

Cellular and Transcriptional Responses of Human Bronchial Epithelial Cells to Delta-9-Tetrahydrocannabinol In Vitro


 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
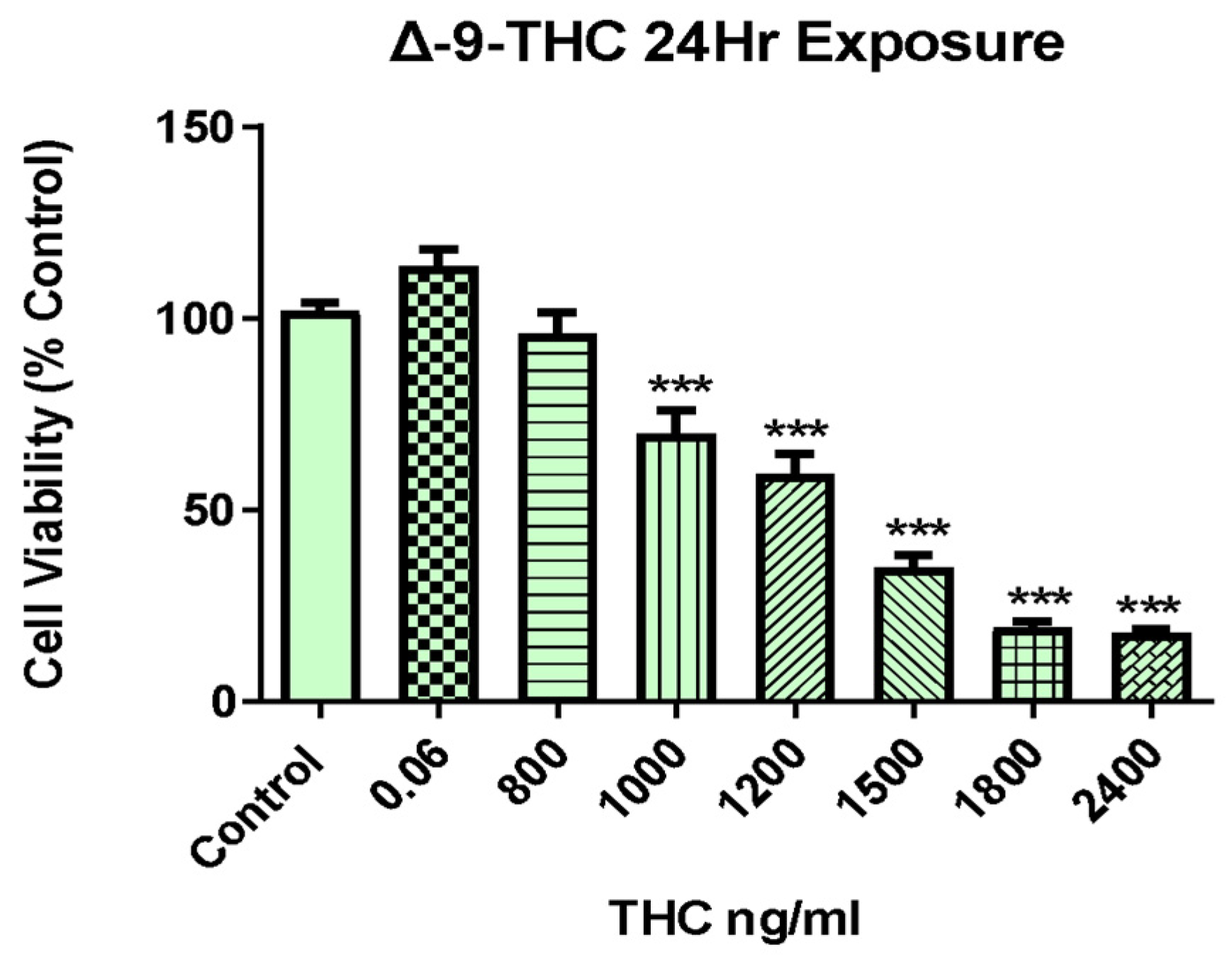
2.1. Δ-9-THC Dose-Dependent Viability of BEAS-2B Cells
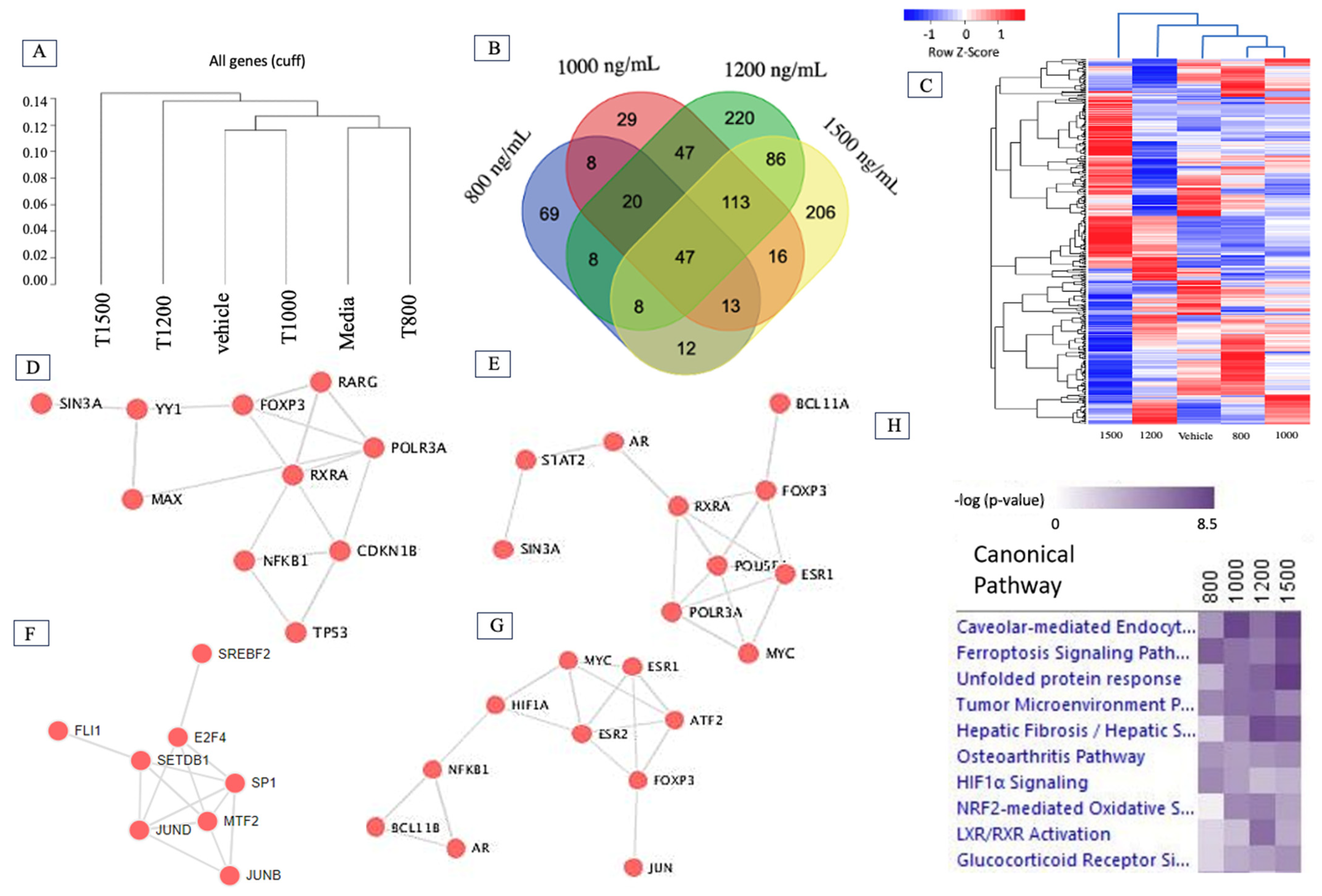
2.2. Δ-9-THC Dose-Mediated Global Alterations in Gene Expression Profile, Biomarkers, Transcription Factors, and Canonical Pathways
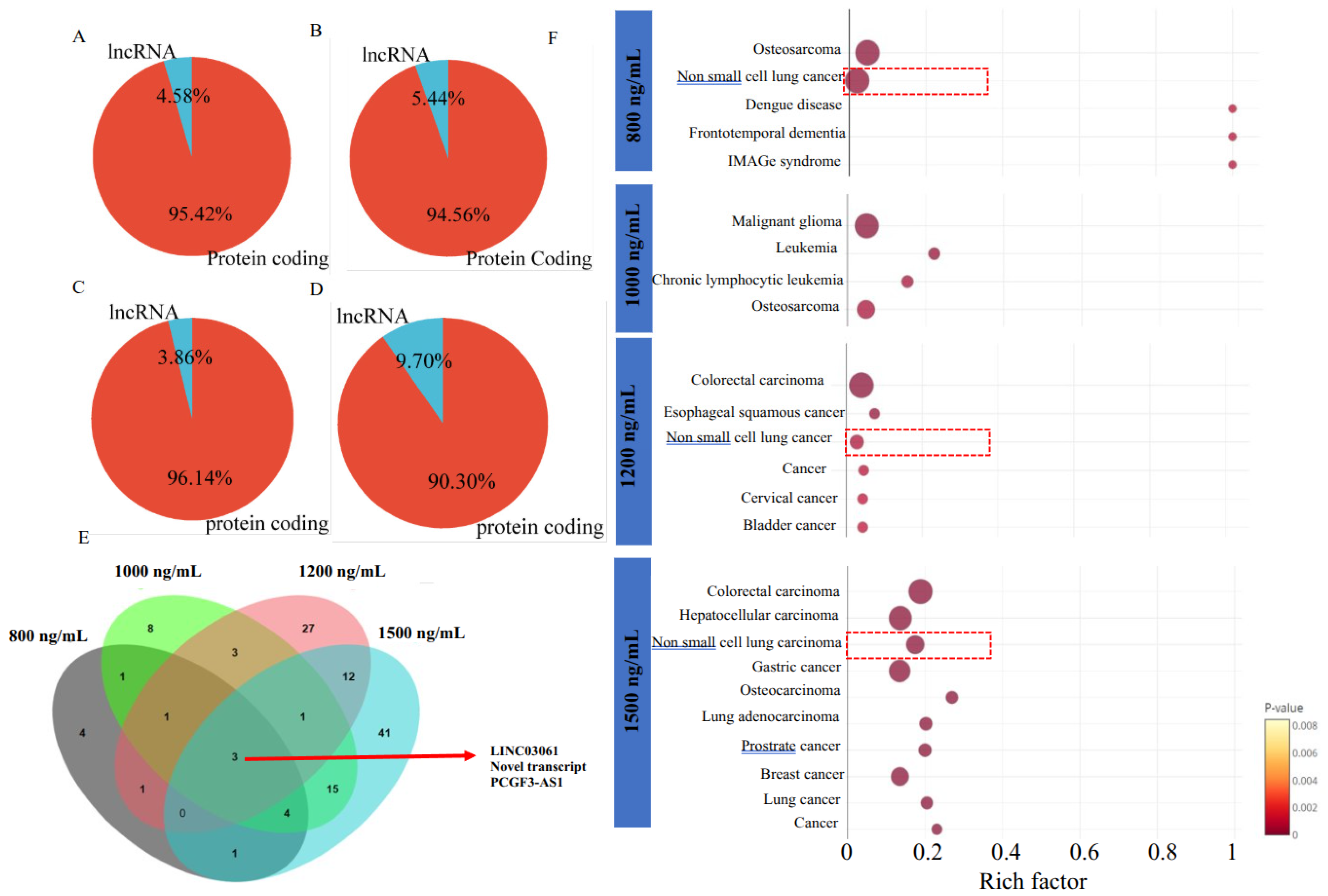
2.3. Δ-9-THC-Mediated Enrichment of Long Non-Coding RNA (lncRNA) in BEAS-2B Cells Associated with Activation of Lung Cancer Pathways
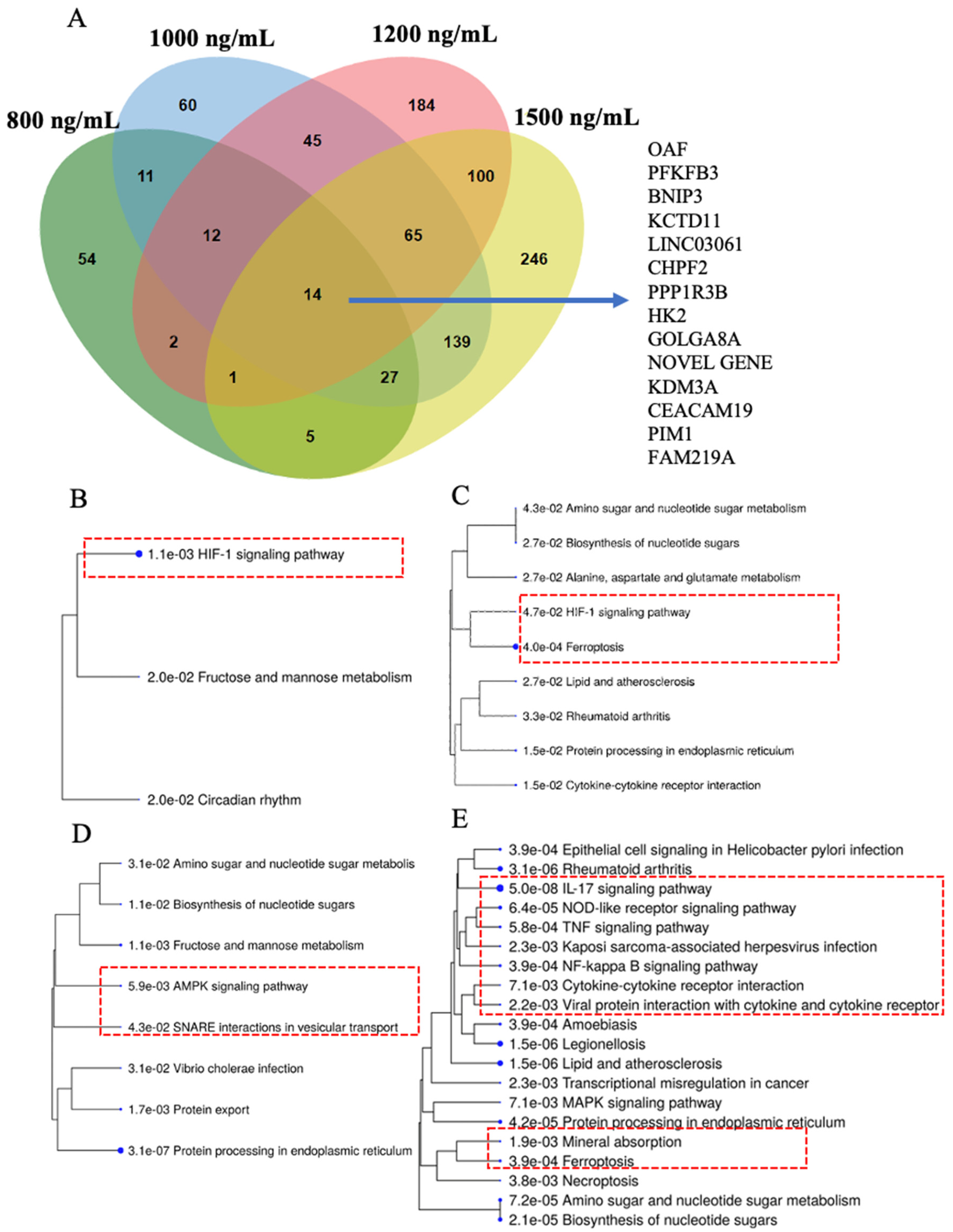
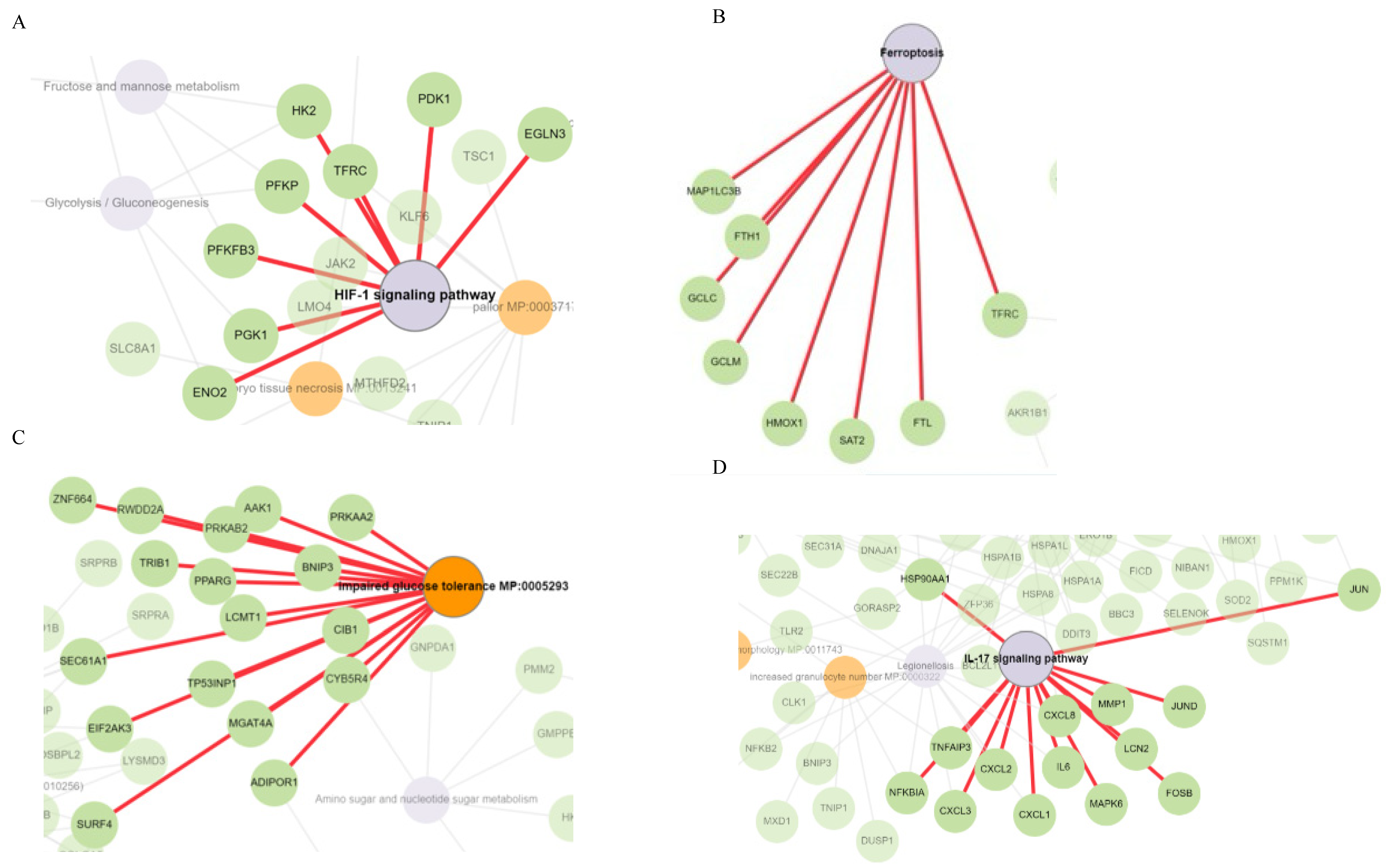
2.4. HIF-1 Signaling, Ferroptosis, AMPK Signaling, and Immunogenic Pathways Were Enriched in Genes Dose-Dependently Upregulated by Δ-9-THC in BEAS-2B Cells
2.5. Cysteine–Methionine Metabolism, Glutathione Metabolism, Amino Acid Metabolism, and Fatty Acid Metabolism Were Significantly Enriched in THC-Mediated Downregulated Genes
2.6. Functional Network Analysis and Enrichment of Mammalian Phenotypes Associated with DEGs
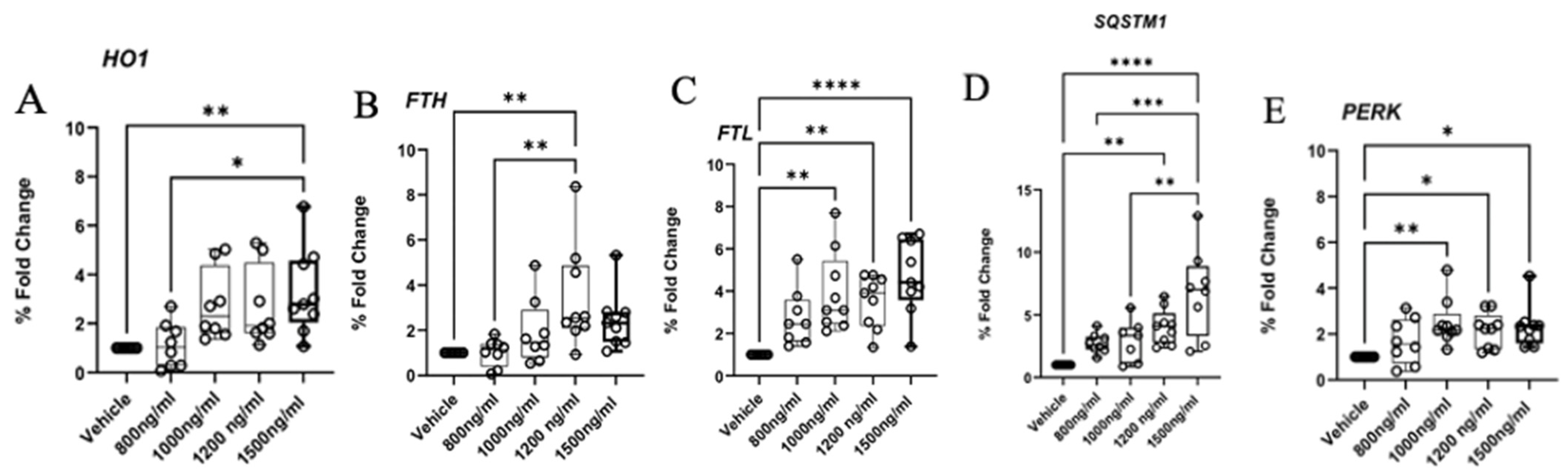
2.7. Aberrant Expression of Ferroptosis, Autophagy, and ER Response Genes
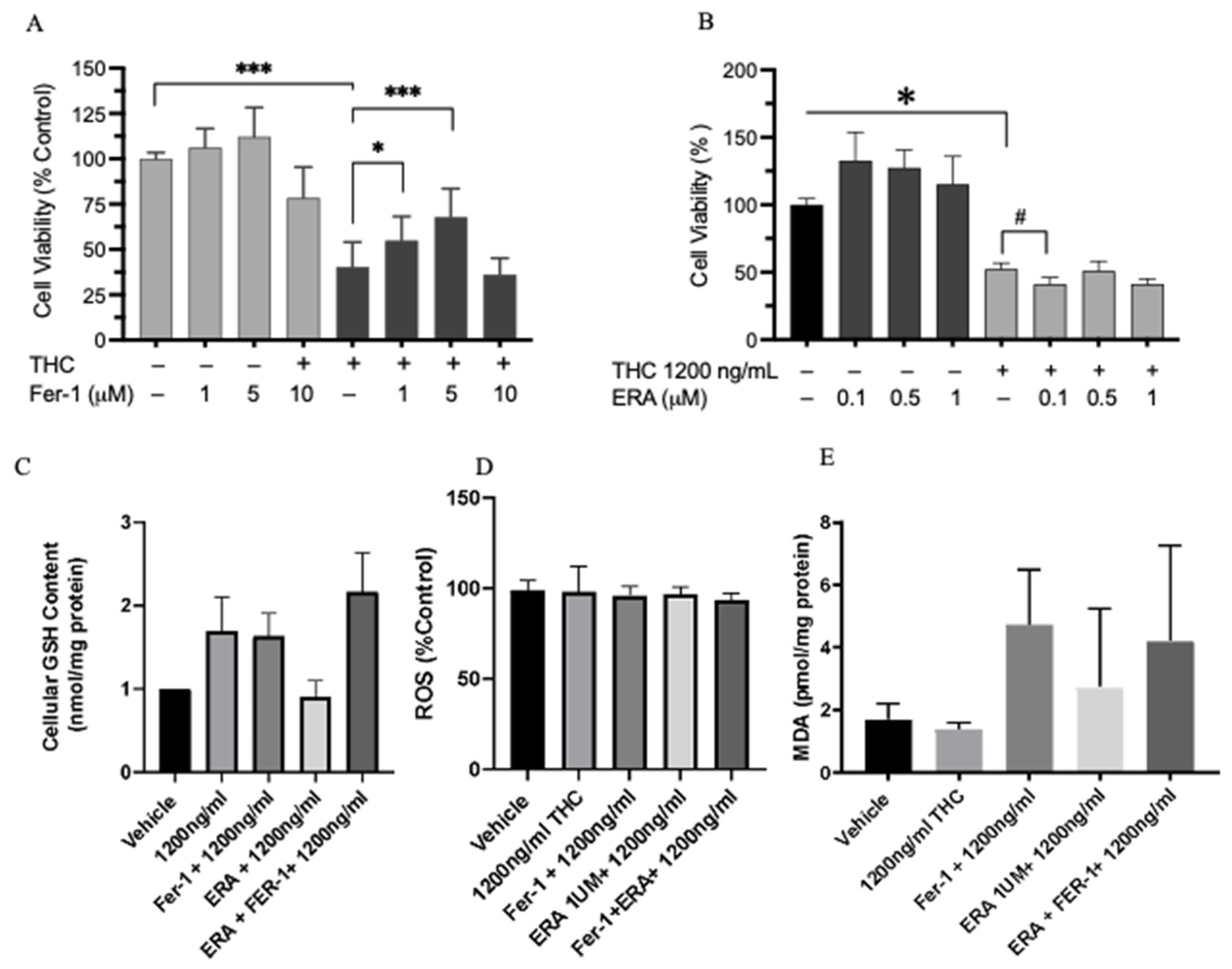
2.8. Cell Viability and Ferroptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture, THC Exposure, and Sample Collection
4.2. Cell Viability Analysis
4.3. RNA/DNA Extraction and cDNA Synthesis
4.4. Library Preparation, RNA Sequencing, and Bioinformatics
4.5. Pathway Analysis
4.6. Gene Validation by Real-Time Quantitative PCR (qPCR)
| HO1 | Forward: 5′ CTC TGA AGT TTA GGC CAT TG 3′ Reverse: 5′ AGT TGC TGT AGG GCT TTA TG 3′ |
| FTH | Forward: 5′ GCA CGA GCA AGT CAA GAC CAT 3′ Reverse: 5′ CTT GTC GAA CAG GTA CTC AGC 3′ |
| FTL | Forward: 5′ CAG CCT GGT CAA TTT GTA CCT 3′ Reverse: 5′ GCC AAT TCG CGG AAG AAG TG 3 |
| SQSTM1 | Forward: 5′ GCC ATA CCC TCT TCG ACT ACG 3′ Reverse: 5′ GAT TCT GGC ATC TGT AGG 3′ |
| PERK | Forward: 5′ CTT ATG CCA GAC ACA CAG AA 3′ Reverse: 5′ TCC ATC GTG CTG AAT GGA ATA C 3′ |
4.7. Testing for the Role of Ferroptosis in Cell Viability
4.8. GSH Measurement
4.9. Intracellular ROS Measurement
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| THC | Delta-9-tetrahydrocannabinol |
| RNAseq | RNA sequencing |
| qPCR | Quantitative teal-time polymerase chain reaction |
| BEAS2B | Epithelial cells isolated from normal human bronchial epithelium derived from autopsies of noncancerous individuals |
References
- Mead, A. Legal and regulatory issues governing cannabis and cannabis-derived products in the United States. Front. Plant Sci. 2019, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, Y.N. Current legal status of medical marijuana and cannabidiol in the United States. Epilepsy Behav. 2020, 112, 107452. [Google Scholar] [CrossRef] [PubMed]
- Lapey, J.D. Medicinal uses of marijuana. Ann. Intern. Med. 1997, 127, 1134–1135. [Google Scholar] [CrossRef]
- Lukhele, S.T.; Motadi, L.R. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement. Altern. Med. 2016, 16, 335. [Google Scholar] [CrossRef]
- Mucke, M.; Weier, M.; Carter, C.; Copeland, J.; Degenhardt, L.; Cuhls, H.; Radbruch, L.; Hauser, W.; Conrad, R. Systematic review and meta-analysis of cannabinoids in palliative medicine. J. Cachexia Sarcopenia Muscle 2018, 9, 220–234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sarafian, T.A.; Magallanes, J.A.M.; Shau, H.; Tashkin, D.; Roth, M.D. Oxidative stress produced by marijuana smoke: An adverse effect enhanced by cannabinoids. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1286–1293. [Google Scholar] [CrossRef]
- Pattnaik, F.; Nanda, S.; Mohanty, S.; Dalai, A.K.; Kumar, V.; Ponnusamy, S.K.; Naik, S. Cannabis: Chemistry, extraction and therapeutic applications. Chemosphere 2022, 289, 133012. [Google Scholar] [CrossRef]
- Bayazit, H.; Selek, S.; Karababa, I.F.; Cicek, E.; Aksoy, N. Evaluation of Oxidant/Antioxidant Status and Cytokine Levels in Patients with Cannabis Use Disorder. Clin. Psychopharmacol. Neurosci. 2017, 15, 237–242. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Campbell, V.A. Tetrahydrocannabinol-induced apoptosis of cultured cortical neurones is associated with cytochrome c release and caspase-3 activation. Neuropharmacology 2001, 40, 702–709. [Google Scholar] [CrossRef]
- Cooper, Z.D.; Haney, M. Actions of delta-9-tetrahydrocannabinol in cannabis: Relation to use, abuse, dependence. Int. Rev. Psychiatry 2009, 21, 104–112. [Google Scholar] [CrossRef]
- Aldington, S.; Williams, M.; Nowitz, M.; Weatherall, M.; Pritchard, A.; McNaughton, A.; Robinson, G.; Beasley, R. The effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007, 62, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef]
- Cudaback, E.; Marrs, W.; Moeller, T.; Stella, N. The Expression Level of CB1 and CB2 Receptors Determines Their Efficacy at Inducing Apoptosis in Astrocytomas. PLoS ONE 2010, 5, e8702. [Google Scholar] [CrossRef]
- Howlett, A.C.; Abood, M.E. Chapter Five—CB1 and CB2 Receptor Pharmacology. In Advances in Pharmacology; Kendall, D., Alexander, S.P.H., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 80, pp. 169–206. [Google Scholar]
- Tetrault, J.M.; Crothers, K.; Moore, B.A.; Mehra, R.; Concato, J.; Fiellin, D.A. Effects of marijuana smoking on pulmonary function and respiratory complications: A systematic review. Arch. Intern. Med. 2007, 167, 221–228. [Google Scholar] [CrossRef]
- Tashkin, D.P. Effects of marijuana smoking on the lung. Ann. Am. Thorac. Soc. 2013, 10, 239–247. [Google Scholar] [CrossRef]
- Ribeiro, L.I.; Ind, P.W. Effect of cannabis smoking on lung function and respiratory symptoms: A structured literature review. NPJ Prim. Care Respir. Med. 2016, 26, 1–8. [Google Scholar] [CrossRef]
- Wolff, V.; Schlagowski, A.-I.; Rouyer, O.; Charles, A.-L.; Singh, F.; Auger, C.; Schini-Kerth, V.; Marescaux, C.; Raul, J.-S.; Zoll, J.; et al. Tetrahydrocannabinol Induces Brain Mitochondrial Respiratory Chain Dysfunction and Increases Oxidative Stress: A Potential Mechanism Involved in Cannabis-Related Stroke. BioMed Res. Int. 2015, 2015, 323706. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Yang, Y.; Yang, Y. Multifaceted Roles of Ferroptosis in Lung Diseases. Front. Mol. Biosci. 2022, 9, 919187. [Google Scholar] [CrossRef]
- Nie, Z.; Chen, M.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Wang, F.; Zhang, S. Ferroptosis and Tumor Drug Resistance: Current Status and Major Challenges. Front. Pharmacol. 2022, 13, 879317. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, Z.; Yu, Z.; Li, J.; Zhang, J.; Wan, N.; Zhang, J.; Cao, J. Effect of curcumin on lung epithelial injury and ferroptosis induced by cigarette smoke. Hum. Exp. Toxicol. 2021, 40, S753–S762. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.H.; Weyer-Nichols, C.E.; Garcia-Sanchez, J.I.; Wilson, K.; Nagarkatti, P.; Nagarkatti, M. Delta-9-Tetrahydrocannabinol Blocks Bone Marrow-Derived Macrophage Differentiation through Elimination of Reactive Oxygen Species. Antioxidants 2024, 13, 887. [Google Scholar] [CrossRef]
- Abuse, S. Key Substance Use and Mental Health Indicators in the United States: Results from the 2019 National Survey on Drug Use and Health; HHS Publication No. PEP20-07-01-001, NSDUH Series H-55; Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2020. [Google Scholar]
- Sarafian, T.A.; Tashkin, D.P.; Roth, M.D. Marijuana Smoke and Δ9-Tetrahydrocannabinol Promote Necrotic Cell Death but Inhibit Fas-Mediated Apoptosis. Toxicol. Appl. Pharmacol. 2001, 174, 264–272. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.; Choi, C. Mitochondrial Network Determines Intracellular ROS Dynamics and Sensitivity to Oxidative Stress through Switching Inter-Mitochondrial Messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef]
- Richter, C.; Schweizer, M.; Cossarizza, A.; Franceschi, C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996, 378, 107–110. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.; Harris, A. Relation of hypoxia inducible factor 1α and 2α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp. Ther. Med. 2018, 16, 4553–4561. [Google Scholar] [CrossRef]
- Xu, Y.R.; Wang, A.L.; Li, Y.Q. Hypoxia-inducible factor 1-alpha is a driving mechanism linking chronic obstructive pulmonary disease to lung cancer. Front. Oncol. 2022, 12, 984525. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ao, Q.; Hu, H.; Huang, Y. Ferroptosis and endoplasmic reticulum stress in rheumatoid arthritis. Front. Immunol. 2024, 15, 1438803. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, X.; Jin, X.; Ye, F.; Liu, X.; Yu, B.; Li, Z.; Zhao, T.; Chen, W.; Liu, X.; Di, C.; et al. Ferroptosis: A novel regulated cell death participating in cellular stress response, radiotherapy, and immunotherapy. Exp. Hematol. Oncol. 2023, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Zhao, Y.; Wang, J.; Yang, X.; Li, S.; Wang, Y.; Yang, X.; Fei, J.; Hao, X.; Zhao, Y.; et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int. J. Biol. Sci. 2021, 17, 2703–2717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Birrell, M.A.; De Alba, J.; Catley, M.C.; Hardaker, E.; Wong, S.; Collins, M.; Clarke, D.L.; Farrow, S.N.; Willson, T.M.; Collins, J.L.; et al. Liver X Receptor Agonists Increase Airway Reactivity in a Model of Asthma via Increasing Airway Smooth Muscle Growth1. J. Immunol. 2008, 181, 4265–4271. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, S.D.; Tontonoz, P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 2015, 242, 29–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morichika, D.; Miyahara, N.; Fujii, U.; Taniguchi, A.; Oda, N.; Senoo, S.; Kataoka, M.; Tanimoto, M.; Kakuta, H.; Kiura, K.; et al. A retinoid X receptor partial agonist attenuates pulmonary emphysema and airway inflammation. Respir. Res. 2019, 20, 2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fujii, U.; Miyahara, N.; Taniguchi, A.; Oda, N.; Morichika, D.; Murakami, E.; Nakayama, H.; Waseda, K.; Kataoka, M.; Kakuta, H.; et al. Effect of a retinoid X receptor partial agonist on airway inflammation and hyperresponsiveness in a murine model of asthma. Respir. Res. 2017, 18, 23. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Park, E.-J.; Park, Y.-J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, X.; Zhang, C.; Yang, T.; Deng, Z.; Song, Y.; Huang, L.; Li, F.; Li, Q.; Lin, S. EF24 induces ferroptosis in osteosarcoma cells through HMOX1. Biomed. Pharmacother. 2021, 136, 111202. [Google Scholar] [CrossRef]
- Meng, D.; Zhu, C.; Jia, R.; Li, Z.; Wang, W.; Song, S. The molecular mechanism of ferroptosis and its role in COPD. Front. Med. 2023, 9, 1052540. [Google Scholar] [CrossRef]
- Remels, A.H.V.; Gosker, H.R.; Langen, R.C.J.; Schols, A.M.W.J. The mechanisms of cachexia underlying muscle dysfunction in COPD. J. Appl. Physiol. 2013, 114, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis (Review). Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free. Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, J.; Cai, L.; Wang, S.; Liu, C.; Zhang, Y.; You, L.; Fu, Y.; Shi, Z.; Yin, Z. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 2775–2783. [Google Scholar] [CrossRef]
- Cocco, E.; Porrini, V.; Derosas, M.; Nardi, V.; Biasiotto, G.; Maccarinelli, F.; Zanella, I. Protective effect of mitochondrial ferritin on cytosolic iron dysregulation induced by doxorubicin in HeLa cells. Mol. Biol. Rep. 2013, 40, 6757–6764. [Google Scholar] [CrossRef]
- Sarafian, T.; Habib, N.; Mao, J.T.; Tsu, I.H.; Yamamoto, M.L.; Hsu, E.; Tashkin, D.P.; Roth, M.D. Gene expression changes in human small airway epithelial cells exposed to Δ9-tetrahydrocannabinol. Toxicol. Lett. 2005, 158, 95–107. [Google Scholar] [CrossRef]
- Aguiar, J.A.; Huff, R.D.; Tse, W.; Stämpfli, M.R.; McConkey, B.J.; Doxey, A.C.; Hirota, J.A. Transcriptomic and barrier responses of human airway epithelial cells exposed to cannabis smoke. Physiol. Rep. 2019, 7, e14249. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, Y.; Chen, X.; Zhao, J.; Han, J.; Dong, H.; Zheng, Q.; Nie, G. Ferrostatin-1 protects auditory hair cells from cisplatin-induced ototoxicity in vitro and in vivo. Biochem. Biophys. Res. Commun. 2020, 533, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free. Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.; Li, Z.; Wang, Y.; Lin, H.; Wu, B. TLSEA: A tool for lncRNA set enrichment analysis based on multi-source heterogeneous information fusion. Front. Genet. 2023, 14, 1181391. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell proliferation and cytotoxicity assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Xie, F.; Liu, K.; Liu, H.; Xie, J. Evaluation of whole cigarette smoke induced oxidative stress in A549 and BEAS–2B cells. Environ. Toxicol. Pharmacol. 2017, 54, 40–47. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doldron, M.S.; Chakraborty, S.; Anand, S.; Faheem, M.; Reh, B.; Wang, X.; Mallik, S.; Jia, Z.; Bhandari, R.K. Cellular and Transcriptional Responses of Human Bronchial Epithelial Cells to Delta-9-Tetrahydrocannabinol In Vitro. Int. J. Mol. Sci. 2025, 26, 5212. https://doi.org/10.3390/ijms26115212
Doldron MS, Chakraborty S, Anand S, Faheem M, Reh B, Wang X, Mallik S, Jia Z, Bhandari RK. Cellular and Transcriptional Responses of Human Bronchial Epithelial Cells to Delta-9-Tetrahydrocannabinol In Vitro. International Journal of Molecular Sciences. 2025; 26(11):5212. https://doi.org/10.3390/ijms26115212
Chicago/Turabian StyleDoldron, Megan S., Sourav Chakraborty, Santosh Anand, Mehwish Faheem, Beh Reh, Xuegeng Wang, Saurav Mallik, Zhenquan Jia, and Ramji Kumar Bhandari. 2025. "Cellular and Transcriptional Responses of Human Bronchial Epithelial Cells to Delta-9-Tetrahydrocannabinol In Vitro" International Journal of Molecular Sciences 26, no. 11: 5212. https://doi.org/10.3390/ijms26115212
APA StyleDoldron, M. S., Chakraborty, S., Anand, S., Faheem, M., Reh, B., Wang, X., Mallik, S., Jia, Z., & Bhandari, R. K. (2025). Cellular and Transcriptional Responses of Human Bronchial Epithelial Cells to Delta-9-Tetrahydrocannabinol In Vitro. International Journal of Molecular Sciences, 26(11), 5212. https://doi.org/10.3390/ijms26115212