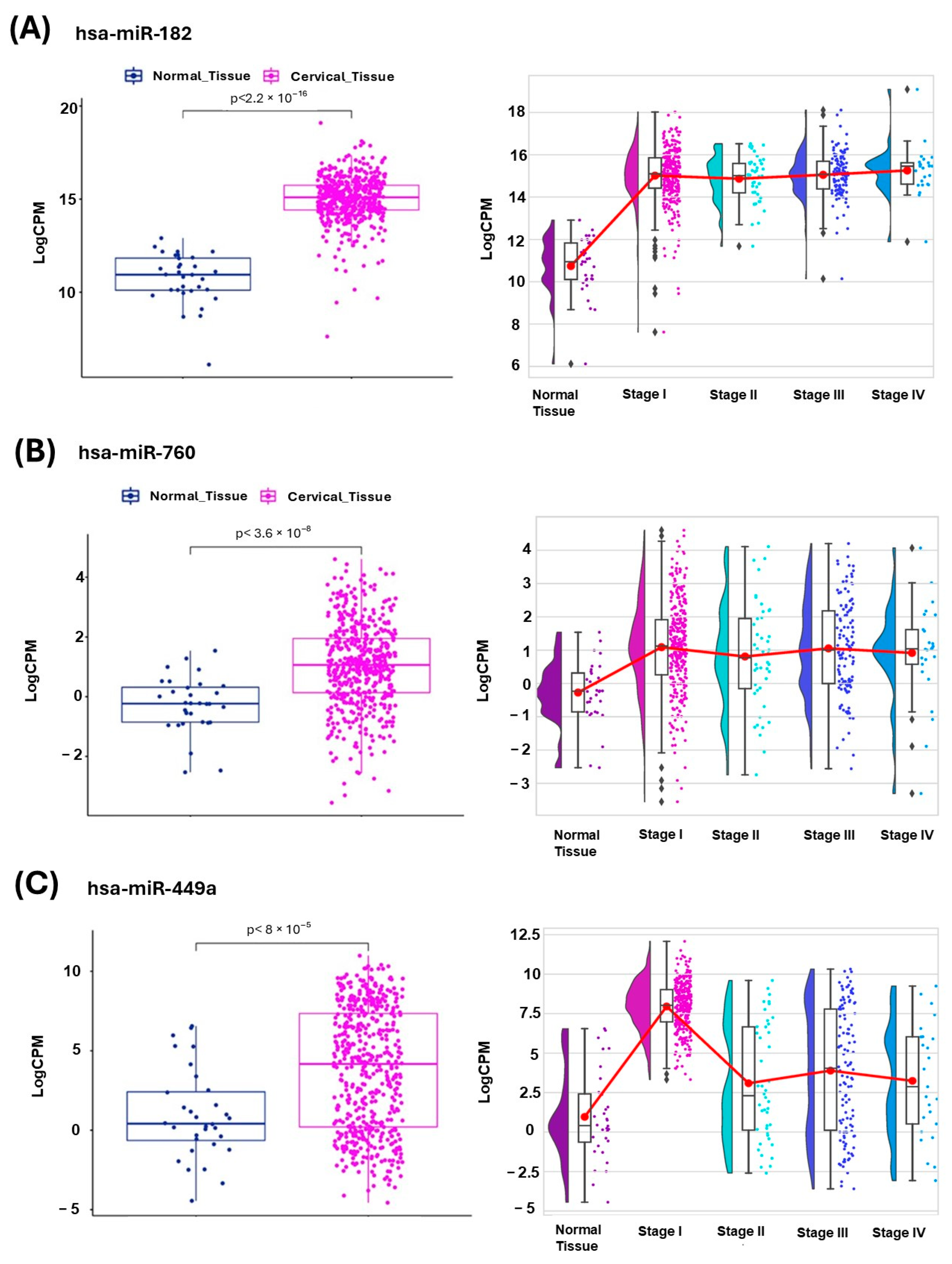
2.2. Validation of Differentially Expressed miRNAs in Endometrial Cancer Subtypes
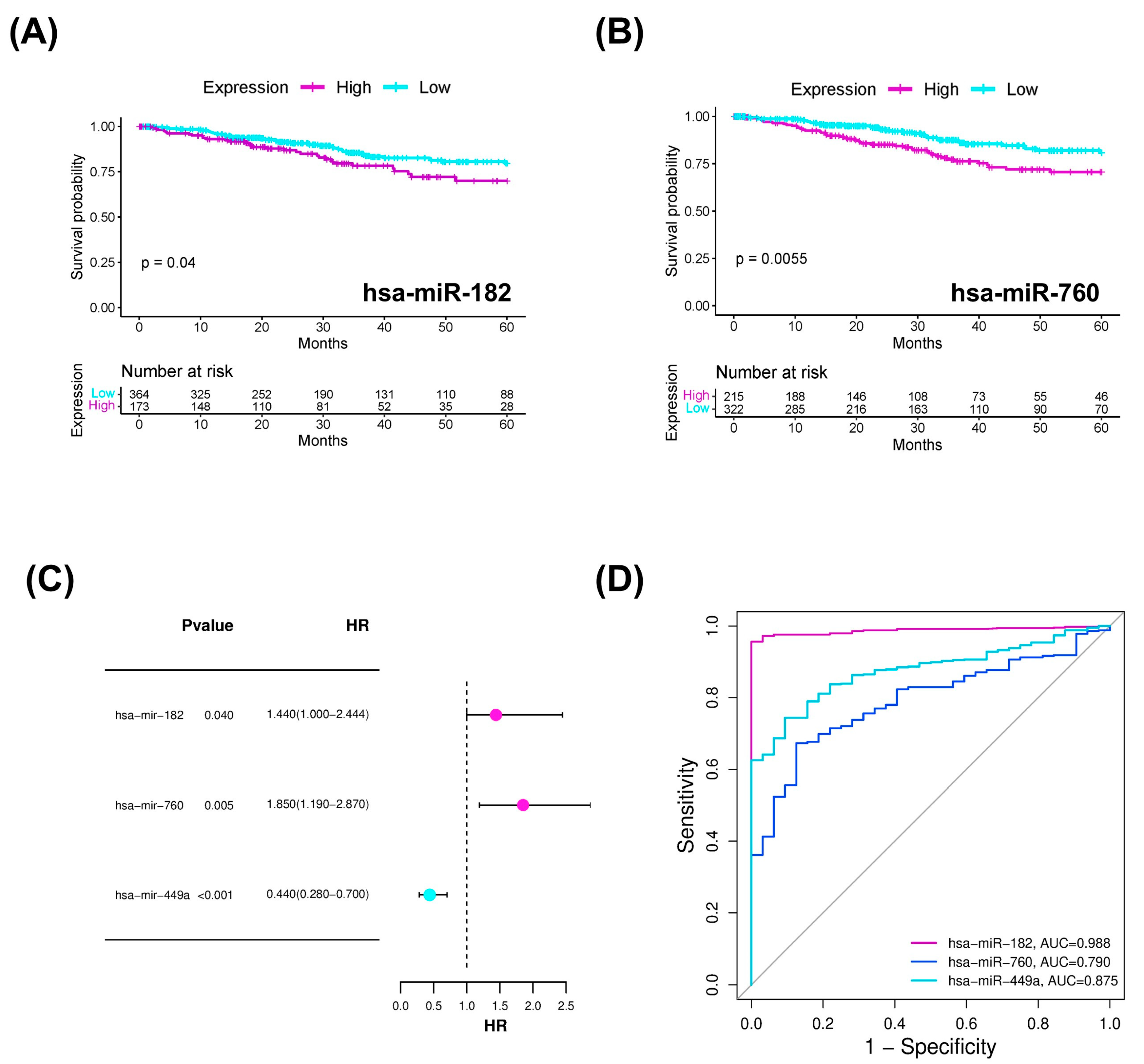
2.3. miRNAs with Diagnostic and Prognostic Value in Endometrial Cancer
2.4. Target Prediction and Molecular Analysis
2.5. Two-Dimensional and Three-Dimensional Graphical Representation of the Structure of Hsa-miR-182, Hsa- miR 760, Hsa-miR 449a, and TP53INP1
3. Discussion
4. Materials and Methods
4.1. DEMs Analysis in Endometrial Cancer Dataset
4.2. Expression of DEMs in Endometrium from the TCGA Dataset
4.3. Overall Survival, Univariate Cox Regression, and ROC Analysis
4.4. Target Gene Prediction of DEMs
4.5. Enrichment Analysis
4.6. Two-Dimensional and Three-Dimensional Structure Topology Prediction
4.7. Correlation Analysis
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EC | endometrial cancer |
| DEMs | differentially expressed miRNAs |
| TCGA | The Cancer Genome Atlas |
| CPM | counts per million |
| Log2FC | log-transformed gene expression change with base 2 |
| AUC | area under the curve |
| ROC | receiver operating characteristic |
| HR | hazard ratio |
| MFE | minimum free energy |
| NGS | next-generation sequencing |
| qPCR | real-time polymerase chain reaction |
| FPKM | fragments per million kilobases |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.A.; Althouse, A.D.; Freese, K.E.; Soisson, S.; Edwards, R.P.; Welburn, S.; Sukumvanich, P.; Comerci, J.; Kelley, J.; LaPorte, R.E.; et al. USA Endometrial Cancer Projections to 2030: Should We Be Concerned? Future Oncol. 2014, 10, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, W.; Liu, J.; Zhang, Y. Minimally Invasive Approaches for the Early Detection of Endometrial Cancer. Mol. Cancer 2023, 22, 53. [Google Scholar] [CrossRef]
- Piergentili, R.; Gullo, G.; Basile, G.; Gulia, C.; Porrello, A.; Cucinella, G.; Marinelli, E.; Zaami, S. Circulating MiRNAs as a Tool for Early Diagnosis of Endometrial Cancer—Implications for the Fertility-Sparing Process: Clinical, Biological, and Legal Aspects. Int. J. Mol. Sci. 2023, 24, 11356. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 199–227. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Rani, M.; Kumari, R.; Singh, S.P.; Devi, A.; Bansal, P.; Siddiqi, A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H.; Rizvi, M.M.A. MicroRNAs as Master Regulators of FOXO Transcription Factors in Cancer Management. Life Sci. 2023, 321, 121535. [Google Scholar] [CrossRef]
- Hermyt, E.; Zmarzły, N.; Grabarek, B.; Kruszniewska-Rajs, C.; Gola, J.; Jęda-Golonka, A.; Szczepanek, K.; Mazurek, U.; Witek, A. Interplay between MiRNAs and Genes Associated with Cell Proliferation in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 6011. [Google Scholar] [CrossRef]
- Bogaczyk, A.; Zawlik, I.; Zuzak, T.; Kluz, M.; Potocka, N.; Kluz, T. The Role of MiRNAs in the Development, Proliferation, and Progression of Endometrial Cancer. Int. J. Mol. Sci. 2023, 24, 11489. [Google Scholar] [CrossRef]
- Abdelmaksoud, N.M.; El-Mahdy, H.A.; Ismail, A.; Elsakka, E.G.E.; El-Husseiny, A.A.; Khidr, E.G.; Ali, E.M.; Rashed, M.H.; El-Demerdash, F.E.-S.; Doghish, A.S. The Role of MiRNAs in the Pathogenesis and Therapeutic Resistance of Endometrial Cancer: A Spotlight on the Convergence of Signaling Pathways. Pathol. Res. Pract. 2023, 244, 154411. [Google Scholar] [CrossRef]
- Androvic, P.; Benesova, S.; Rohlova, E.; Kubista, M.; Valihrach, L. Small RNA-Sequencing for Analysis of Circulating MiRNAs. J. Mol. Diagn. 2022, 24, 386–394. [Google Scholar] [CrossRef]
- Hegde, S.; Wagh, K.; Narayana, S.M.; Abikar, A.; Nambiar, S.; Ananthamurthy, S.; Narayana, N.H.; Reddihalli, P.V.; Chandraiah, S.; Ranganathan, P. MicroRNA Profile of Endometrial Cancer from Indian Patients-Identification of Potential Biomarkers for Prognosis. Biochem. Biophys. Rep. 2024, 39, 101812. [Google Scholar] [CrossRef] [PubMed]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial Cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Matias-Guiu, X.; Creutzberg, C.; Fotopoulou, C.; Gaffney, D.; Kehoe, S.; Lindemann, K.; Mutch, D.; Concin, N. FIGO Staging of Endometrial Cancer: 2023. Int. J. Gynecol. Obstet. 2023, 162, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Boroń, D.; Zmarzły, N.; Wierzbik-Strońska, M.; Rosińczuk, J.; Mieszczański, P.; Grabarek, B.O. Recent Multiomics Approaches in Endometrial Cancer. Int. J. Mol. Sci. 2022, 23, 1237. [Google Scholar] [CrossRef]
- Nagamitsu, Y.; Nishi, H.; Sasaki, T.; Takaesu, Y.; Terauchi, F.; Isaka, K. Profiling Analysis of Circulating MicroRNA Expression in Cervical Cancer. Mol. Clin. Oncol. 2016, 5, 189–194. [Google Scholar] [CrossRef]
- Donkers, H.; Hirschfeld, M.; Weiß, D.; Erbes, T.; Jaeger, M.; Pijnenborg, J.M.A.; Bekkers, R.; Galaal, K. Usefulness of MicroRNA Detection in the Diagnostics of Endometrial Cancer. Acta Obstet. Gynecol. Scand. 2021, 100, 1148–1154. [Google Scholar] [CrossRef]
- Myatt, S.S.; Wang, J.; Monteiro, L.J.; Christian, M.; Ho, K.-K.; Fusi, L.; Dina, R.E.; Brosens, J.J.; Ghaem-Maghami, S.; Lam, E.W.-F. Definition of MicroRNAs That Repress Expression of the Tumor Suppressor Gene FOXO1 in Endometrial Cancer. Cancer Res. 2010, 70, 367–377. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct Target Ther 2021, 6, 425. [Google Scholar] [CrossRef]
- Ulianich, L.; Insabato, L. Endoplasmic Reticulum Stress in Endometrial Cancer. Front. Med. 2014, 1, 55. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA Modifications in Cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Ahuja, N. The Emerging Roles of ATP-Dependent Chromatin Remodeling Complexes in Pancreatic Cancer. Cancers 2019, 11, 1859. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ruiz, S.; Garrido-Cano, I.; Lameirinhas, A.; Burgués, O.; Hernando, C.; Martínez, M.T.; Rojo, F.; Bermejo, B.; Tapia, M.; Carbonell-Asins, J.A.; et al. MiRNA-449 Family Is Epigenetically Repressed and Sensitizes to Doxorubicin through ACSL4 Downregulation in Triple-Negative Breast Cancer. Cell Death Discov. 2024, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Vajen, B.; Bhowmick, R.; Greiwe, L.; Schäffer, V.; Eilers, M.; Reinkens, T.; Stalke, A.; Schmidt, G.; Fiedler, J.; Thum, T.; et al. MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation. Int. J. Mol. Sci. 2022, 23, 5131. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 October 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Turkes, E.; Ostendorf, B.; Grioni, A.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R Package Version 1.26.0. Available online: https://www.rdocumentation.org/packages/EnhancedVolcano/versions/1.11.3/topics/EnhancedVolcano (accessed on 20 October 2024).
- Malcolm Perry Flexible Heatmaps for Functional Genomics and Sequence Features. 2015. Available online: https://bioconductor.org/packages/release/bioc/html/heatmaps.html (accessed on 6 October 2024).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinformatics 2011, 12, 77. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and Customizable Prediction of RNA–RNA Interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M.; Stiegler, P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Universität Wien, I. for T.C. RNAFold WebServer. 2003. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on 10 January 2025).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapata García, J.A. Alterations in the Expression of a Set of miRNAs in Endometrial Cancer and Their Correlation with Clinical Variables and the p53 Signaling Pathway. Int. J. Mol. Sci. 2025, 26, 5215. https://doi.org/10.3390/ijms26115215
Zapata García JA. Alterations in the Expression of a Set of miRNAs in Endometrial Cancer and Their Correlation with Clinical Variables and the p53 Signaling Pathway. International Journal of Molecular Sciences. 2025; 26(11):5215. https://doi.org/10.3390/ijms26115215
Chicago/Turabian StyleZapata García, Jessica Alejandra. 2025. "Alterations in the Expression of a Set of miRNAs in Endometrial Cancer and Their Correlation with Clinical Variables and the p53 Signaling Pathway" International Journal of Molecular Sciences 26, no. 11: 5215. https://doi.org/10.3390/ijms26115215
APA StyleZapata García, J. A. (2025). Alterations in the Expression of a Set of miRNAs in Endometrial Cancer and Their Correlation with Clinical Variables and the p53 Signaling Pathway. International Journal of Molecular Sciences, 26(11), 5215. https://doi.org/10.3390/ijms26115215