
Identification of Novel Genetic Variants in a Cohort of Congenital Hypogonadotropic Hypogonadism: Computational Analysis of Pathogenicity Predictions

 ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Results
2.1. Clinical Features of the HH Cohort
2.2. Genetic Variants and Their Pathogenicity
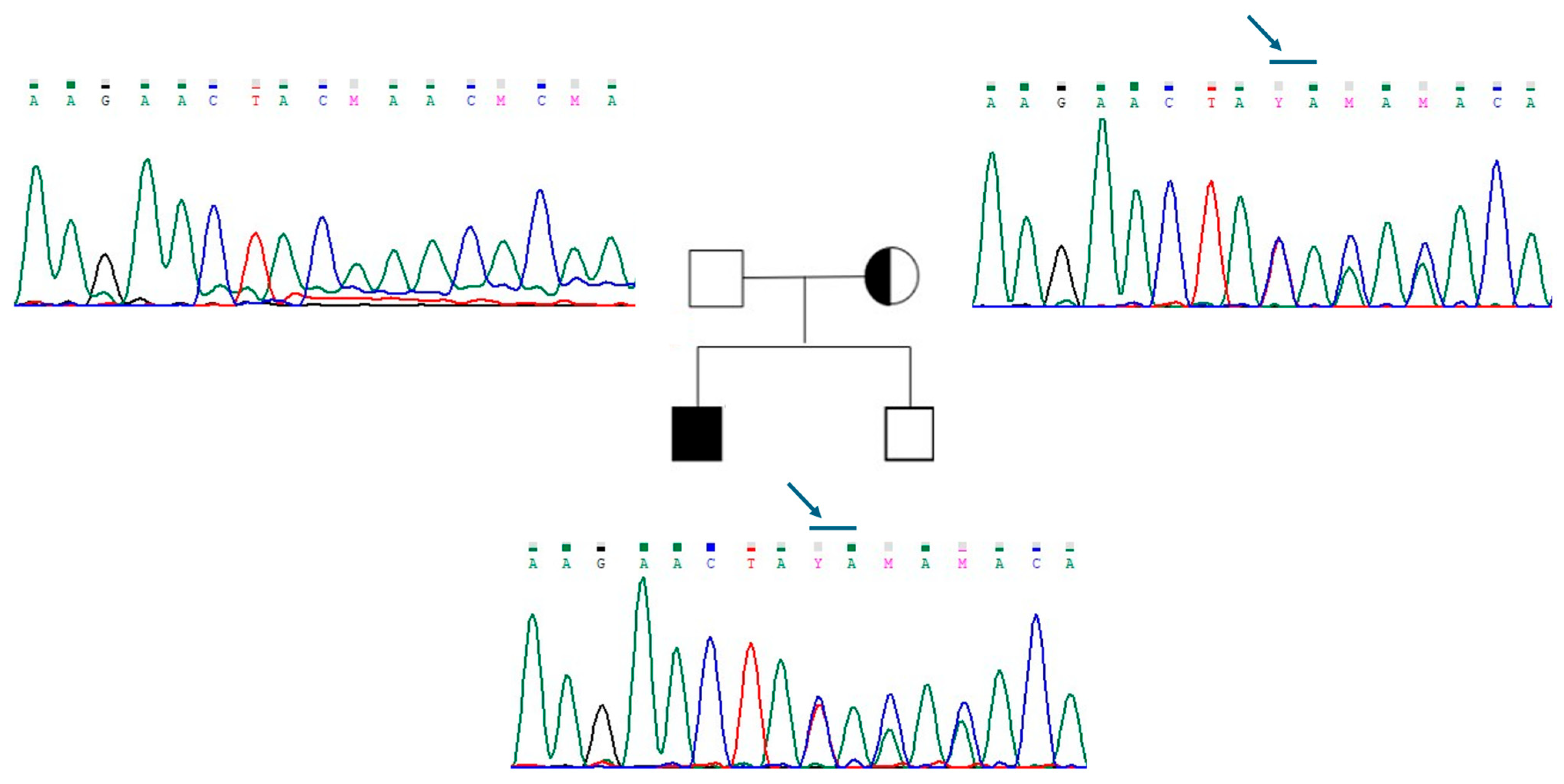
2.3. Oligogenic Transmission in Three Patients with CHH
2.4. Frequency of Variants According to Phenotype
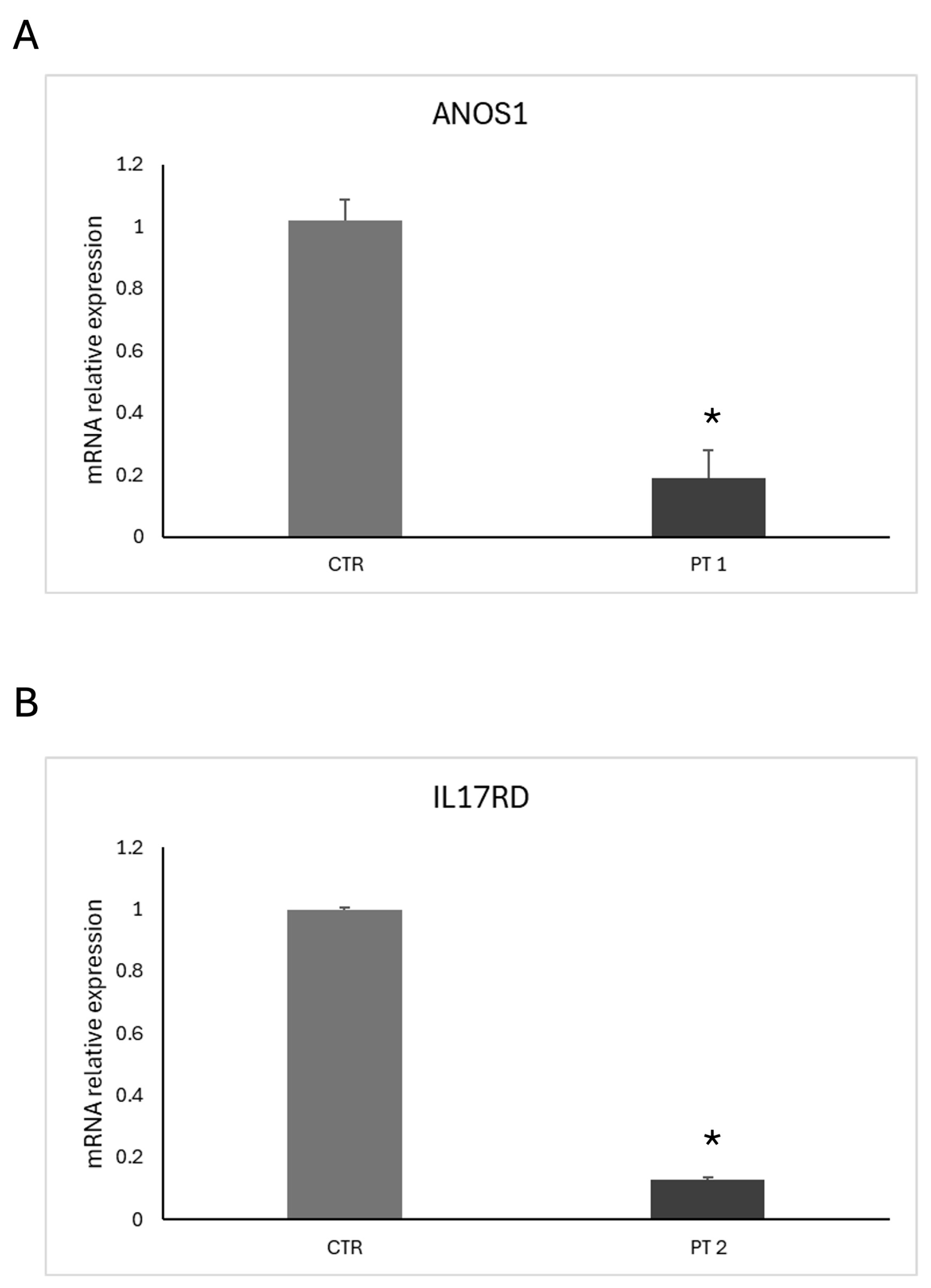
2.5. Functional Characterization of ANOS1 and IL17RD Mutations
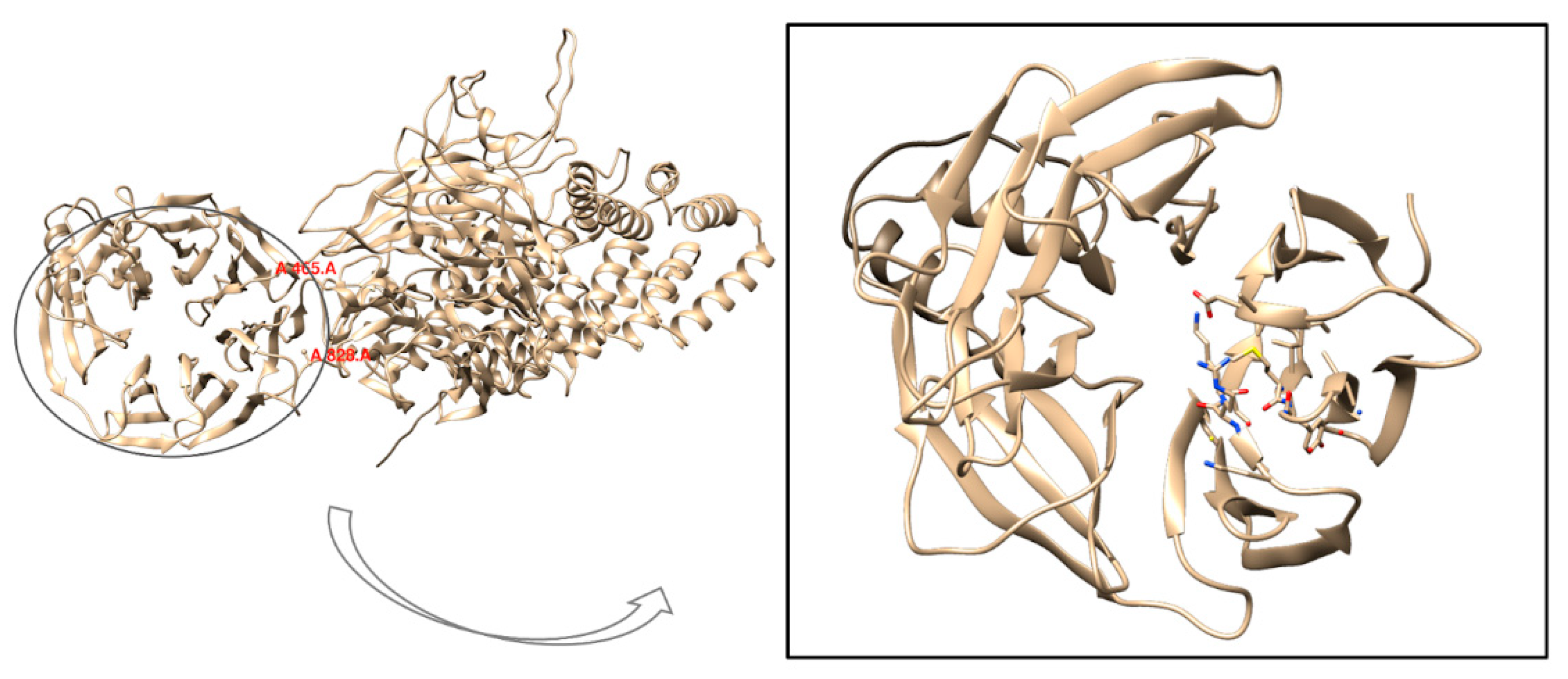
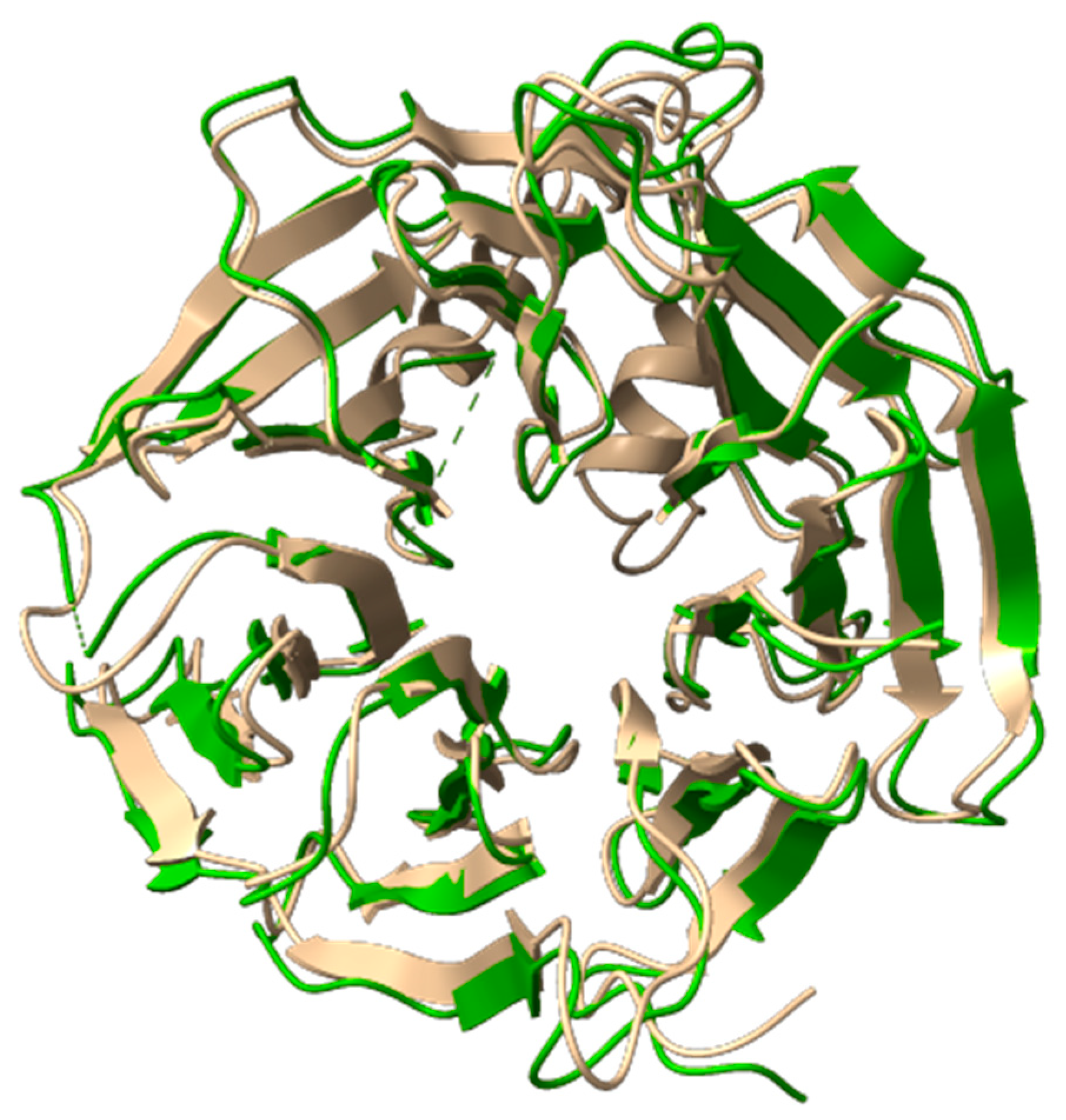
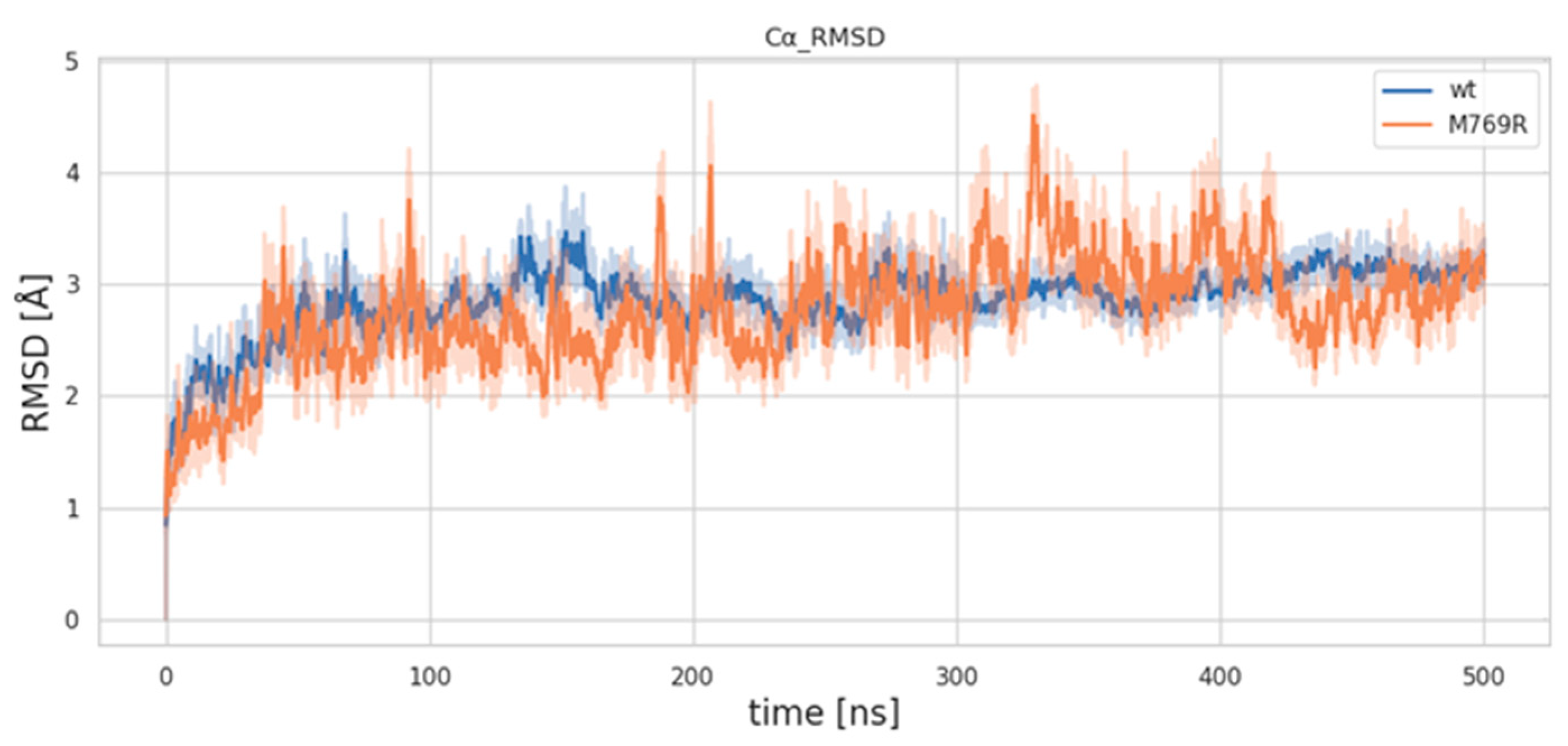
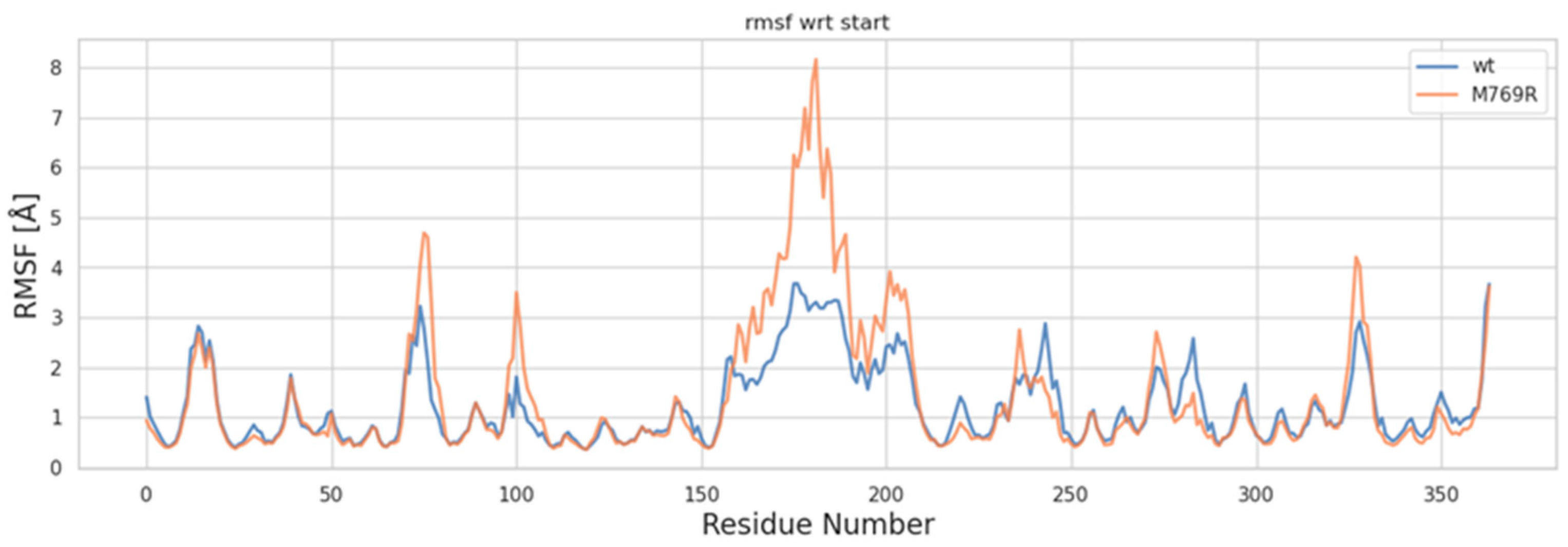
2.6. Molecular Dynamics Simulations of WDR11 Variant
3. Discussion
4. Materials and Methods
- -
- low serum testosterone concentrations in the setting of low gonadotropin concentrations (<264 ng/dL in adults);
- -
- delayed/absent pubertal development, defined by lack of testicular enlargement (volume < 4 mL with Prader orchidometer) in boys at age 14 and absence of breast development in girls at age 13; and/or
- -
- infertility/sterility; and/or
- -
- criptorchidism and micropenis in the neonatal period; and/or
- -
- anosmia/hyposmia assessed by Sniffin’ test (Sense Trading BV, Groningen, The Netherlands), a validated measure of olfactory function; and/or
- -
- developmental anomalies, such as cleft lip and palate, dental agenesis, renal agenesis, hypospadias, bimanual synkinesis, sensorineural deafness, and skeletal anomalies; and/or
- -
- no identifiable cause of hypothalamic or pituitary dysfunction, such as chronic diseases, pituitary tumors, head trauma; and/or
- -
- presence of family cases of CHH and/or infertility sine causa.
4.1. Instrumental and Molecular Analyses
- i.
- Expression of the variant in a biological sample.
- ii.
- Investigation of the variant through MDs and dynamic cross-correlation analysis.
4.2. Reverse Transcription and Quantitative Real-Time Amplification (qRT-PCR)
4.3. Molecular Dynamics Simulations and Dynamic Cross-Correlation Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Howard, S.R. Interpretation of Reproductive Hormones before, during and after the Pubertal Transition-Identifying Health and Disordered Puberty. Clin. Endocrinol. 2021, 95, 702–715. [Google Scholar] [CrossRef]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann Syndrome: Phenotype and Genotype of Hypogonadotropic Hypogonadism. Metabolism 2018, 86, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, B.; Swee, D.S.; Quinton, R.; Bonomi, M. Genetics of Congenital Hypogonadotropic Hypogonadism: Peculiarities and Phenotype of an Oligogenic Disease. Hum. Genet. 2021, 140, 77–111. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; et al. Oligogenic Basis of Isolated Gonadotropin-Releasing Hormone Deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar] [CrossRef]
- Maione, L.; Dwyer, A.A.; Francou, B.; Guiochon-Mantel, A.; Binart, N.; Bouligand, J.; Young, J. GENETICS IN ENDOCRINOLOGY: Genetic Counseling for Congenital Hypogonadotropic Hypogonadism and Kallmann Syndrome: New Challenges in the Era of Oligogenism and next-Generation Sequencing. Eur. J. Endocrinol. 2018, 178, R55–R80. [Google Scholar] [CrossRef]
- Boehm, U.; Bouloux, P.-M.; Dattani, M.T.; de Roux, N.; Dodé, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.-P.; Juul, A.; et al. Expert Consensus Document: European Consensus Statement on Congenital Hypogonadotropic Hypogonadism--Pathogenesis, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 Repeat Domain Proteins: A Novel Target Class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Deng, H.; Jia, G.; Li, P.; Tang, Y.; Zhao, L.; Yang, Q.; Zhao, J.; Wang, J.; Tu, Y.; Yong, X.; et al. The WDR11 Complex Is a Receptor for Acidic-Cluster-Containing Cargo Proteins. Cell 2024, 187, 4272–4288.e20. [Google Scholar] [CrossRef]
- Patil, V.A.; Lila, A.R.; Shah, N.; Arya, S.; Ekbote, A.V.; Sarathi, V.; Shah, R.; Jadhav, S.S.; Memon, S.S.; Bandgar, T. Regional Genotypic Variations in Normosmic Congenital Hypogonadotropic Hypogonadism: Our Experience and Systematic Review. Pituitary 2022, 25, 444–453. [Google Scholar] [CrossRef]
- Patil, V.A.; Lila, A.R.; Shah, N.; Arya, S.; Sarathi, V.; Shah, R.; Jadhav, S.S.; Memon, S.S.; Karlekar, M.; Bandgar, T. Genetic Spectrum of Kallmann Syndrome: Single-Center Experience and Systematic Review. Clin. Endocrinol. (Oxf.) 2022, 97, 804–813. [Google Scholar] [CrossRef]
- Caminsky, N.; Mucaki, E.J.; Rogan, P.K. Interpretation of mRNA Splicing Mutations in Genetic Disease: Review of the Literature and Guidelines for Information-Theoretical Analysis. F1000Research 2014, 3, 282. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep Intronic Mutations and Human Disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-G.; Ahn, J.-W.; Kurth, I.; Ullmann, R.; Kim, H.-T.; Kulharya, A.; Ha, K.-S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD Protein That Interacts with Transcription Factor EMX1, Is Mutated in Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Osborn, D.P.; Lee, J.-Y.; Araki, M.; Araki, K.; Mohun, T.; Känsäkoski, J.; Brandstack, N.; Kim, H.-T.; Miralles, F.; et al. WDR11-Mediated Hedgehog Signalling Defects Underlie a New Ciliopathy Related to Kallmann Syndrome. EMBO Rep. 2018, 19, 269–289. [Google Scholar] [CrossRef]
- Yamada, R.; Yamakita, N.; Yasuda, K.; Imai, A. Adult-Onset Reversible Idiopathic Hypogonadotropic Hypogonadism in Male Adult Carrying a WDR11 Missense Mutation. BMJ Case Rep. 2022, 15, e250444. [Google Scholar] [CrossRef]
- Bouilly, J.; Messina, A.; Papadakis, G.; Cassatella, D.; Xu, C.; Acierno, J.S.; Tata, B.; Sykiotis, G.; Santini, S.; Sidis, Y.; et al. DCC/NTN1 Complex Mutations in Patients with Congenital Hypogonadotropic Hypogonadism Impair GnRH Neuron Development. Hum. Mol. Genet. 2018, 27, 359–372. [Google Scholar] [CrossRef]
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tétreault, M.; Takanohashi, A.; Carosso, G.; Clément, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A Encoding a Catalytic Subunit of RNA Polymerase Pol III Cause a Recessive Hypomyelinating Leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423. [Google Scholar] [CrossRef]
- Tétreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive Mutations in POLR3B, Encoding the Second Largest Subunit of Pol III, Cause a Rare Hypomyelinating Leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef]
- Synofzik, M.; Kernstock, C.; Haack, T.B.; Schöls, L. Ataxia Meets Chorioretinal Dystrophy and Hypogonadism: Boucher-Neuhäuser Syndrome Due to PNPLA6 Mutations. J. Neurol. Neurosurg. Psychiatry 2015, 86, 580–581. [Google Scholar] [CrossRef]
- Laitinen, E.M.; Hero, M.; Vaaralahti, K.; Tommiska, J.; Raivio, T. Bone Mineral Density, Body Composition and Bone Turnover in Patients with Congenital Hypogonadotropic Hypogonadism. Int. J. Androl. 2012, 35, 534–540. [Google Scholar] [CrossRef]
- Margolin, D.H.; Kousi, M.; Chan, Y.-M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, Dementia, and Hypogonadotropism Caused by Disordered Ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- De Roux, N.; Carel, J.-C.; Léger, J. Congenital Hypogonadotropic Hypogonadism: A Trait Shared by Several Complex Neurodevelopmental Disorders. Endocr. Dev. 2016, 29, 72–86. [Google Scholar] [CrossRef]
- Rochira, V.; Faustini-Fustini, M.; Balestrieri, A.; Carani, C. Estrogen Replacement Therapy in a Man with Congenital Aromatase Deficiency: Effects of Different Doses of Transdermal Estradiol on Bone Mineral Density and Hormonal Parameters. J. Clin. Endocrinol. Metab. 2000, 85, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Simmonett, A.C.; Brooks, B.R. Analytical Hessians for Ewald and Particle Mesh Ewald Electrostatics. J. Chem. Phys. 2021, 154, 104101. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt 1 | Pt 2 | Pt 3 | Pt 4 | Pt 5 | Pt 6 | Pt 7 | Pt 8 | Pt 9 | Pt 10 | Pt 11 | Pt 12 | Pt 13 | Pt 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at diagnosis | 52 years | 18 years | 22 years | 18 years | 42 years | 7 months | 18 years | 17 years | 24 years | 16 years | 18 years | 30 years | 17 years | 17 years |
| Sex | M | M | F | M | F | M | M | M | M | F | M | F | M | M |
| Phenotype | KS | nHH | nHH | KS | KS | KS | nHH | KS | nHH | KS | KS | nHH | nHH | nHH |
| Clinical characteristics | Hypogonadism Anosmia | Hypogonadism | Hypogonadism Secondary amenorrhea Ataxia | Hypogonadism Anosmia Eunuchoidal proportions | Hypogonadism Primary amenorrhea Anosmia | Cryptorchidism Micropenis Hypogonadism Anosmia | Hypogonadism | Hypogonadism Anosmia Eunuchoidal proportions | Hypogonadism Osteoporosis | Hypogonadism Primary amenorrhea Iposmia | Hypogonadism Gynecomastia Anosmia Eunuchoidal proportions | Hypogonadism Secondary amenorrhea | Cryptorchidism Hypogonadism Eunuchoidal proportions | Hypogonadism Eunuchoidal proportions |
| Puberty | Absent | Absent | Normal | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Normal | Arrest | Absent |
| CHH reversal | No | No | No | No | No | No | No | No | Yes | No | No | No | No | No |
| Gene | ANOS1 | IL17RD | WDR11 DCC | PROK2 | FGF8 IL17RD | - | IL17RD | PROK2 | DCC | CHD7 TACR3 | FGFR11 | TACR3 | - | DUSP6 |
| LH (IU/L) | 0.05 | 0.16 | 0.80 | 0 | 0.10 | 0.20 | 0.38 | 0.39 | 0.39 | 0.40 | 0.18 | 0.25 | 1.58 | 0.30 |
| FSH (IU/L) | 1.48 | 3.05 | 0.90 | 0.68 | 0.34 | 0.70 | 1.56 | 0.54 | 0.54 | 2.15 | 1.30 | 0.90 | 1.89 | 1.05 |
| Testosterone (ng/mL) or estradiol (pg/mL) | 113 | 20 | 10 | 0.30 | 10 | 0.03 | 0.18 | 14 | 14 | 11.80 | 14 | 15.2 | 19 | 12 |
| Inhibin B (pg/mL) | 156 | 210 | 175 | 165 | 182 | 200 | 180 | 156 | 223 | 210 | ||||
| AMH (ng/mL) | 3 | 1.2 | 4.2 | 2.6 | ||||||||||
| Testicular volume (cm3) or ovarian volume (cm3) | 3.0 | 2.0 | 5.0 | 2.0 | 2.0 | 1.0 | <2.0 | 2.0 | 4.0 | <2.0 | 2.0 | 2.5 | 6.0 | 2.0 |
| Brain MRI | Normal | Normal | Cerebellar atrophy | Normal | Hypoplasia of olfactory bulbs | Normal | Normal | Normal | Normal | Hypoplasia of olfactory bulbs | Normal | Normal | Normal | Normal |
| Therapy | Testosterone/Gonadotropin | Gonadotropin/Testosterone | Estrogen/Gonadotropin/EP | Gonadotropin/Testosterone | Estrogen/Gonadotropin/EP | Gonadotropin/Testosterone | Testosterone/Gonadotropin | Gonadotropin | Testosterone | Estrogen | Gonadotropin/Testosterone | EP | Gonadotropin | Testosterone |
| Gene | RefSeq/Variant Identified | Location | Consequence | Impact | Genotype | Inheritance | Clinical Significance |
|---|---|---|---|---|---|---|---|
| ANOS1 | NM_000216.4: c.541+1G>A GRCh37: ChrX:g.8565074C>T | Intron 4 | - | LOF | Hemizygous | AD | Probably pathogenic |
| IL17RD | NM_017563.5: c.1303_1304dup GRCh37: Ch3:g.57132427_5713 | Exon 12 | (p.Lys436ThrfsTer58) | Frameshift LOF | Heterozygous | AR/AD/Oligo | Probably pathogenic |
| DCC | NM_005215.4 c.3533C>T GRCh37: Chr18:g.50985742C>T | Exon 24 | (p.Ser1178Phe) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| WDR11 | NM_018117.12 c.2306T>G GRCh37 | Exon 18 | (p.Met769Arg) | Missense | Heterozygous | AD | VUS |
| PROK2 | NM_001126128.2 c.146G>T | Exon 2 | (p.Ser49Ile) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| FGF8 | NM_033163.5 Chr1o: g.103530393C>T GFCh37 | Intron 5 | - | Missense | Heterozygous | AD/Oligo | VUS |
| IL17RD | NM_017563.5 Chr3: c.1622C>T | Exon 12 | (p.Ser541Phe) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| IL17RD | NM_017563.5 c.692A>G | Exon 7 | (p.Tyr231CYS) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| PROK2 | NM_001126128.2 c.121G>A | Exon 2 | (p.Gly41Ser) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| DCC | NM_005215.4 c.1435A>G | Exon 9 | (p.Thr479Ala) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| CHD7 | NM_017780.4 | (p.Met340Val) | Missense | Heterozygous | AR | Benign | |
| TACR3 | NM_001059 | (p.Lys286Arg) | Missense | Heterozygous | Benign | ||
| FGFR11 | NM_023110.3 c.1058C>G | Exon 8 | (p.S353C) | Missense | Heterozygous | AR/AD/Oligo | VUS |
| TACR3 | NM_001059.3 Chr4: c.101A>T | Exon 4 | (p.Lys361Ter) | LOF | Heterozygous | AR | Probably pathogenic |
| TACR3 | NM_001959.3 Chr4: c.824G>A | Exon 3 | (p.Trp275Ter) | LOF | Heterozygous | AR | Pathogenic |
| DUSP6 | NM_001946.4 c.838+3G>C Chr12:g.89744362C>G hg19/GRCh37 | Intron 2 | - | Missense | Heterozygous | AR | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarello, P.; Gualtieri, G.; Bossio, S.; Seminara, G.; Molinaro, M.; Antonucci, G.; Perri, A.; Rocca, V.; Cannarella, R.; La Vignera, S.; et al. Identification of Novel Genetic Variants in a Cohort of Congenital Hypogonadotropic Hypogonadism: Computational Analysis of Pathogenicity Predictions. Int. J. Mol. Sci. 2025, 26, 5207. https://doi.org/10.3390/ijms26115207
Chiarello P, Gualtieri G, Bossio S, Seminara G, Molinaro M, Antonucci G, Perri A, Rocca V, Cannarella R, La Vignera S, et al. Identification of Novel Genetic Variants in a Cohort of Congenital Hypogonadotropic Hypogonadism: Computational Analysis of Pathogenicity Predictions. International Journal of Molecular Sciences. 2025; 26(11):5207. https://doi.org/10.3390/ijms26115207
Chicago/Turabian StyleChiarello, Paola, Gianmarco Gualtieri, Sabrina Bossio, Giuseppe Seminara, Marianna Molinaro, Gemma Antonucci, Anna Perri, Valentina Rocca, Rossella Cannarella, Sandro La Vignera, and et al. 2025. "Identification of Novel Genetic Variants in a Cohort of Congenital Hypogonadotropic Hypogonadism: Computational Analysis of Pathogenicity Predictions" International Journal of Molecular Sciences 26, no. 11: 5207. https://doi.org/10.3390/ijms26115207
APA StyleChiarello, P., Gualtieri, G., Bossio, S., Seminara, G., Molinaro, M., Antonucci, G., Perri, A., Rocca, V., Cannarella, R., La Vignera, S., Calogero, A. E., Greco, E. A., Iuliano, R., Alcaro, S., & Aversa, A. (2025). Identification of Novel Genetic Variants in a Cohort of Congenital Hypogonadotropic Hypogonadism: Computational Analysis of Pathogenicity Predictions. International Journal of Molecular Sciences, 26(11), 5207. https://doi.org/10.3390/ijms26115207