

Integrated Transcriptomic and Metabolomic Analysis Reveals the Regulation Network of CEBiP in Rice Defense Against Magnaporthe oryzae
 , , , and
, , , and 
Abstract
1. Introduction
2. Results
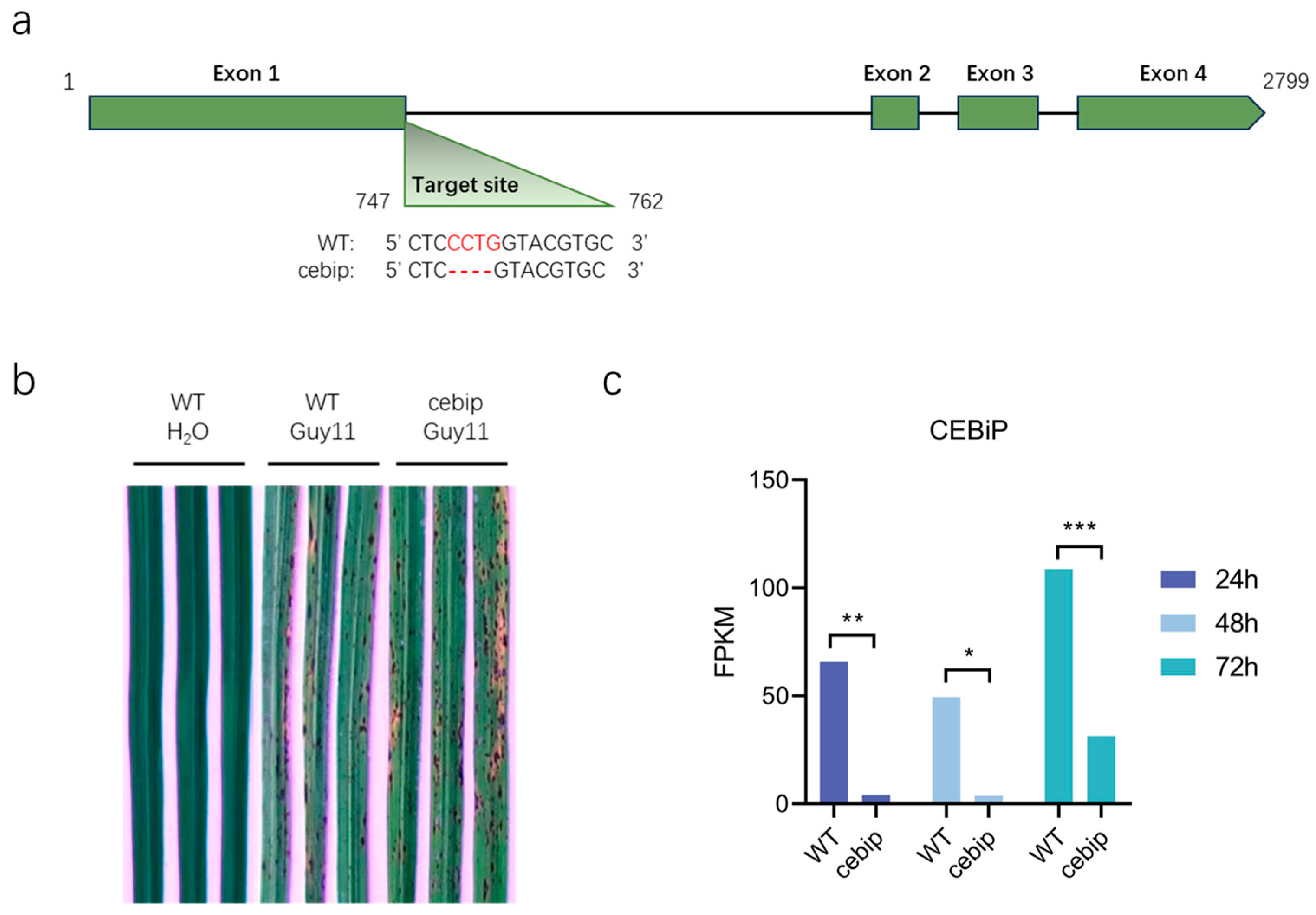
2.1. Knockout of the CEBiP Led to a Mild Increase in the Severity of Disease Symptoms upon M. oryzae Infection
2.2. Transcriptome Analysis Reveals a Set of Differentially Expressed Genes Involved in CEBiP Immune Function
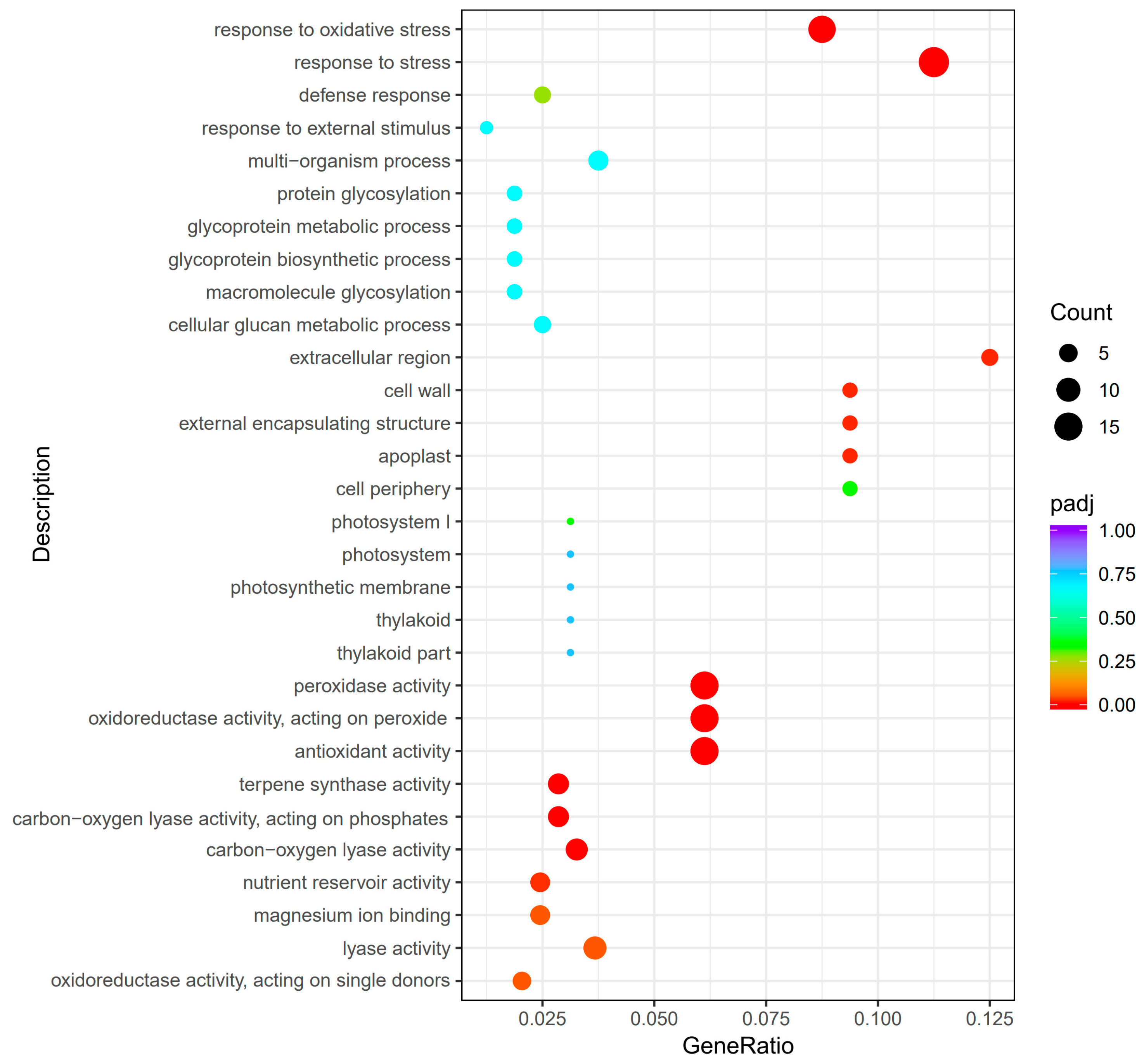
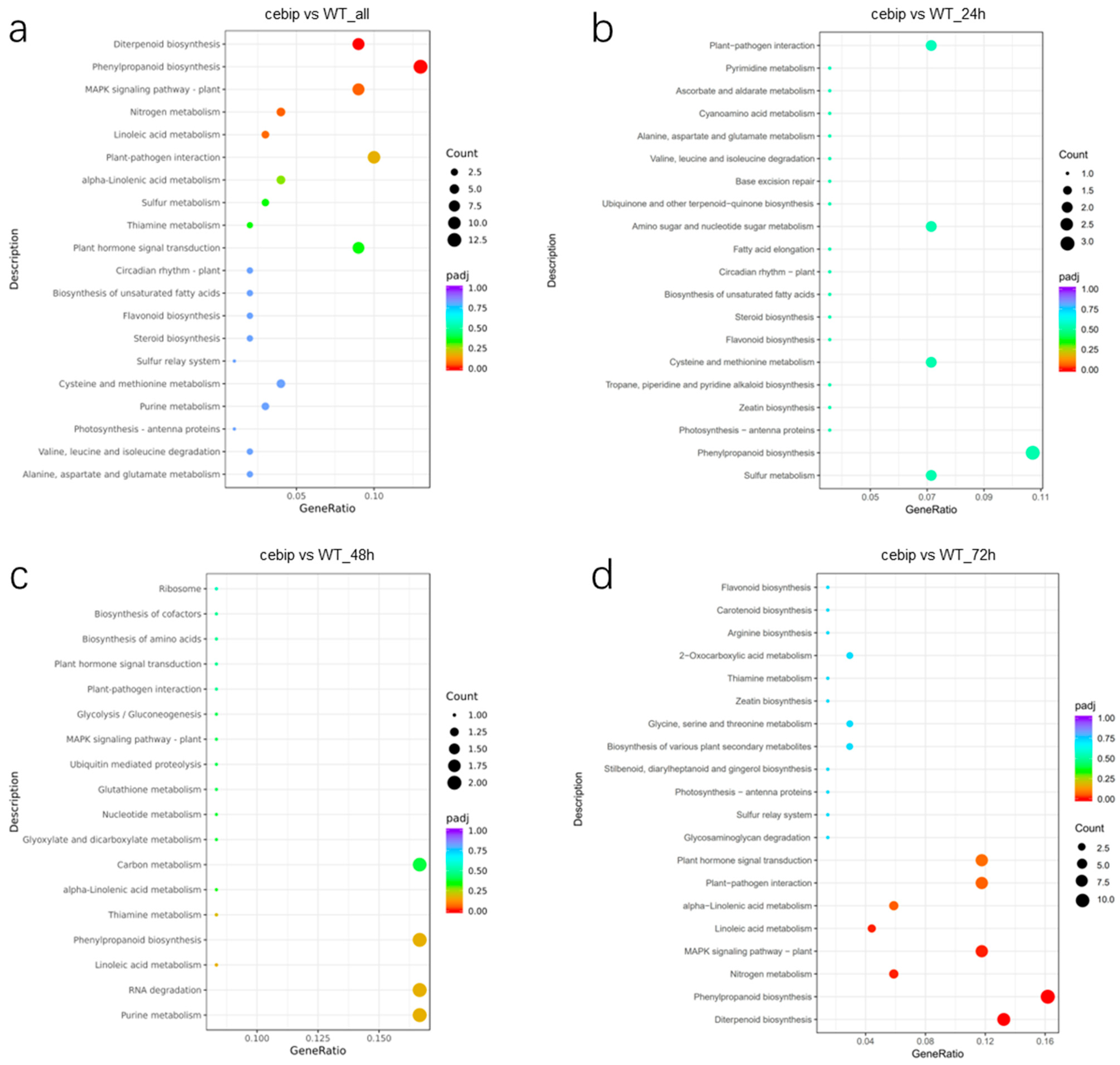
2.3. GO and KEGG Enrichment Analysis of Significantly Differentially Expressed Genes
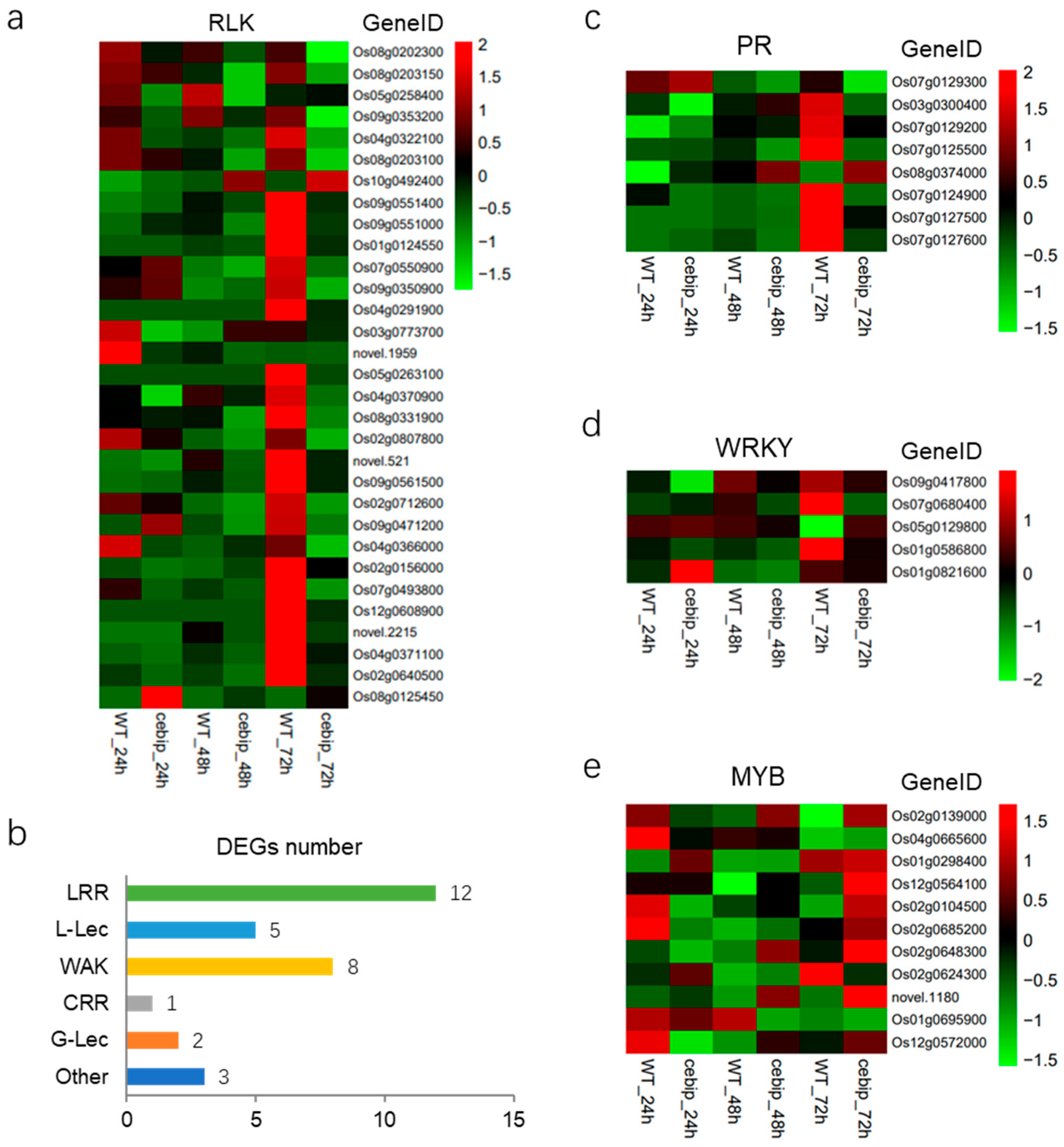
2.4. Defense-Related DEGs Analysis
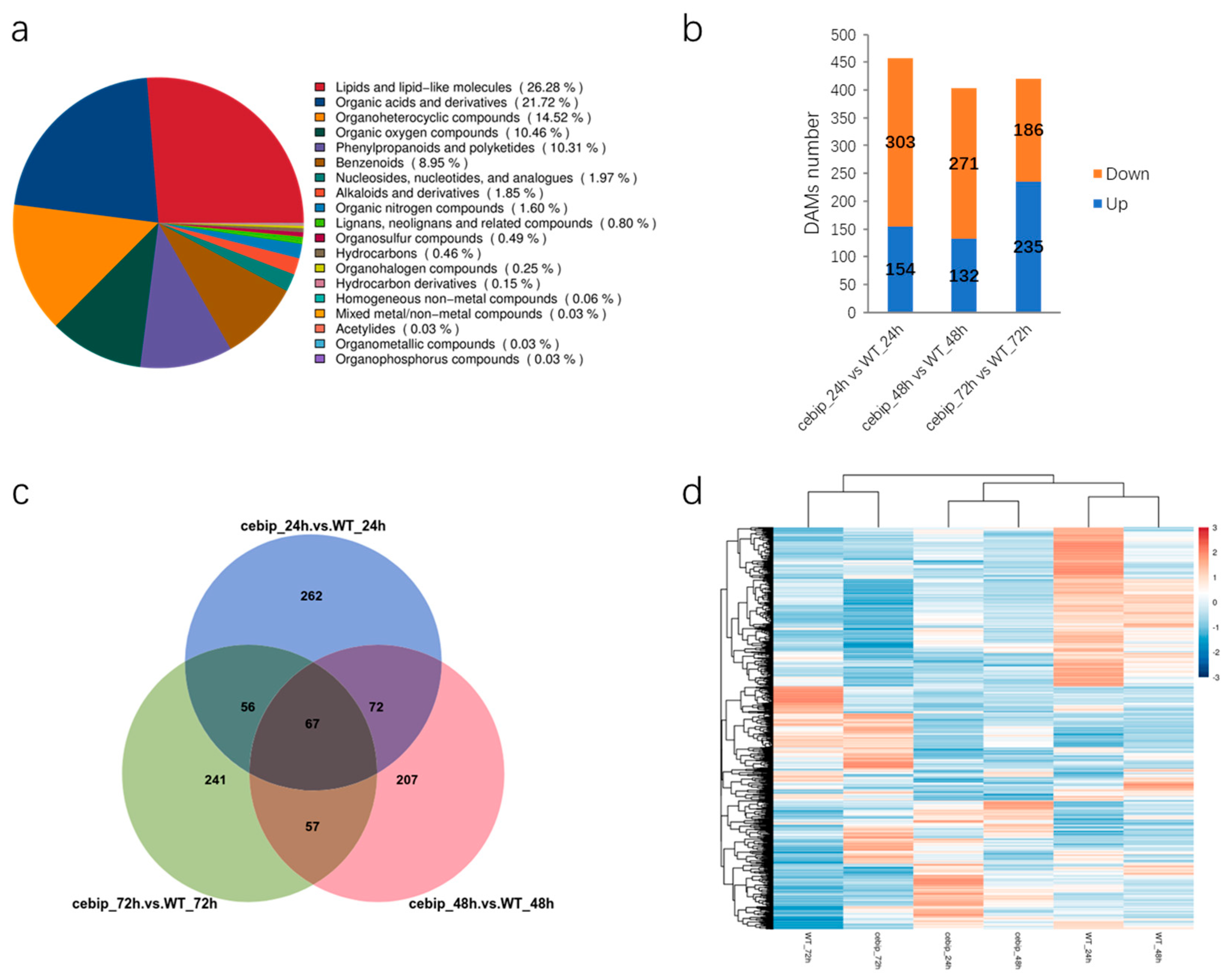
2.5. Differentially Accumulated Metabolites Analysis Reveals a Set of Metabolites Involved in CEBiP-Mediated Defense Response
2.6. KEGG Enrichment Analysis of Significantly Expressed DAMs
2.7. Correlation Analysis Between Transcriptomic and Metabonomic Data
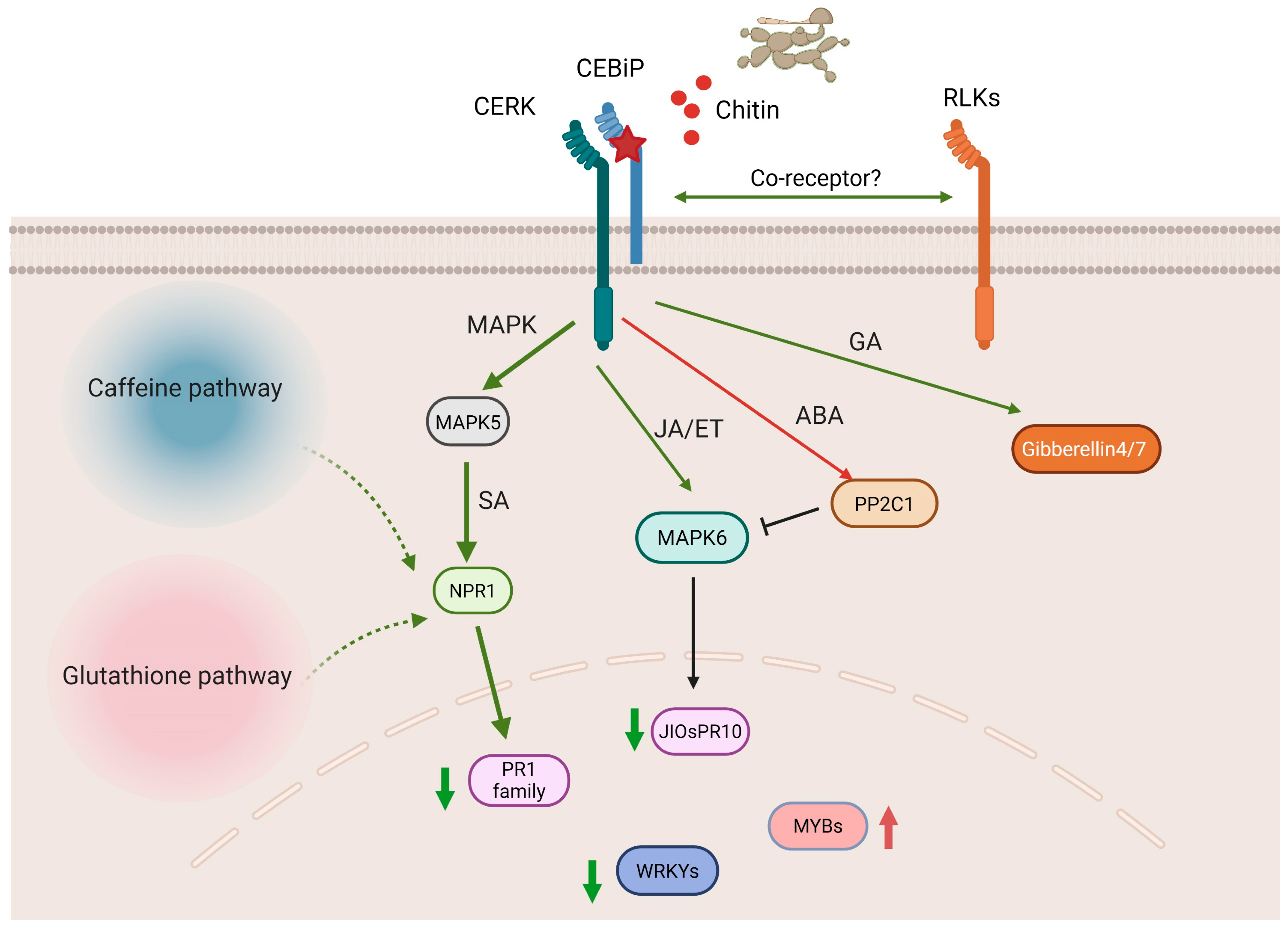
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Magnaporthe Oryzae Infection Assay
4.3. RNA Extraction, Library Preparation, and Sequencing
4.4. Transcriptome Analysis
4.5. Metabolite Extraction and MS Analysis
4.6. Data Process and Metabolomics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Talbot, N.J. On the Trail of a Cereal Killer: Exploring the Biology of Magnaporthe Grisea. Annu. Rev. Microbiol. 2003, 57, 177–202. [Google Scholar] [CrossRef]
- Couch, B.C.; Fudal, I.; Lebrun, M.H.; Tharreau, D.; Valent, B.; Van Kim, P.; Nottéghem, J.L.; Kohn, L.M. Origins of Host-Specific Populations of the Blast Pathogen Magnaporthe Oryzae in Crop Domestication with Subsequent Expansion of Pandemic Clones on Rice and Weeds of Rice. Genetics 2005, 170, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Saha, S.; Ray, D.P.; Bag, M.K. Effect of Different Active Fungicides Molecules on the Management of Rice Blast Disease. Int. J. Agric. Environ. Biotechnol. 2012, 5, 247–251. [Google Scholar]
- Danso Ofori, A.; Zheng, T.; Titriku, J.K.; Appiah, C.; Xiang, X.; Kandhro, A.G.; Ahmed, M.I.; Zheng, A. The Role of Genetic Resistance in Rice Disease Management. Int. J. Mol. Sci. 2025, 26, 956. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The Plant Immune System. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ngou, B.P.M.; Jones, J.D.G.; Ding, P. Plant Immune Networks. Trends Plant Sci. 2022, 27, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Plant Pattern-Recognition Receptors. Trends Immunol. 2014, 35, 345–351. [Google Scholar] [CrossRef]
- Ranf, S. Sensing of Molecular Patterns through Cell Surface Immune Receptors. Curr. Opin. Plant Biol. 2017, 38, 68–77. [Google Scholar] [CrossRef]
- van der Burgh, A.M.; Joosten, M.H.A.J. Plant Immunity: Thinking Outside and Inside the Box. Trends Plant Sci. 2019, 24, 587–601. [Google Scholar] [CrossRef]
- Felix, G.; Duran, J.D.; Volko, S.; Boller, T. Plants Have a Sensitive Perception System for the Most Conserved Domain of Bacterial Flagellin. Plant J. 1999, 18, 265–276. [Google Scholar] [CrossRef]
- Miya, A.; Albert, P.; Shinya, T.; Desaki, Y.; Ichimura, K.; Shirasu, K.; Narusaka, Y.; Kawakami, N.; Kaku, H.; Shibuya, N. CERK1, a LysM Receptor Kinase, Is Essential for Chitin Elicitor Signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 19613–19618. [Google Scholar] [CrossRef]
- DeFalco, T.A.; Zipfel, C. Molecular Mechanisms of Early Plant Pattern-Triggered Immune Signaling. Mol. Cell 2021, 81, 3449–3467. [Google Scholar] [CrossRef]
- Huang, W.R.H.; Joosten, M.H.A.J. Immune Signaling: Receptor-like Proteins Make the Difference. Trends Plant Sci. 2024, 30, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chern, M.; Yin, J.; Wang, J.; Chen, X. Recent Advances in Broad-Spectrum Resistance to the Rice Blast Disease. Curr. Opin. Plant Biol. 2019, 50, 114–120. [Google Scholar] [CrossRef]
- Liu, W.; Liu, J.; Ning, Y.; Ding, B.; Wang, X.; Wang, Z.; Wang, G.L. Recent Progress in Understanding PAMP-and Effector-Triggered Immunity against the Rice Blast Fungus Magnaporthe Oryzae. Mol. Plant 2013, 6, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Kaku, H.; Nishizawa, Y.; Ishii-minami, N.; Akimoto-tomiyama, C.; Dohmae, N.; Takio, K.; Minami, E.; Shibuya, N. Plant Cells Recognize Chitin Fragments for Defense Signaling through a Plasma Membrane Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 11086–11091. [Google Scholar] [CrossRef]
- Shibuya, N.; Minami, E. Oligosaccharide Signalling for Defence Responses in Plant. Physiol. Mol. Plant Pathol. 2001, 59, 223–233. [Google Scholar] [CrossRef]
- Saberi Riseh, R.; Gholizadeh Vazvani, M.; Vatankhah, M.; Kennedy, J.F. Chitin-Induced Disease Resistance in Plants: A review. Int. J. Biol. Macromol. 2024, 266, 131105. [Google Scholar] [CrossRef]
- Kishimoto, K.; Kouzai, Y.; Kaku, H.; Shibuya, N.; Minami, E.; Nishizawa, Y. Perception of the Chitin Oligosaccharides Contributes to Disease Resistance to Blast Fungus Magnaporthe Oryzae in Rice. Plant J. 2010, 64, 343–354. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakano, T.; Takamizawa, D.; Desaki, Y.; Ishii-Minami, N.; Nishizawa, Y.; Minami, E.; Okada, K.; Yamane, H.; Kaku, H.; et al. Two LysM Receptor Molecules, CEBiP and OsCERK1, Cooperatively Regulate Chitin Elicitor Signaling in Rice. Plant J. 2010, 64, 204–214. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, A. The Pathogenesis Related Class 10 proteins in Plant Defense against Biotic and Abiotic Stresses. Adv. Plants Agric. Res. 2016, 3, 00077. [Google Scholar] [CrossRef]
- Islam, M.M.; El-Sappah, A.H.; Ali, H.M.; Zandi, P.; Huang, Q.; Soaud, S.A.; Alazizi, E.M.Y.; Wafa, H.A.; Hossain, M.A.; Liang, Y. Pathogenesis-Related Proteins (PRs) Countering Environmental Stress in Plants: A Review. S. Afr. J. Bot. 2023, 160, 414–427. [Google Scholar] [CrossRef]
- Birkenbihl, R.P.; Liu, S.; Somssich, I.E. Transcriptional Events Defining Plant Immune Responses. Curr. Opin. Plant Biol. 2017, 38, 1–9. [Google Scholar] [CrossRef]
- Ng, D.W.K.; Abeysinghe, J.K.; Kamali, M. Regulating the Regulators: The Control of Transcription Factors in Plant Defense Signaling. Int. J. Mol. Sci. 2018, 19, 3737. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Somssich, I.E. Transcriptional Networks in Plant Immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.F.; Ao, Y.; Qu, J.; Li, Z.; Su, J.; Zhang, Y.; Liu, J.; Feng, D.; Qi, K.; et al. Lysin Motif-Containing Proteins LYP4 and LYP6 Play Dual Roles in Peptidoglycan and Chitin Perception in Rice Innate Immunity. Plant Cell 2012, 24, 3406–3419. [Google Scholar] [CrossRef]
- Ao, Y.; Li, Z.; Feng, D.; Xiong, F.; Liu, J.; Li, J.F.; Wang, M.; Wang, J.; Liu, B.; Wang, H. Bin OsCERK1 and OsRLCK176 Play Important Roles in Peptidoglycan and Chitin Signaling in Rice Innate Immunity. Plant J. 2014, 80, 1072–1084. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yamada, K.; Ishikawa, K.; Yoshimura, S.; Hayashi, N.; Uchihashi, K.; Ishihama, N.; Kishi-Kaboshi, M.; Takahashi, A.; Tsuge, S.; et al. A Receptor-like Cytoplasmic Kinase Targeted by a Plant Pathogen Effector Is Directly Phosphorylated by the Chitin Receptor and Mediates Rice Immunity. Cell Host Microbe 2013, 13, 347–357. [Google Scholar] [CrossRef]
- Li, Z.; Ao, Y.; Feng, D.; Liu, J.; Wang, J.; Wang, H.-B.; Liu, B. OsRLCK 57, OsRLCK107 and OsRLCK118 Positively Regulate Chitin- and PGN-Induced Immunity in Rice. Rice 2017, 10, 6. [Google Scholar] [CrossRef]
- Gong, B.Q.; Guo, J.; Zhang, N.; Yao, X.; Wang, H.-B.; Li, J.F. Cross-Microbial Protection via Priming a Conserved Immune Co-Receptor through Juxtamembrane Phosphorylation in Plants. Cell Host Microbe 2019, 26, 810–822.e7. [Google Scholar] [CrossRef]
- Li, B.; Ferreira, M.A.; Huang, M.; Camargos, L.F.; Yu, X.; Teixeira, R.M.; Carpinetti, P.A.; Mendes, G.C.; Gouveia-Mageste, B.C.; Liu, C.; et al. The Receptor-like Kinase NIK1 Targets FLS2/BAK1 Immune Complex and Inversely Modulates Antiviral and Antibacterial Immunity. Nat. Commun. 2019, 10, 4996. [Google Scholar] [CrossRef]
- Derksen, H.; Rampitsch, C.; Daayf, F. Signaling Cross-Talk in Plant Disease Resistance. Plant Sci. 2013, 207, 79–87. [Google Scholar] [CrossRef]
- Bürger, M.; Chory, J. Stressed Out About Hormones: How Plants Orchestrate Immunity. Cell Host Microbe 2019, 26, 163–172. [Google Scholar] [CrossRef]
- Mao, P.; Duan, M.; Wei, C.; Li, Y. WRKY62 Transcription Factor Acts Downstream of Cytosolic NPR1 and Negatively Regulates Jasmonate-Responsive Gene Expression. Plant Cell Physiol. 2007, 48, 833–842. [Google Scholar] [CrossRef]
- Jung, C.; Nguyen, N.H.; Cheong, J.J. Transcriptional Regulation of Protein Phosphatase 2c Genes to Modulate Abscisic Acid Signaling. Int. J. Mol. Sci. 2020, 21, 9517. [Google Scholar] [CrossRef]
- Schweighofer, A.; Kazanaviciute, V.; Scheikl, E.; Teige, M.; Doczi, R.; Hirt, H.; Schwanninger, M.; Kant, M.; Schuurink, R.; Mauch, F.; et al. The PP2C-Type Phosphatase AP2C1, Which Negatively Regulates MPK4 and MPK6, Modulates Innate Immunity, Jasmonic Acid, and Ethylene Levels in Arabidopsis. Plant Cell 2007, 19, 2213–2224. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y. MAP Kinase Cascades in Plant Development and Immune Signaling. EMBO Rep. 2022, 23, e53817. [Google Scholar] [CrossRef]
- Jwa, N.S.; Kumar Agrawal, G.; Rakwal, R.; Park, C.H.; Prasad Agrawal, V. Molecular Cloning and Characterization of a Novel Jasmonate Inducible Pathogenesis-Related Class 10 Protein Gene, JIOsPR10, from Rice (Oryza sativa L.) Seedling Leaves. Biochem. Biophys. Res. Commun. 2001, 286, 973–983. [Google Scholar] [CrossRef]
- Sinha, R.K.; Verma, S.S.; Rastogi, A. Role of Pathogen-Related Protein 10 (PR 10) under Abiotic and Biotic Stresses in Plants. Phyton 2020, 89, 167–182. [Google Scholar] [CrossRef]
- Hall, B.P.; Shakeel, S.N.; Schaller, G.E. Ethylene Receptors: Ethylene Perception and Signal Transduction. J. Plant Growth Regul. 2007, 26, 118–130. [Google Scholar] [CrossRef]
- Merchante, C.; Alonso, J.M.; Stepanova, A.N. Ethylene Signaling: Simple Ligand, Complex Regulation. Curr. Opin. Plant Biol. 2013, 16, 554–560. [Google Scholar] [CrossRef]
- Chakraborty, R.; Rehman, R.U.; Siddiqui, M.W.; Liu, H.; Seth, C.S. Phytohormones: Heart of Plants’ Signaling Network under Biotic, Abiotic, and Climate Change Stresses. Plant Physiol. Biochem. 2025, 223, 109839. [Google Scholar] [CrossRef]
- Monte, I. Jasmonates and Salicylic Acid: Evolution of Defense Hormones in Land Plants. Curr. Opin. Plant Biol. 2023, 76, 102470. [Google Scholar] [CrossRef]
- Ashihara, H.; Sano, H.; Crozier, A. Caffeine and Related Purine Alkaloids: Biosynthesis, Catabolism, Function and Genetic Engineering. Phytochemistry 2008, 69, 841–856. [Google Scholar] [CrossRef]
- Critchley, E.M.R. Caffeine and Related Methylxanthines: Possible Naturally Occurring Pesticides. Science 1970, 296, 1082–1083. [Google Scholar]
- Tsubouchi, H.; Terada, H.; Yamamoto, K.; Hisada, K.; Sakabe, Y. Caffeine Degradation and Increased Ochratoxin A Production by Toxigenic Strains of Aspergillus Ochraceus Isolated from Green Coffee Beans. Mycopathologia 1985, 90, 181–186. [Google Scholar] [CrossRef]
- Aneja, M.; Gianfagna, T. Induction and Accumulation of Caffeine in Young, Actively Growing Leaves of Cocoa (Theobroma Cacao L.) by Wounding or Infection with Crinipellis Perniciosa. Physiol. Mol. Plant Pathol. 2001, 59, 13–16. [Google Scholar] [CrossRef]
- Hollingsworth, R.G.; Armstrong, J.W.; Campbell, E. Caffeine as a Repellent for Slugs and Snails. Nature 2002, 417, 915–916. [Google Scholar] [CrossRef]
- Kim, Y.S.; Sano, H. Pathogen Resistance of Transgenic Tobacco Plants Producing Caffeine. Phytochemistry 2008, 69, 882–888. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, Y.E.; Sano, H. Plant Vaccination: Stimulation of Defense System by Caffeine Production in Planta. Plant Signal. Behav. 2010, 5, 489–493. [Google Scholar] [CrossRef]
- Park, J.C.; Yoo, Y.; Lim, H.; Yun, S.; Win, K.T.Y.S.; Kim, K.M.; Lee, G.S.; Cho, M.H.; Lee, T.H.; Sano, H.; et al. Intracellular Ca2+ Accumulation Triggered by Caffeine Provokes Resistance against a Broad Range of Biotic Stress in Rice. Plant Cell Environ. 2022, 45, 1049–1064. [Google Scholar] [CrossRef]
- Király, L.; Király, Z. To Die or Not to Die—Is Cell Death Dispensable for Resistance during the Plant Hypersensitive Response? Acta Phytopathol. Entomol. Hungarica 2006, 41, 11–21. [Google Scholar] [CrossRef]
- Gullner, G.; Zechmann, B.; Künstler, A.; Király, L. The Signaling Roles of Glutathione in Plant Disease Resistance. In Glutathione in Plant Growth, Development, and Stress Tolerance; Springer: Berlin/Heidelberg, Germany, 2017; pp. 331–357. [Google Scholar] [CrossRef]
- Ayub, A.; Rahayu, F.; Gacem, A.; Muzammil, K.; Yadav, K.K.; Antarlina, S.S.; Saidah, S.; Anggoro, G.W.; Sunarto, D.A.; Alqahtani, T.A.; et al. Glutathione and Biosensor Technologies: Enhancing Plant Resilience to Environmental Stressors. Physiol. Mol. Plant Pathol. 2025, 136, 102570. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEGs | Gene Name | Short Annotation | Plant–Pathogen Interaction (dosa04626) | MAPK Signaling Pathway (dosa04016) | Plant Hormone Signal Transduction (dosa04075) |
|---|---|---|---|---|---|
| Os03g0133400 | CEBiP | Chitin elicitor-binding protein | ✓ | ||
| Os01g0529800 | Os01g0529800 | 3-ketoacyl-CoA synthase 6 | ✓ | ||
| Os07g0129200 | PR1A | Pathogenesis-related protein PRB1–3 | ✓ | ✓ | ✓ |
| Os07g0125500 | Os07g0125500 | Pathogenesis-related protein PR-1 type | ✓ | ✓ | ✓ |
| Os07g0124900 | PR1#071 | Pathogenesis-related protein PR-1 type | ✓ | ✓ | ✓ |
| Os07g0127500 | PR1#072 | Pathogenesis-related protein PRB1–2 | ✓ | ✓ | ✓ |
| Os07g0127600 | PR1#073 | Pathogenesis-related protein 1 | ✓ | ✓ | ✓ |
| Os02g0504700 | Os02g0504700 | - | ✓ | ||
| Os06g0191500 | Os06g0191500 | Mitogen-activated protein kinase 5 | ✓ | ✓ | ✓ |
| Os07g0129300 | PR1 | Pathogenesis-related protein 1 | ✓ | ✓ | ✓ |
| Os05g0155200 | ERS2 | Probable ethylene response sensor 2 | ✓ | ✓ | |
| Os09g0325700 | PP2C1 | Probable protein phosphatase 2 | ✓ | ✓ |
| Pathway | DEGs | p-Value | DAMs | p-Value |
|---|---|---|---|---|
| Zeatin biosynthesis | Os10g0178500 | 0.2450 | 5′-Methylthioadenosine; O-beta-D-Glucosylzeatin | 0.1746 |
| Flavonoid biosynthesis | Os03g0819600 | 0.3625 | Naringenin chalcone; Homoeriodictyol | 0.2249 |
| Nitrogen metabolism | Os08g0470200/Os08g0468100/ Os08g0423600/Os08g0423500 | 0.0008 | L-Glutamic acid | 0.2418 |
| Sulfur metabolism | Os02g0167100 | 0.4465 | O-Succinyl-L-homoserine | 0.2418 |
| Arginine biosynthesis | Os03g0279400 | 0.3625 | L-Ornithine; L-Glutamic acid | 0.3275 |
| Starch and sucrose metabolism | Os01g0930800/Os02g0753000 | 0.6791 | 6-Phospho-beta-D-glucosyl-(1,4)-D-glucose | 0.3402 |
| Cysteine and methionine metabolism | Os05g0475400/Os02g0167100 | 0.4928 | 5′-Methylthioadenosine; Glutathione; O-Succinyl-L-homoserine | 0.4050 |
| Terpenoid backbone biosynthesis | Os07g0190000 | 0.5525 | 2-C-Methyl-D-erythritol 2,4-cyclodiphosphate | 0.5009 |
| Biosynthesis of unsaturated fatty acids | Os07g0417200 | 0.3714 | alpha-Linolenic acid | 0.5662 |
| Plant hormone signal transduction | Os07g0127600/Os07g0127500/ Os07g0129300/Os07g0125500/ Os06g0191500/Os07g0124900/ Os07g0129200/Os05g0155200 | 0.0125 | Gibberellin A4 | 0.5662 |
| Diterpenoid biosynthesis | Os11g0474800/Os04g0179200/ Os12g0491800/Os04g0178300/ Os04g0179700/Os04g0178400/ Os02g0570400/Os02g0569900/ Os06g0568600 | 2.22E-09 | Gibberellin A4; Gibberellin A7 | 0.6244 |
| 2-Oxocarboxylic acid metabolism | Os05g0475400/Os03g0279400 | 0.3376 | L-Ornithine; 3-(5′-Methylthio)pentylmalic acid; cis-(Homo)3-aconitate; Citraconic acid | 0.7603 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.; Bao, J.; Li, L.; Shen, Z.; Wang, J.; Daskalov, A.; Zhu, X.; Lin, F. Integrated Transcriptomic and Metabolomic Analysis Reveals the Regulation Network of CEBiP in Rice Defense Against Magnaporthe oryzae. Int. J. Mol. Sci. 2025, 26, 5194. https://doi.org/10.3390/ijms26115194
Zheng Q, Bao J, Li L, Shen Z, Wang J, Daskalov A, Zhu X, Lin F. Integrated Transcriptomic and Metabolomic Analysis Reveals the Regulation Network of CEBiP in Rice Defense Against Magnaporthe oryzae. International Journal of Molecular Sciences. 2025; 26(11):5194. https://doi.org/10.3390/ijms26115194
Chicago/Turabian StyleZheng, Qi, Jiandong Bao, Lin Li, Zifang Shen, Jiaoyu Wang, Asen Daskalov, Xueming Zhu, and Fucheng Lin. 2025. "Integrated Transcriptomic and Metabolomic Analysis Reveals the Regulation Network of CEBiP in Rice Defense Against Magnaporthe oryzae" International Journal of Molecular Sciences 26, no. 11: 5194. https://doi.org/10.3390/ijms26115194
APA StyleZheng, Q., Bao, J., Li, L., Shen, Z., Wang, J., Daskalov, A., Zhu, X., & Lin, F. (2025). Integrated Transcriptomic and Metabolomic Analysis Reveals the Regulation Network of CEBiP in Rice Defense Against Magnaporthe oryzae. International Journal of Molecular Sciences, 26(11), 5194. https://doi.org/10.3390/ijms26115194