1. Introduction
Circular RNA (circRNA) has emerged as a novel and versatile class of RNA molecules, distinguished by their covalently closed loop structure. This topology grants circRNA remarkable stability, as it lacks the 5′ and 3′ ends that make linear RNA susceptible to exonuclease-mediated degradation. Furthermore, the use of the internal ribosome entry site (IRES) in circRNAs allows them to be translated [
1], which has increased the attention they have received in various fields, including gene therapy [
2], vaccine development [
3,
4], and synthetic biology [
5]. Its ability to sustain prolonged protein expression makes circRNA an attractive alternative to traditional linear RNA [
6], particularly in therapeutic applications where long-lasting effects are desired without the risks associated with genomic integration.
The production of circRNA typically involves in vitro transcription (IVT) with a subsequent circularization and a complex purification, a process to separate circular from linear RNA species [
7]. The circularization process has recently evolve from the use of low effective T4 ligase-mediated reactions [
8] to the utilization of the highly efficient engineered permuted intron–exon (PIE) self-splicing strategy to promote the circularization of RNA molecules [
9,
10]. However, the efficient purification of circRNA from precursor, introns, and nicked RNA remains a significant challenge. Those RNA contaminants can compromise the immunity and functionality of circRNA [
11,
12], which is particularly problematic in applications that require high fidelity and minimal interference, such as in RNA vaccines and gene therapies [
13]. Traditional methods for RNA purification, such as gel electrophoresis [
5] and high-performance liquid chromatography (HPLC) [
9], have been used to isolate circRNA. Nevertheless, RNA yields from these methods are typically less than 1% of the input RNA [
2,
9], making large-scale circRNA production economically challenging.
The lipid nanoparticle (LNP) system is one of the more effective for in vivo delivery of circRNA-based therapies. LNPs are typically composed of four key components: ionizable lipids, phospholipids, cholesterol, and polyethylene glycol (PEG)-lipids. Among these, ionizable lipids are crucial for the efficient release of RNA cargo into the cytoplasm. These ionizable lipids acquire a positive charge in acidic environments, such as within endosomes, facilitating the release of RNA into the cytoplasm. Despite their pivotal role, current ionizable lipids present several limitations that hinder their broader application. Firstly, endosomal escape is highly inefficient, with only 1–2% of internalized LNPs successfully releasing their RNA cargo into the cytoplasm [
14]. This represents a major bottleneck in the intracellular delivery process, significantly limiting the therapeutic potency of RNA-based interventions [
15]. Additionally, the vast majority of ionizable lipid screenings have been conducted using mRNA as the model cargo, resulting in limited data on the performance and functionality of LNPs when encapsulating alternative RNA modalities, such as self-amplifying RNA or circRNA [
16]. Toxicity is another concern; some ionizable lipids have been associated with elevated inflammatory responses [
17]. Moreover, the limited number of clinically approved ionizable lipids, such as MC3, SM-102, and ALC-0315, poses challenges related to complex synthesis process [
18] and licensing restrictions and high costs, thereby complicating access and increasing the expense of RNA-based treatments [
19]. In this context, the development of novel lipid formulations that are accessible at an affordable cost is not only scientifically necessary but also critical from a public health and equity standpoint [
20].
Additionally to the lipid components, the physical and chemical stability of LNP formulations represent another significant challenge for their widespread application [
21]. Current RNA-LNP therapies, including the approved mRNA vaccines Comirnaty and Spikevax, require cryogenic preservation and transportation between −80 °C and −60 °C for Comirnaty and at −20 °C for Spikevax [
22]. The stringent storage requirements are primarily due to the intrinsic sensitivity of mRNA to environmental factors such as oxygen, enzymatic degradation, and fluctuations in pH. Additionally, complex interactions between the lipid components and degradation by-products from oxidation and hydrolysis further compromise the stability of mRNA. As water, oxygen, and lipids are included in mRNA-LNP formulations, ensuring high stability in liquid mRNA-LNP systems has historically presented some difficulties [
23].
Enhancing the stability of mRNA-LNP therapies has shown promise through lyophilization, a process that removes water by sublimation under vacuum at low temperatures [
24,
25]. One study reported successful lyophilization of mannose-modified LNPs containing SM-102 as the ionizable lipid, resulting in a durable immune response against rabies and SARS-CoV-2 viruses [
26]. While lyophilization has been primarily developed for mRNA-based LNPs, there is limited research on its application to circRNA-LNP formulations. Consequently, developing a circRNA-LNP platform that incorporates novel ionizable lipids capable of withstanding the lyophilization process will be crucial for making these therapies more accessible and practical for public health applications.
In this study, we introduce a complete circRNA platform designed to overcome the economic and industrial challenges associated with circRNA-LNP-based therapies. To address the critical issue of low purification yields, we propose an innovative purification method utilizing poly(A) affinity chromatography. This approach significantly enhances purification efficiency, achieving yields comparable to those of mRNA, thus rendering the process feasible for industrial-scale production. Furthermore, we address the challenge of ionizable lipid selection, a key component in LNP formulations, by exploring novel lipid candidates’ performance with circRNA [
27,
28]. Finally, we integrate a lyophilization technique tailored to circRNA-LNP formulations [
29], ensuring the practicality and accessibility of these therapies for widespread health applications.
3. Discussion
Circular RNA has gained significant attention in therapeutic applications due to its exceptional stability and resistance to exonucleases. However, achieving efficient and scalable purification remains a considerable challenge. Traditional methods, such as PAGE electrophoresis and RNase R digestion, although effective, have significant limitations in terms of throughput, scalability, and the ability to achieve high purity [
36]. PAGE, commonly employed for RNA separation based on size, conformation, and total molecular charge, provides high resolution but is labor-intensive and prone to contamination, which significantly reduces its practicality for large-scale operations [
37]. Likewise, RNase R digestion, which selectively degrades linear RNA while preserving circRNA, often leaves structured linear contaminants and proves cost-prohibitive for large-scale purification. Consequently, while these methods are suitable for initial characterization, they fall short in terms of scalability and widespread industrial application [
38].
High-performance liquid chromatography has also been explored for its potential to remove impurities such as dsRNA [
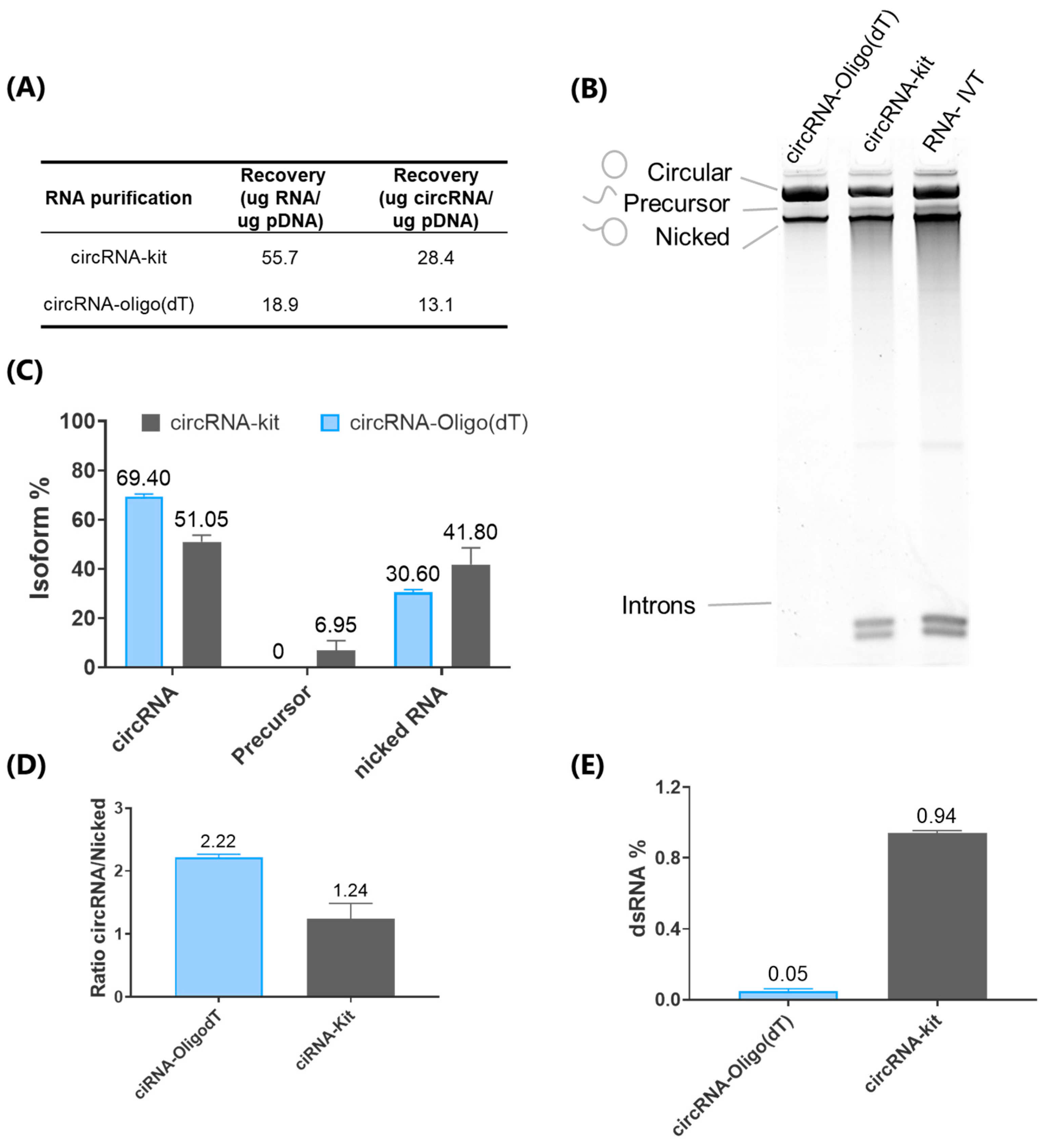
9]. However, the HPLC technique reported for circRNA suffer from low recovery yields, as only a small fraction of the total RNA is collected, thus making the method challenging to scale for industrial use [
9]. To overcome this scale restrictions, we have developed an innovative affinity chromatography method that for the first time employs Oligo (dT) columns to purify circRNA for therapy purposes. Additionally, we have eliminated the need for RNase R digestion, which provides a cost-effective, scalable alternative to HPLC.
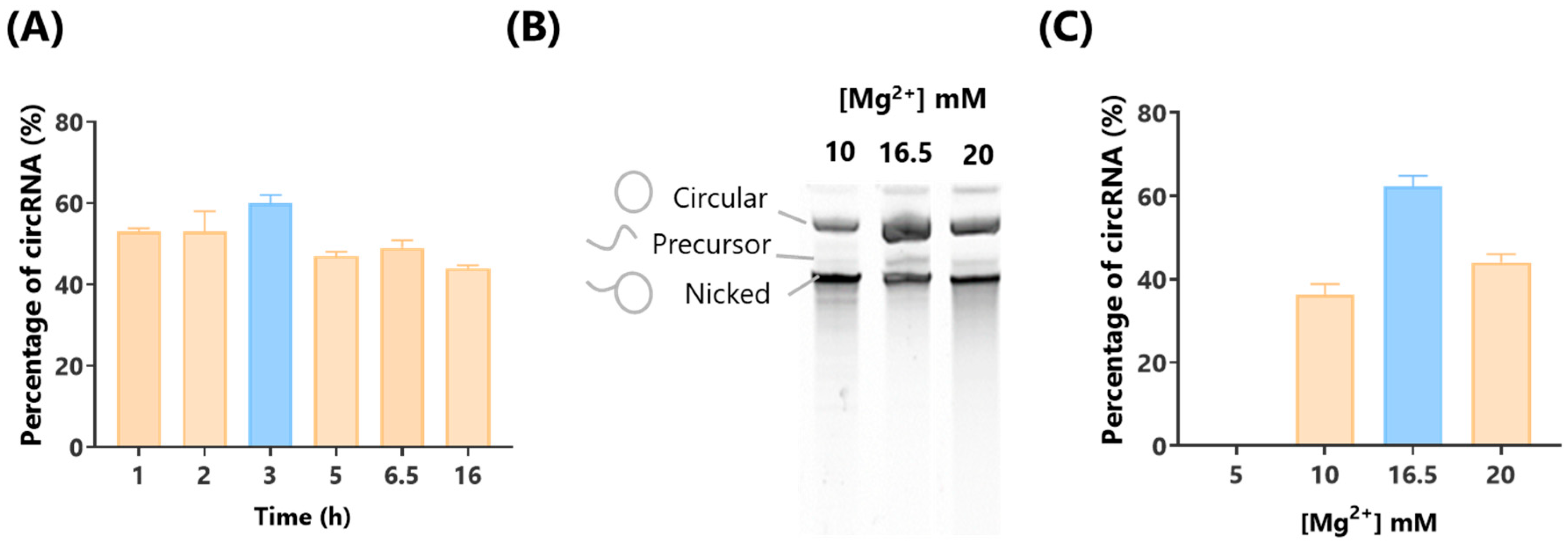
In relation to Mg
2+ concentration during the IVT process, previous reports largely rely on commercial kits where Mg
2+ concentrations are undisclosed, making it impossible to modify or optimize [
9,
11,
13]. In this study, we demonstrated that by adjusting the Mg
2+ concentration, we can significantly increase the proportion of circRNA produced, providing an important optimization strategy for circRNA synthesis.
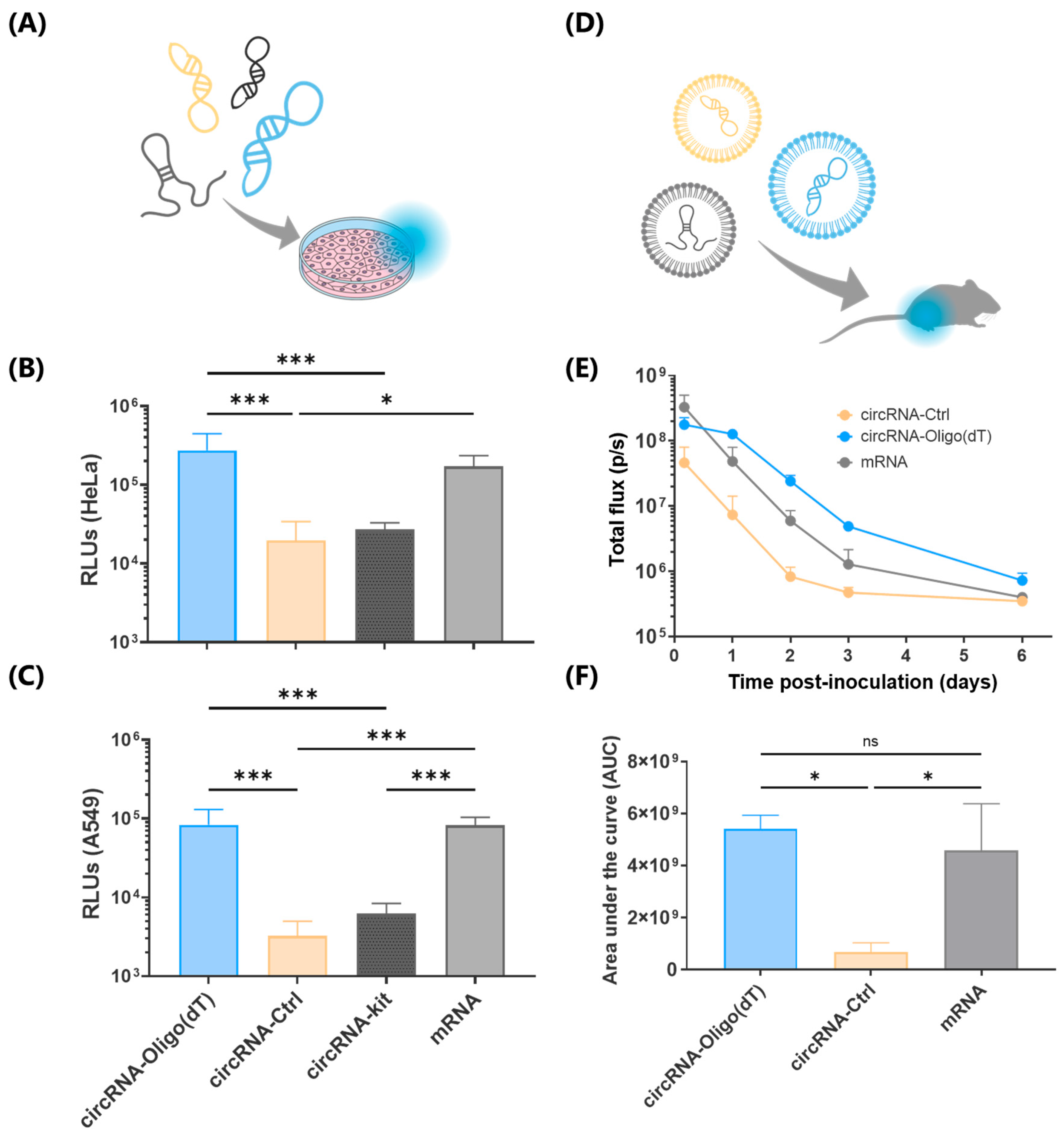
In terms of in vivo delivery of circRNA, only a limited number of studies have reported successful strategies. Some approaches have employed cationic polymers such as in vivo-jetPEI [
11] or O6-stat-N6 CARTs, [
3,
6] while others have focused on LNPs using ionizable lipids like cKK-E12 [
11], MC3 [
2,
39], SM-102, and ALC-0315 [
38]. Nonetheless, several studies that utilize LNPs do not specify the ionizable lipid used [
25]. Here, we report the best-performing ionizable lipid described to date, which demonstrated a remarkable 92-fold increase in in vivo protein expression at 6 days post-injection compared to SM-102. Moreover, while previous studies typically employed 5 to 20 μg of RNA for in vivo delivery [
2,
3,
4,
26,
39], we achieved robust protein expression with just 1 μg of circRNA. This demonstrates the exceptional efficiency of our novel lipid formulations and their potential to significantly reduce the required therapeutic circRNA dosage.
A critical factor in evaluating circRNA for clinical applications is the duration of gene expression, as circRNA is designed to provide more sustained and stable expression compared to mRNA. To our knowledge, the longest previously reported in vivo signal duration is 42 h using LNPs [
13] and 168 h (7 days) using O6-stat-N6 CARTs [
6]. In this study, we report an extended signal duration of 336 h (14 days), doubling the 7-day expression window reported in the literature [
6]. This prolonged expression suggests that the combination of Oligo (dT) purification and novel ionizable lipids provides superior performance over existing methods, making this approach particularly promising for long-term therapeutic applications.
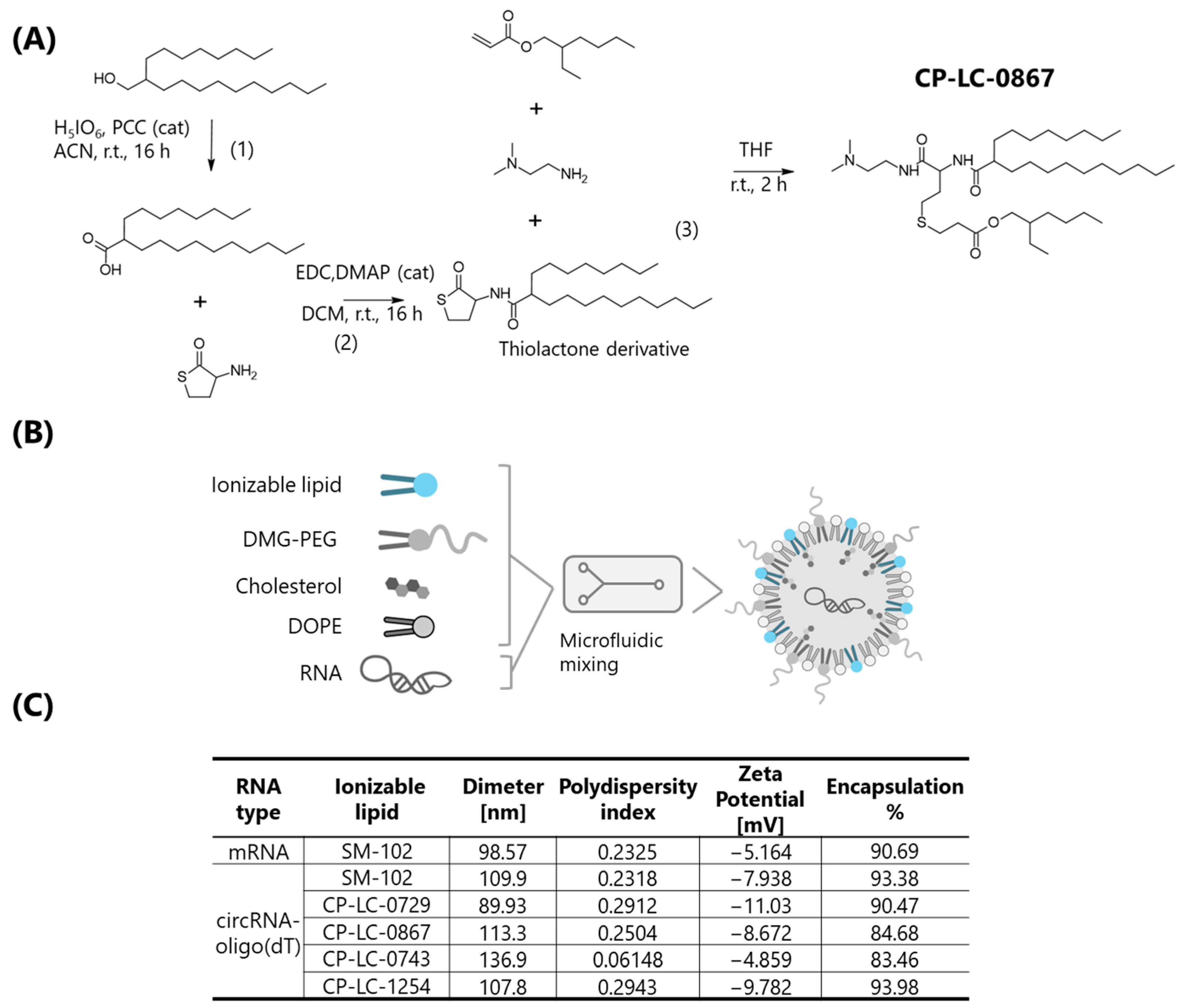
Among the ionizable lipids evaluated in this study, CP-LC-0867 and CP-LC-0729 demonstrated not only superior in vivo efficacy but also notable advantages in terms of synthetic accessibility and scalability. The previously described synthesis of CP-LC-0729 involves just two steps, with the final step carried out as a straightforward one-pot reaction [
27]. Similarly, CP-LC-0867, although requiring three synthetic steps, is produced under mild conditions without the need for stringent parameters [
28]. In contrast, clinically established ionizable lipids such as SM-102 [
40] and ALC-0315 [
41] require four and three synthetic steps, respectively, and typically involve more demanding conditions, including high temperatures, reactions more prone to byproduct formation, and strictly anhydrous environments. These features highlight the practical advantages of CP-LC-0867 and CP-LC-0729 for large-scale and cost-effective manufacturing.
Lyophilization has been extensively studied to enhance the stability of mRNA vaccines [
29], but its application to circRNA-LNPs remains largely unexplored, with only one prior study reported to date [
26]. However, this study does not specify the ionizable lipid used and focuses on mannose-modified PEG for circRNA-LNP delivery to lymphocytes, without evaluating the impact of lyophilization on LNPs containing conventional, unmodified PEG. To our knowledge, our study provides the first demonstration of successful lyophilization of circRNA-LNPs using novel ionizable lipids [
42] and standard PEGylated formulations. By evaluating both the physical integrity and functional preservation of the LNPs post-lyophilization, we offer a more comprehensive and translationally relevant analysis of this strategy’s potential to support broader storage and distribution of circRNA therapeutics.
Despite the promising results, our study has some limitations that warrant further investigation. First, while we have demonstrated enhanced circRNA yield and prolonged in vivo expression using Oligo (dT) purification and novel ionizable lipids, the evaluation was based on intramuscular injection. This route of administration is widely used for vaccines and other therapeutic applications, providing valuable insights into the platform’s effectiveness. However, it may not fully capture how the system would perform with alternative delivery routes, such as intravenous or subcutaneous injections. Exploring these routes in future studies will offer a broader understanding of the platform’s therapeutic potential and expand its applicability across various clinical scenarios. Additionally, we successfully lyophilized circRNA-LNPs, preserving their functionality, and developed an optimized and streamlined process for generating high-quality circRNA. Together, these advancements mark a qualitative breakthrough, setting the stage for more efficient and scalable circRNA therapeutic development. Looking ahead, we aim to explore the long-term stability of lyophilized circRNA-LNPs, building on our previous success with mRNA-LNPs stored for over a year at 4 °C [
29], which included the use of CP-LC-0867 and other top-performing lipids. In addition, we will delve deeper into our extensive library of over 1500 ionizable lipids to identify even more effective candidates for circRNA delivery [
42]. Furthermore, a thorough evaluation of the safety profile of our novel ionizable lipids will be essential in future studies to determine how they compare with clinically established standards.
In summary, our study provides an important step forward in the scalable production and in vivo delivery of circular RNA (circRNA), utilizing optimized purification strategies and novel ionizable lipids. We demonstrated that the use of Oligo (dT) affinity chromatography enhances the yield and purity of circRNA, allowing for efficient downstream applications without the need for additional enzymatic treatments. Furthermore, we successfully encapsulated circRNA into LNPs formulated with promising ionizable lipids, including CP-LC-0867 and CP-LC-0729, which resulted in superior protein expression profiles compared to conventional lipids such as SM-102. Additionally, our exploration of lyophilization revealed that lyophilized circRNA-LNP formulations retain their functionality, featuring their potential for long-term storage and global distribution. These results demonstrate the substantial potential of this platform to advance the clinical translation of circRNA therapies by enhancing production efficiency, stability, and delivery, which are critical factors for achieving reliable and scalable therapeutic outcomes.
4. Materials and Methods
4.1. Template Plasmid Construction for circRNA
The DNA template for circRNA synthesis was constructed by cloning the Anabaena 3.0 ribozyme, the internal ribosome entry site IRES-CVB3, both as previously described [
9], and a firefly luciferase coding sequence optimized for human expression into a pUC-based plasmid under the control of a T7 promoter. Gene synthesis, cloning, and plasmid preparations were outsourced to Genscript (Piscataway, NJ, USA).
Subsequently, the plasmid was linearized with the BspQI restriction enzyme (Hongene, Shanghai, China) at 50 °C for 2 h. The linearized plasmid was then purified using the Wizard® SV gel and PCR clean-up system kit (Promega, Madison, WI, USA) following the manufacturer’s instructions.
4.2. Synthesis of RNA and Enrichment of circRNA
Modified linear mRNA and unmodified circRNA were synthesized by in vitro transcription (IVT) using T7 RNA polymerase (Hongene). The transcription reaction was set up with 1 µg of linearized plasmid DNA, 100 U of T7 RNA polymerase, 5 mM rNTPs, and 1× transcription buffer in a total volume of 20 µL. IVT reaction was incubated at 37 °C for 3 h. Following the IVT reaction, the DNA template was digested with 4 U of DNase I (Hongene) at 37 °C for 15 min.
For circRNA synthesis optimization, we systematically varied the IVT incubation times (1, 2, 3, 5, 7, or 16 h) and adjusted the concentration of Mg2+ in the transcription buffer to 5, 10, 16.5, or 20 mM.
To enrich the circRNA, the IVT product was denatured at 70° C for 5 min and then immediately placed on ice for 3 min. Subsequently, GTP was added to a final concentration of 2 mM, and the mixture was incubated at 55 °C for 15 min. The reaction was then quenched by the addition of 5 mM EDTA at pH 8 and storage for its subsequent purification.
The circRNA used in this study has an approximate molecular weight of 829.1 kDa, corresponding to 2573 nucleotides. The control circular RNA (circRNA-Ctrl) used in this study was obtained from GenScript (SC8888), where it was purified via high-performance liquid chromatography (HPLC) [
35]. This control circRNA contains the same elements as the experimental circRNA constructs produced in our laboratory, ensuring consistency in evaluating the effects of various conditions on circRNA functionality and stability [
35].
4.3. Purification and Identification of circRNA
Resulting RNA transcripts were purified by affinity chromatography using POROS Oligo (dT) 25 (ThermoFisher, Bedford, MA, USA). The columns were equilibrated with 5 column volumes of binding buffer (50 mM sodium phosphate, 0.5 M NaCl, 5 mM EDTA, pH = 7.0) before use. The RNA solution was applied to the equilibrated Oligo dT column. The column was washed with 10 column volumes of wash buffer (50 mM sodium phosphate, 5 mM EDTA, pH = 7.0) and was eluted by applying 2 column volumes of double-deionized water. The eluate was collected and immediately placed on ice. A representative chromatogram is presented in
Figure S2, illustrating the separation profile achieved during the purification process.
Subsequently, a polishing treatment of the RNAs was performed to enrich the circular isoform relative to the other species. Resulting RNA fractions were visualized on a 4% polyacrylamide gel (PAGE) containing 8 M urea and analyzed using the FIJI software (version Image J2).
The circRNA-Kit was purified using the Monarch® Spin RNA Cleanup Kit (NEB, Ipswich, MA, USA) (50 μg), following the manufacturer’s instructions for optimal RNA recovery.
The quality and size of the resulting RNA samples were evaluated using a Bioanalyzer Agilent 2100 (Agilent Technologies, Waldbronn, Germany), with the Agilent RNA 6000 Nano kit. The resulting data were processed and evaluated with 2100 Expert Software (Version: B.02.08.SI648 (SR2)) RNA quality was further evaluated using high-performance liquid chromatography (HPLC). Samples were processed through a CIMac™ SDVB 0.1 mL (BiA Separations, Ajdouscina, Slovenia) analytical reverse-phase column (2 µm channels, Sartorius) connected to an HPLC WATERS e2695. CIMac SDVB column was equilibrated at 65 °C in mobile phase A (50 mM TEAA and 7.5% ACN, pH 7.0). Approximately 0.4 µg RNA was injected on the column. After an initial equilibration step (1 min at 100% MPA), a linear gradient was performed, 0–100% B (50 mM TEAA in 18% CAN pH = 7.0) in 27 min at a flow rate 1 mL min–1. The column was eluted with 100% MPC (50 mM TEAA in 75% CAN pH = 8.2) for 1 min before re-equilibration in 100% MPA.
4.4. Determination of dsRNA Content
The circRNAs were spotted onto positively charged nylon membranes (Nytran SC, Cytiva, München, Germany). The membranes were then blocked with 5% (w/v) non-fat dried milk in TBS-T buffer (20 mM Tris–HCl, 150 mM NaCl, 0.1% [v/v] Tween-20, pH 7.4), and incubated with dsRNA-specific mAb J2 (Jena Bioscience, Jena, Germany) at 4 °C overnight. Membranes were washed with TBS-T buffer and incubated with HRP-conjugated goat anti-mouse IgG (Abcam, Cambridge, UK). Following additional wash with TBS-T, the membranes were treated with ECL Plus Western blot detection reagent (Amersham, Chicago, IL, USA). Images were captured using an iBright 750 digital imaging system.
4.5. Synthesis and Characterization of Ionizable Lipids
The ionizable lipids CP-LC-0743, CP-LC-0729, and CP-LC-1254 were synthesized following established protocols based on the Sequential Thiolactone Amine Acrylate Reaction [
27,
42]. In brief, thiolactone derivatives (0.15 mmoL, 1 equiv.) and acrylate (0.15 mmoL, 1 equiv.) were dissolved in 300 μL of tetrahydrofuran (THF) at room temperature (RT), followed by the addition of the amine (0.15 mmoL, 1 equiv). After two hours, the THF was removed under vacuum. Notably, CP-LC-0743 was previously referred to as A4B2C1, and CP-LC-1254 was previously designated as CP-LC-0729-04 [
27].
The synthesis of CP-LC-0867 was carried out as follows: 2-Octyldecanoic acid: 2-octyl-1-dodecanol (1077 mg, 3.5 mmoL) and periodic acid (4144 mg, 18 mmoL) were dissolved in acetonitrile (28 mL). Then, pyridinium chlorochromate (23 mg, 0.030 mmoL) was added to the previous solution. The reaction mixture was stirred 16 h at room temperature. Then, the solvent was evaporated under reduced pressure. Subsequently, the crude was dissolved in AcOEt and first washed twice with water and finally with brine. The organic phase was dried with anhydrous MgSO4, filtered, and evaporated under reduced pressure. The residue was used in the next step without any further purification.
Thiolactone derivative (2-octyl-N-(2-oxotetrahydrothiophen-3-yl) dodecanamide): DL-homocysteine thiolactone hydrochloride (1.8 mmoL) was dissolved in 5 mL of anhydrous dichloromethane at room temperature. Then, triethylamine (1.8 mmoL) was added followed by EDC hydrochloride (1.3 mmoL), 4-(dimethylamino) pyridine (0.20 mmoL), and 2-octyldecanoic acid (1.0 mmoL). The reaction mixture was stirred at room temperature overnight under an argon atmosphere. The organic layer was dried with anhydrous MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by flash chromatography (gradient of hexane/ethyl acetate: 100/0 to 0/100) to afford as a pure product.
Thiolactone derivative [
27] (0.15 mmoL, 1 equiv.) and 2-ethylhexyl acrylate (0.15 mmoL, 1 equiv.) were dissolved in 300 μL of THF at RT, followed by the addition of N,N-dimethylethylenediamine (0.15 mmoL, 1 equiv.). After two hours, the THF was removed under vacuum, and CP-LC-0867 was purified using a CombiFlash NextGen 300+ with gradient elution, starting from 100% dichloromethane to 50% of an 80/20/1 mixture of DCM/MeOH/NH
4OH (aq).
All synthesized lipids were analyzed by high-performance liquid chromatography (HPLC) equipped with a charged aerosol detector (CAD) and characterized by mass spectrometry (ISQ ThermoFisher, Waltham, MA, USA). The structures of the synthesized lipids are available in
Figure S4.
1H-RMN data are shown in
Figures S8 and S9 and summarized below:
CP-LC-0867: MS-QDa: theoretical [M + H]+ = 684.57, experimental [M + H]+ = 684.58; 1H NMR (500 MHz, CDCl3) δ 6.63 (t, J = 5.1 Hz, 1H), 6.25 (d, J = 7.9 Hz, 1H), 4.59 (dt, J= 7.2 Hz J = 7.2 Hz, 1H), 4.07–3.96 (m, 2H), 3.32 (dt, J = 6.4, 5.5 Hz, 2H), 2.80 (t, J = 7.3 Hz, 2H), 2.67–2.49 (m, 4H), 2.40 (t, J = 6.1 Hz, 2H), 2.22 (s, 6H), 2.11–1.99 (m, 2H), 1.94 (m, 1H), 1.57 (m, 3H), 1.45–1.32 (m, 4H), 1.25 (m, 34H), 0.88 (m, 12H).
CP-LC-0743: MS-QDa: theoretical [M + H]+ = 766.25, experimental [M + H]+ = 766.81; 1H NMR (400 MHz, CDCl3) δ 6.64 (t, J = 4.8 Hz, 1H), 6.26 (d, J = 8.0 Hz, 1H), 5.39–5.29 (m, 2H), 4.59 (dt, J = 7.2 Hz J = 7.2 Hz, 1H), 4.08 (t, J = 6.8 Hz, 2H), 3.32 (dt, J = 5.9 Hz J = 5.9 Hz, 2H), 2.79 (t, J = 7.2 Hz, 2H), 2.67–2.48 (m, 4H), 2.40 (t, J = 6.1 Hz, 2H), 2.22 (s, 6H), 2.11–1.88 (m, 7H), 1.59 (m, 4H), 1.46–1.15 (m, 44H), 0.87 (m, 9H).
CP-LC-0729: MS-QDa: theoretical [M + H]+ = 740.63, experimental [M + H]+ = 740.85; 1H NMR (400 MHz, CDCl3) δ: 6.76 (m, 1H); 6.37 (d, J = 7.9 Hz, 1H); 4.60 (dt, J = 7.1 Hz J = 7.1 Hz, 1H); 3.98 (d, J = 5.8 Hz, 2H); 3.36 (dt, J = 5.7 Hz J = 5.7 Hz, 2H); 2.78 (t, J = 6.8 Hz, 2H); 2.59 (m, 4H); 2.48 (t, J = 5.9 Hz, 2H); 2.28 (s, 6H); 2.06 (m, 2H); 1.96 (m, 1H); 1.58 (m, 3H); 1.41 (m, 2H); 1.25 (m, 44H); 0.87 (m, 12H).
CP-LC-1254: MS-QDa: theoretical [M + H]+ = 755.63, experimental [M + H]+ = 755.83; 1H NMR (400 MHz, CDCl3) δ: 6.79 (m, 1H); 4.05 (d, J = 5.8 Hz, 2H); 4.01 (d, J = 5.8 Hz, 2H); 3.45 (t, J = 7.1 Hz, 1H); 3.36 (m, 2H); 2.79 (t, J = 7.6 Hz, 2H); 2.69–2.49 (m, 4H); 2.44 (t, J = 6.0 Hz, 2H); 2.26 (s, 6H); 2.21 (m, 2H); 1.65 (m, 2H); 1.29 (m, 48H); 0.90 (m, 12H).
4.6. Lipid Nanoparticle (LNP) Formulation and Characterization
The synthesis of LPNs was performed using a microfluidic method. An ethanolic lipid solution containing the synthetized CP-LC ionizable lipids or the commercial SM-102 (BocSCI, Shirley, NY, USA), DOPE (Corden Pharma, Plankstadt, Germany), cholesterol (Merk, Rahway, NJ, USA), and DMG-PEG2000 (Cayman, Ann Arbor, MI, USA) at a molar ratio of 50:10:38.5:1.5 was prepared. This lipid mixture was then combined with an aqueous phase containing mRNA or circRNA diluted in 10 mM citrate buffer (pH 4), achieving an ionizable lipid/RNA weight ratio of 10:1. LNPs were formulated using an INanoTM microfluidic device set to a total flow rate of 12 mL/min with an aqueous-to-ethanol volume ratio of 3:1. After LNP formation, ethanol was removed by dialysis (Pur-A-Lyzer™ Midi Dialysis Kit, Sigma-Aldrich, St. Louis, MO, USA).
For LNP characterization the average size, polydispersity (PDI), and zeta potential of LNPs were determined using a Malvern Zetasizer Advance Lab Blue Label (Malvern Instruments Ltd., Malvern, UK). Concentration of encapsulated RNA (circRNA or mRNA) was then quantified using the Quant-IT® Ribogreen assay (Invitrogen, Waltham, MA, USA) following the manufacturer’s instructions, and LNP encapsulation efficiency were verified by agarose gel electrophoresis.
The final LNP formulation was adjusted to an RNA (circRNA or mRNA) concentration of 100 μg/mL and was subsequently lyophilized and/or stored at 4 °C for subsequent in vivo and in vitro analysis.
4.7. LNP Lyophilization
Lyophilization was performed following previously reported protocols, [
29] using a Virtis Genesis Pilot Freeze Dryer (SP, Warminster, PA, USA). In brief, LNPs were lyophilized with 20% maltose as a lyoprotectant in TRIS buffer. The process included three stages: (1) freezing at −50 °C, (2) primary drying at −10 °C under 180 mTorr pressure, and (3) secondary drying at 40 °C, gradually reducing the pressure to 120 mTorr and finally to 50 mTorr. Upon completion, the vials were backfilled with pure nitrogen to maintain an inert atmosphere, sealed, and stored at various temperatures for stability assessments. For reconstitution, 300 μL of RNase-free water was added to each vial, and the contents were gently swirled until a homogenous suspension was obtained.
4.8. Cell Culture and circRNA Transfection
The HeLa (DSMZ GmbH, Braunschweig, Germany), HEK293T (ATCC; American Type Culture Collection, Manassas, VA, USA).) and A549 (NIBSC; (National Institute for Biological Standards and Control, South Mimms, Hertfordshire, UK).) cell lines were maintained in DMEM with high glucose (Merk, Darmstadt, Germany), supplemented with 10% Fetal Bovine Serum (Sigma, Darmstadt, Germany), 1% Penicillin-Streptomycin Solution (GibcoTM, Thermo Fisher Scientific, Waltham, MA, USA) and 2 mM Glutamax (Thermo Fisher Scientific, Waltham, MA, USA). All cell lines were grown in 175 cm2 flasks.
The day before transfection, cells were detached from culture flasks using trypsinization and subsequently seeded into 96-well plates at a density of 1 × 104 cells per well. Immediately before the transfections, the culture media in each well was replaced with 90 μL of fresh media. For RNA (circRNA or mRNA) transfection using a cationic lipid-based transfection reagent a mixture containing RNA and Lipofectamine MessengerMAX™ (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) was pre-incubated in OptiMEM (GibcoTM, Thermo Fisher Scientific, Waltham, MA, USA) media according to the manufacturer’s instructions. This RNA-lipofectamine complex was then added to the designated wells in triplicate, achieving a final RNA concentration of 100 ng/well.
For RNA-LNP transfection a suspension of RNA-containing LNPs was added to the designated wells in triplicate to achieve a final RNA concentration of 200,100 and 50 ng/well. The cells were then incubated for 24 h at 37 °C in a 5% CO2 atmosphere to allow translation of the delivered RNA into luciferase protein.
4.9. Luciferase Activity Quantification In Vitro
At 24 or 48 h post-transfection, the cells were lysed by adding 100 μL of PBS containing 0.1% Triton X-100 to each well and incubating for 10 min at room temperature. This lysis step allows the release of luciferase protein translated from the delivered circRNA or mRNA. Following lysis, 98 μL of the cell lysate was transferred to an opaque 96-well white plate. To each well, 100 μL of a buffered d-Luciferin solution (GoldBio, Olivette, MO, USA) containing 100 mM Tris-HCl (pH 7.8), 5 mM MgCl2, 250 μM CoA, and 150 μM ATP was added, resulting in a final d-Luciferin concentration of 150 μg/mL. Luminescence was measured after a 5 min incubation at room temperature using a FLUOstar Omega (version 5.70) plate reader (BMG LABTECH, Ortenberg, Germany). Cells that had not been treated with any mRNA were used as a negative control to assess background luminescence.
4.10. Administration of LNP-circRNAs in Mice
Female BALB/c mice (Janvier), aged 8–10 weeks and weighing 18–23 g, were acclimatized to the experimental facilities for 3–7 days upon arrival. The mice were housed under controlled conditions, maintaining a room temperature of 20–24 °C, humidity levels between 50 and 70%, and a light intensity of 60 lux with a 12 h light–dark cycle.
For in vivo measurement of Firefly Luciferase activity, LNPs containing 1 μg of the specified RNA in a final volume of 30 μL. were administered via intramuscular injection. At the designated post-injection times, mice were anesthetized with 4% isoflurane administered via a vaporizer for induction, and anesthesia was maintained at 1.5% isoflurane. Following anesthesia, D-luciferin (Quimigen, Madrid, Spain) was administered intraperitoneally at a dose of 150 mg/kg. Ten minutes after D-luciferin administration, luminescence images were captured using the IVIS Lumina XRMS Imaging System (Living Image software version 4.8.2).



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}