

Virtual Screening of Novel Benzothiozinone Derivatives to Predict Potential Inhibitors of Mycobacterium Tuberculosis Kinases 2D-QSAR, Molecular Docking, MM-PBSA Dynamics Simulations, and ADMET Properties


 , and
, and
Abstract
1. Introduction
2. Results and Discussions
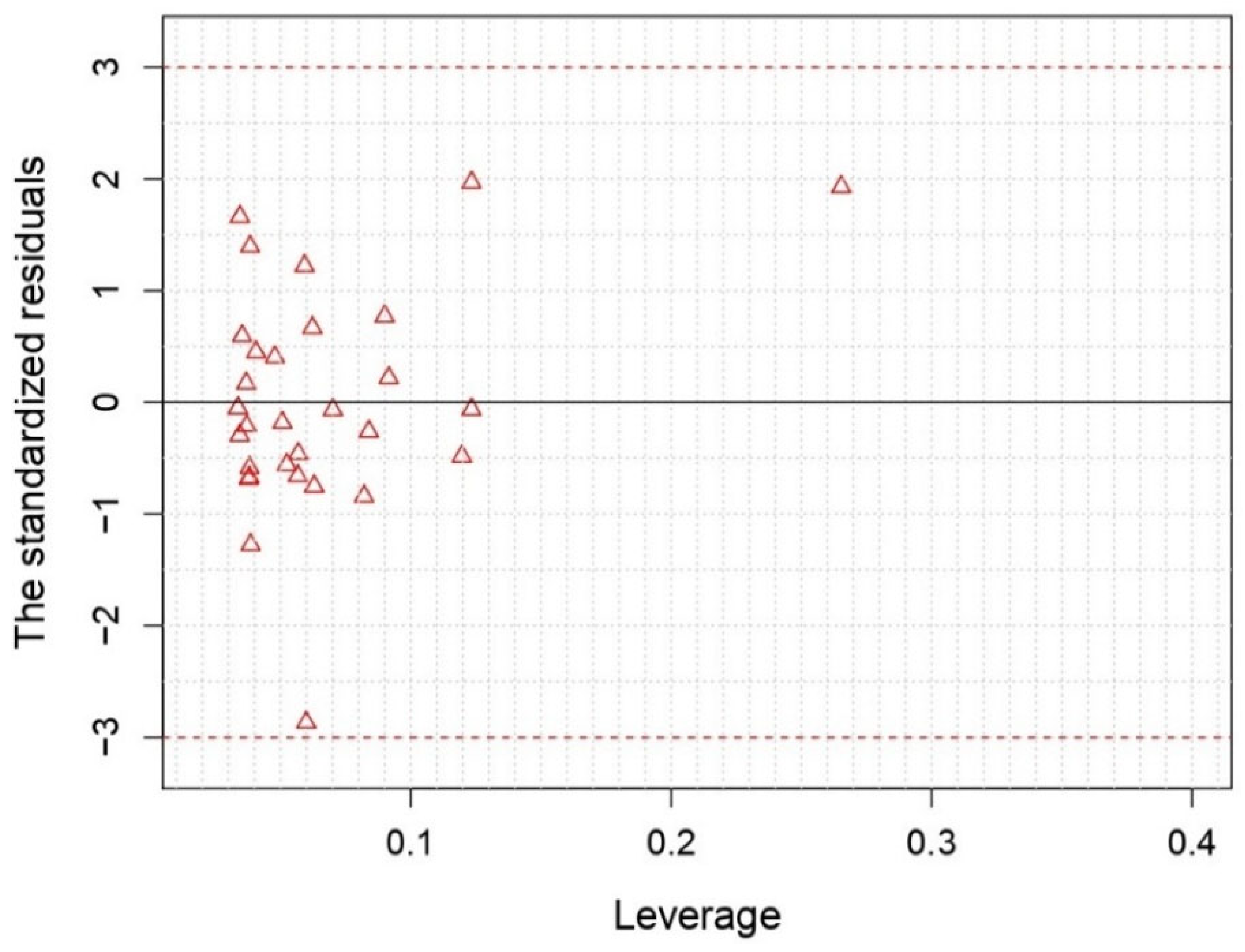
2.1. Two-Dimensional QSAR Analysis
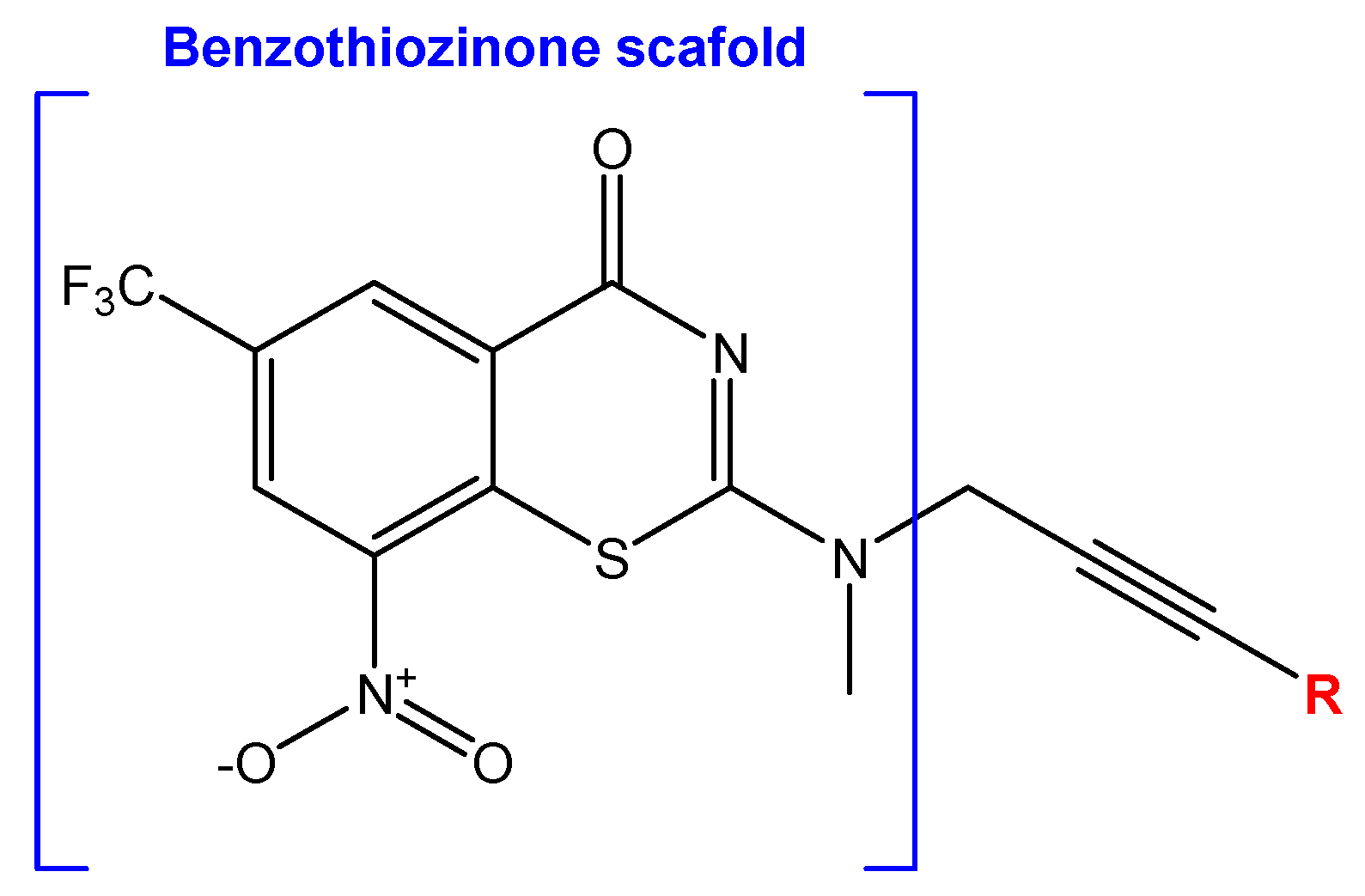
2.2. Library Creation
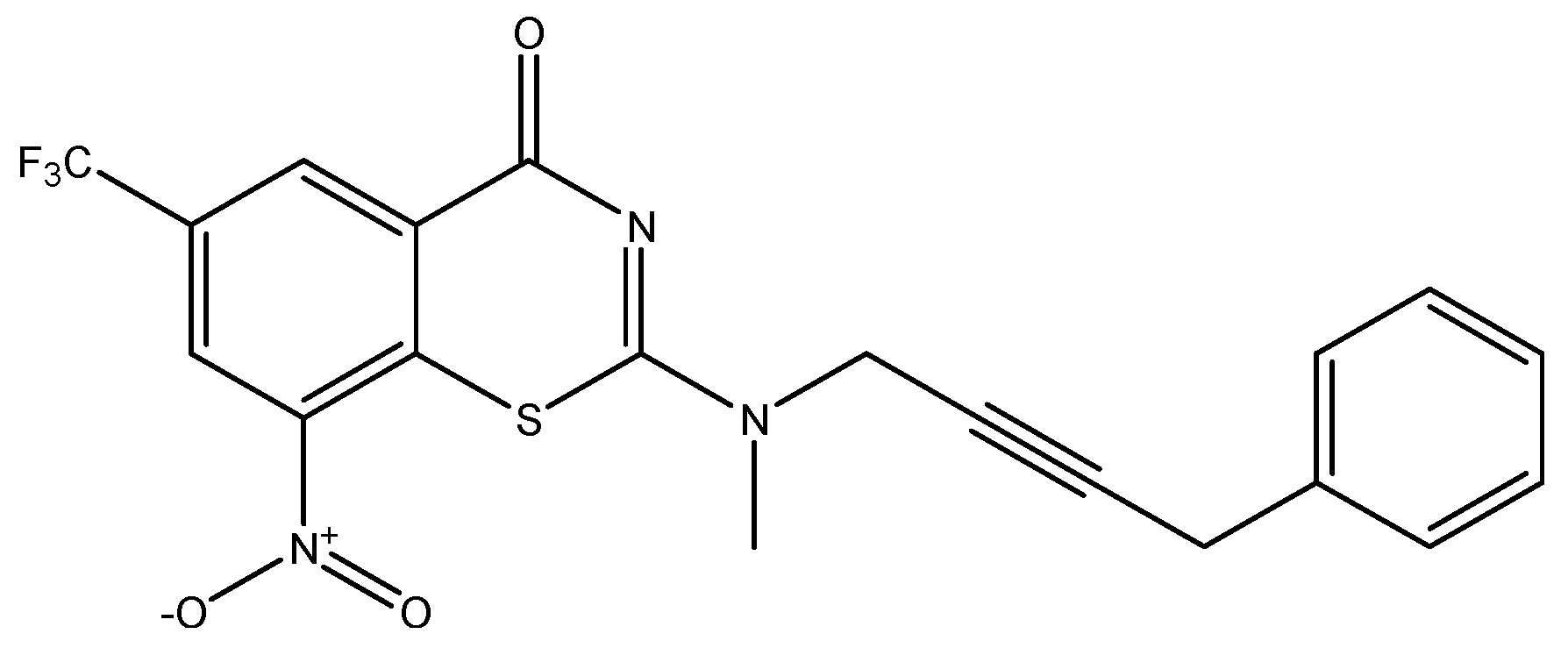
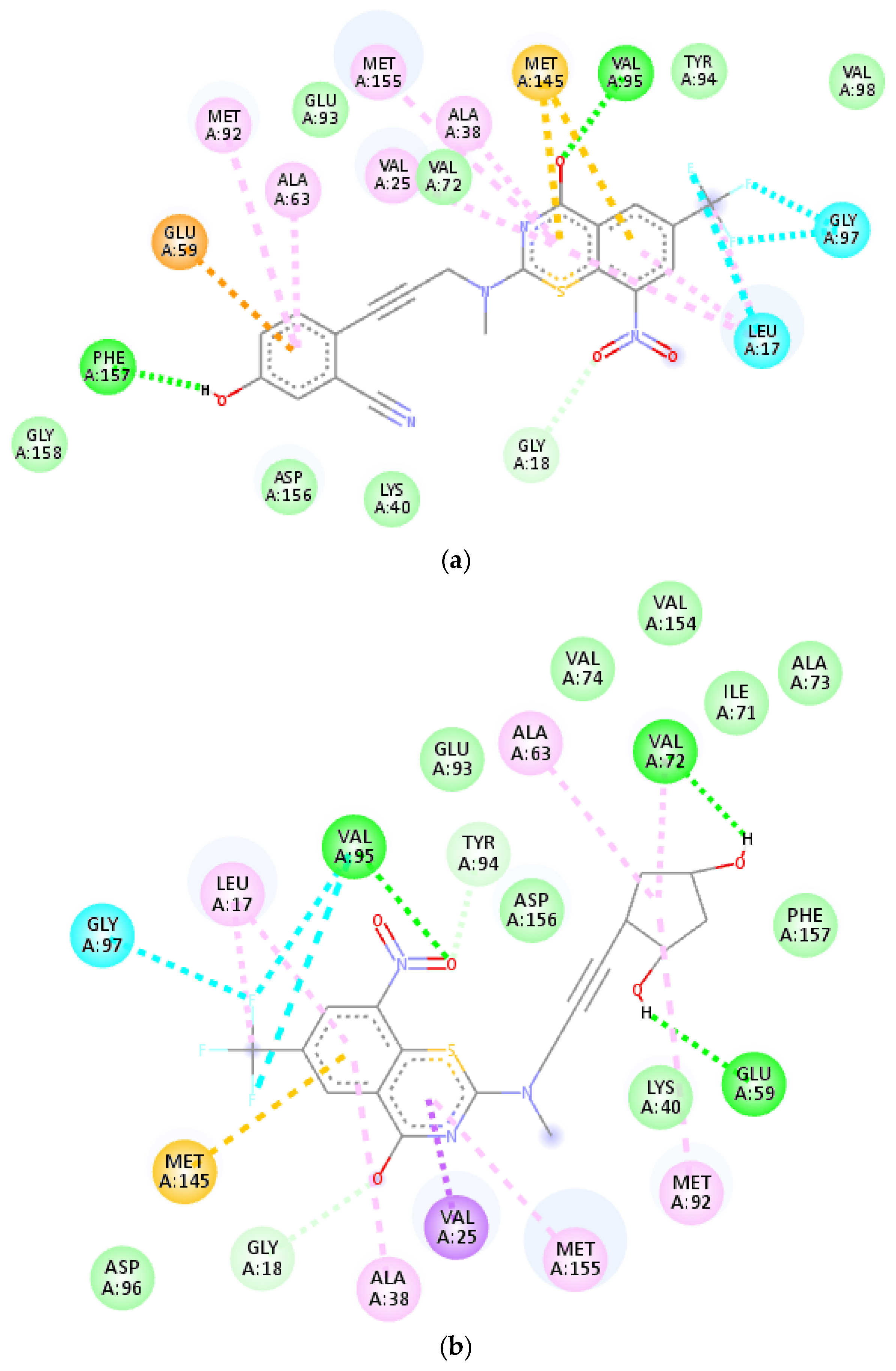
2.3. Virtual Screening and Molecular Docking
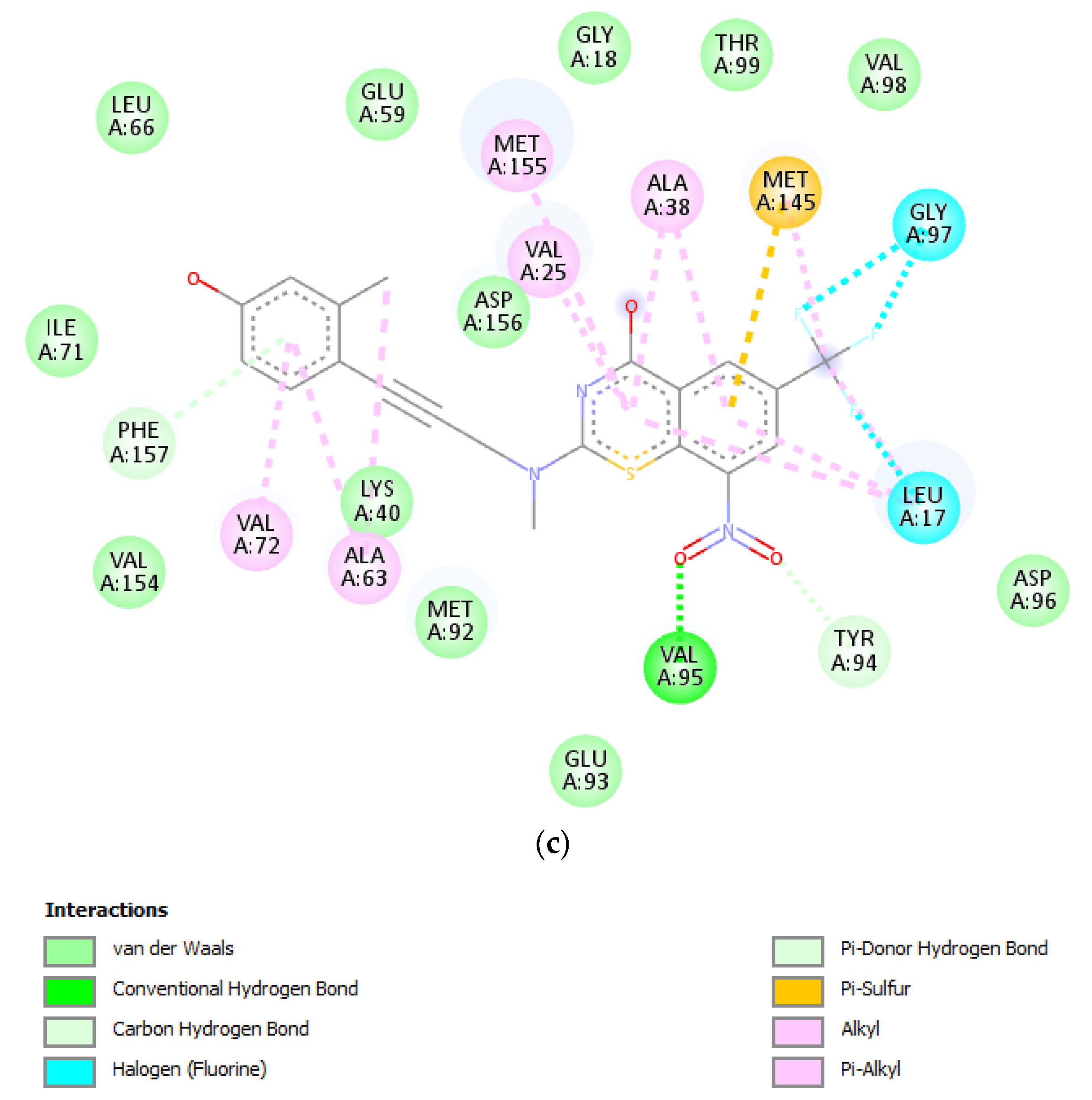
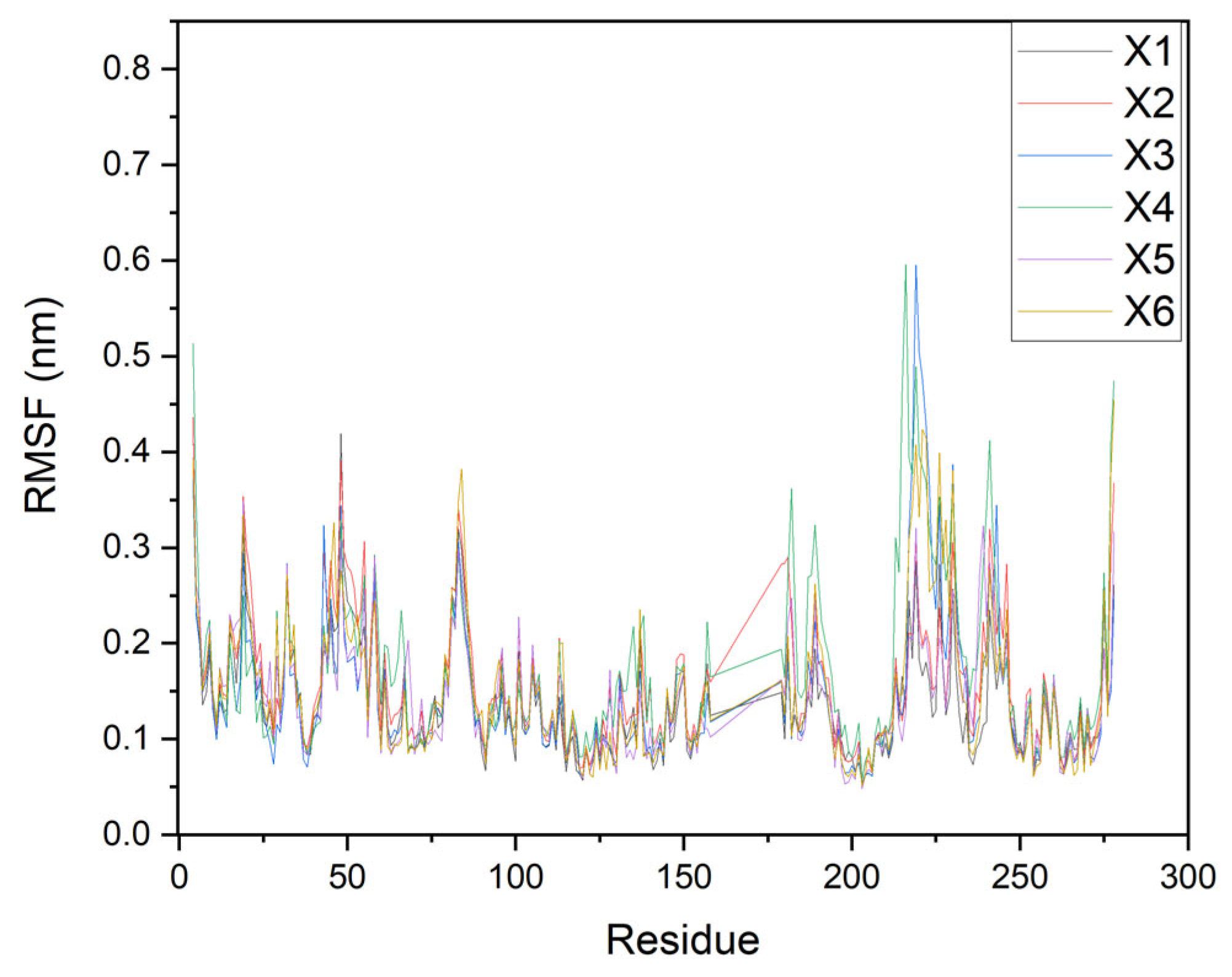
2.4. Molecular Dynamics (MD) Simulations
3. Materials and Methods
3.1. Building the Compound Dataset
3.2. Two-Dimensional QSAR Virtual Screening and Model Development
3.3. Virtual Screening
3.4. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D-QSAR | Two-dimensional quantitative structure–Activity Relationship |
| AD | Applicability Domain |
| ADMET | Absorption, Distribution, Metabolism, Excretion and Toxicity |
| btz | benzothiozinone |
| CADD | Computer-Aided Drug Design |
| HVS | High Virtual Screening |
| LOO | Leave-One-Out |
| MD | Molecular Dynamics |
| MDR | Multidrug-Resistant |
| MIC | Minimum Inhibitory Concentration |
| MLR | Multiple Linear Regression |
| MM-PBSA | Molecular Mechanics Poisson–Boltzmann Surface Area |
| MMFF | Merck Molecular Force Field |
| MSE | Mean Squared Error |
| Mtb | Mycobacterium tuberculosis |
| PBC | Periodic Boundary Conditions |
| PDB | Protein DataBank |
| pMIC | predicted Minimum Inhibitory Concentration |
| QSAR | Quantitative Structure–Activity Relationship |
| Rg | Radius of gyration |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| TB | TuBerculosis |
| vdW | van der Waals |
| VS | Virtual Screening |
| XDR | eXtensively Drug-Resistant |
References
- World Health Organization. Report of the 7th Virtual End TB Strategy Summit for the Highest TB Burden Countries and Countries on the WHO Global Watchlist 16–17 November 2021; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. WHO Operational Handbook on Tuberculosis: Module 5: Management of Tuberculosis in Children and Adolescents; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Ejalonibu, M.A.; Ogundare, S.A.; Elrashedy, A.A.; Ejalonibu, M.A.; Lawal, M.M.; Mhlongo, N.N.; Kumalo, H.M. Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach. Int. J. Mol. Sci. 2021, 22, 13259. [Google Scholar] [CrossRef]
- Guendouzi, A.; Belkhiri, L.; Kebila, Y.; Houari, B.; Djekoune, A.; Boucekkine, A.; Tayyeb, J.Z.; Akash, S.; Abdellattif, M.H.; Guendouzi, A. Identification of tuberculosis inhibitors through QSAR-based virtual screening and molecular dynamics simulation of novel pyrimidine derivatives. J. Indian Chem. Soc. 2024, 101, 101298. [Google Scholar] [CrossRef]
- Hassan, A.M.M.; Mohammed, A.F.; Kumari, J.; Sriram, D.; Abdu-Allah, H.H.; Abdel-Moty, S.G. Hybrids of 4-aminosalicylic acid with dual anti-mycobacterial and anti-inflammatory activities: Synthesis, biological evaluation, in silico investigation and structure-activity relationships exploration. J. Mol. Struct. 2024, 1318, 139217. [Google Scholar] [CrossRef]
- Shah, Y.B.; Mistry, P.S.; Dhameliya, T.M.; Ranch, K. Tuberculosis: Current Treatment Options and Future Scope. In Tubercular Drug Delivery Systems: Advances in Treatment of Infectious Diseases; Shegokar, R., Pathak, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 59–77. [Google Scholar]
- Shaikh, S.A.; Labhade, S.R.; Kale, R.R.; Pachorkar, P.Y.; Meshram, R.J.; Jain, K.S.; Labhade, H.S.; Boraste, D.R.; More, R.A.; Chobe, S.S.; et al. Synthesis, Biological and Molecular Docking Studies of Thiazole-Thiadiazole derivatives as potential Anti-Tuberculosis Agents. Chem. Biodivers. 2024, 21, e202400496. [Google Scholar] [CrossRef]
- Xu, X.; Dong, B.; Peng, L.; Gao, C.; He, Z.; Wang, C.; Zeng, J. Anti-tuberculosis drug development via targeting the cell envelope of Mycobacterium tuberculosis. Front. Microbiol. 2022, 13, 1056608. [Google Scholar] [CrossRef]
- Singh, V.; Chibale, K. Strategies to Combat Multi-Drug Resistance in Tuberculosis. Acc. Chem. Res. 2021, 54, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Ronning, D.R. Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev. Anti-Infect. Ther. 2012, 10, 1023–1036. [Google Scholar] [CrossRef]
- Wu, X.; Wang, W.; Stelitano, G.; Riabova, O.; Wang, B.; Niu, W.; Cocorullo, M.; Shi, R.; Chiarelli, L.R.; Makarov, V.; et al. Benzothiozinone derivatives with anti-tubercular Activity−Further side chain investigation. Eur. J. Med. Chem. 2024, 264, 115976. [Google Scholar] [CrossRef]
- Bajorath, J. Computer-aided drug discovery [version 1; peer review: 3 approved]. F1000Research 2015, 4, 630. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Guendouzi, A.; Belkhiri, L.; Guendouzi, A.; Derouiche, T.M.T.; Djekoun, A. A combined in silico approaches of 2D-QSAR, molecular docking, molecular dynamics and ADMET prediction of anti-cancer inhibitor activity for actinonin derivatives. J. Biomol. Struct. Dyn. 2024, 42, 119–133. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Abchir, O.; Daoui, O.; Belaidi, S.; Ouassaf, M.; Qais, F.A.; ElKhattabi, S.; Belaaouad, S.; Chtita, S. Design of novel benzimidazole derivatives as potential α-amylase inhibitors using QSAR, pharmacokinetics, molecular docking, and molecular dynamics simulation studies. J. Mol. Model. 2022, 28, 106. [Google Scholar] [CrossRef]
- Guendouzi, A.; Mekelleche, S.M. Prediction of the melting points of fatty acids from computed molecular descriptors: A quantitative structure–property relationship study. Chem. Phys. Lipids 2012, 165, 1–6. [Google Scholar] [CrossRef]
- Guendouzi, A.; Belkhiri, L.; Djelti, F.; Zendaoui, Z.M.; Brahim, H.; Djekoun, A.; Boucekkine, A. In-silico design novel phenylsulfonyl furoxan and phenstatin derivatives as multi-target anti-cancer inhibitors based on 2D-QSAR, molecular docking, dynamics and ADMET approaches. Mol. Simul. 2024, 50, 470–492. [Google Scholar] [CrossRef]
- Guendouzi, A.; Belkhiri, L.; Culletta, G.; Tutone, M. Unveiling Novel Hybrids Quinazoline/Phenylsulfonylfuroxan Derivatives with Potent Multi-Anticancer Inhibition: DFT and In Silico Approach Combining 2D-QSAR, Molecular Docking, Dynamics Simulations, and ADMET Properties. ChemistrySelect 2024, 9, e202404283. [Google Scholar] [CrossRef]
- Djelti, F.; Berkane, A.; Alharbi, H.M.; Guendouzi, A.; Ghellai, I.; Nabil, B.; Belkhiri, L.; Brahim, H.; Tarik, C.; Belarbi, M.; et al. Identification of Ajuga Iva Extracts as Potential Candidates for Antidysmenorrhea Targeting Human COX2 and PGE2S-1 through In Vitro and In Silico Drug Repurposing Approach. ChemistrySelect 2024, 9, e202400353. [Google Scholar] [CrossRef]
- Akash, S.; Abdelkrim, G.; Bayil, I.; Hosen, E.; Mukerjee, N.; Shater, A.F.; Saleh, F.M.; Albadrani, G.M.; Al-Ghadi, M.Q.; Abdel-Daim, M.M.; et al. Antimalarial drug discovery against malaria parasites through haplopine modification: An advanced computational approach. J. Cell. Mol. Med. 2023, 27, 3168–3188. [Google Scholar] [CrossRef]
- Jawarkar, R.; Bakal, R.L.; Zaki, M.E.; Al-Hussain, S.; Ghosh, A.; Gandhi, A.; Mukerjee, N.; Samad, A.; Masand, V.H.; Lewaa, I. QSAR based virtual screening derived identification of a novel hit as a SARS CoV-229E 3CLpro Inhibitor: GA-MLR QSAR modeling supported by molecular Docking, molecular dynamics simulation and MMGBSA calculation approaches. Arab. J. Chem. 2022, 15, 103499. [Google Scholar] [CrossRef]
- Talete, S. DRAGON for Windows and Linux. 2007. Available online: http://www.talete.mi.it (accessed on 10 March 2024).
- RStudio, R.T. Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Papa, E.; Dearden, J.C.; Gramatica, P. Linear QSAR regression models for the prediction of bioconcentration factors by physicochemical properties and structural theoretical molecular descriptors. Chemosphere 2007, 67, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Sun, Q.; Berkelbach, T.C.; Blunt, N.S.; Booth, G.H.; Guo, S.; Li, Z.; Liu, J.; McClain, J.D.; Sayfutyarova, E.R.; Sharma, S.; et al. PySCF: The Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci. 2018, 8, e1340. [Google Scholar] [CrossRef]
- Tosco, P.; Stiefl, N.; Landrum, G. Bringing the MMFF force field to the RDKit: Implementation and validation. J. Cheminformatics 2014, 6, 37. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Molegro Virtual Docker for Docking. In Docking Screens for Drug Discovery; de Azevedo, W.F., Jr., Ed.; Springer: New York, NY, USA, 2019; pp. 149–167. [Google Scholar]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef]
- Lengauer, T.; Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Anbuselvam, M.; Easwaran, M.; Meyyazhagan, A.; Anbuselvam, J.; Bhotla, H.K.; Sivasubramanian, M.; Annadurai, Y.; Kaul, T.; Pappusamy, M.; Balasubramanian, B. Structure-based virtual screening, pharmacokinetic prediction, molecular dynamics studies for the identification of novel EGFR inhibitors in breast cancer. J. Biomol. Struct. Dyn. 2021, 39, 4462–4471. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Bemis, G.; Hanzelka, B.; Zuccola, H.; Wynn, M.; Moody, C.S.; Green, J.; Locher, C.; Liu, A.; Gao, H.; et al. Mtb PKNA/PKNB Dual Inhibition Provides Selectivity Advantages for Inhibitor Design To Minimize Host Kinase Interactions. ACS Med. Chem. Lett. 2017, 8, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34 (Suppl. S1), D302–D305. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Studio, D. Discovery Studio, version 2.1, Accelrys: San Diego, CA, USA, 2008.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Orgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Schrödinger, L. The PyMOL Molecular Graphics System, version 1.3 r1. 2010. Available online: https://www.pymol.org/#products (accessed on 10 March 2024).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Exp | MLR | ID | Exp | MLR | ID | Exp | MLR | ID | Exp | MLR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8.15 | 7.95 | 11 ★ | 7.55 | 7.02 | 21 | 6.06 | 6.17 | 31 | 7.55 | 7.03 |
| 2 | 7.95 | 7.58 | 12 | 6.88 | 6.86 | 22 | 5.97 | 6.27 | 32 | 7.53 | 7.23 |
| 3 ★ | 7.88 | 8.56 | 13 | 7.52 | 7.21 | 23 | 4.77 | 5.81 | 33 | 7.61 | 7.70 |
| 4 | 7.88 | 7.56 | 14 | 7.03 | 6.95 | 24 ★ | 5.85 | 5.93 | 34 ★ | 7.34 | 7.73 |
| 5 ★ | 8.15 | 7.26 | 15 | 6.83 | 7.40 | 25 | 8.30 | 7.80 | 35 ★ | 8.69 | 7.86 |
| 6 | 7.03 | 7.65 | 16 | 6.32 | 6.21 | 26 | 8.69 | 9.10 | 36 | 8.04 | 8.12 |
| 7 | 7.53 | 7.26 | 17 | 5.99 | 6.47 | 27 ★ | 7.63 | 7.86 | 37 | 8.69 | 8.40 |
| 8 | 7.27 | 7.24 | 18 | 6.37 | 5.43 | 28 | 6.42 | 6.39 | 38 | 8.00 | 8.28 |
| 9 | 7.79 | 7.83 | 19 | 5.71 | 5.81 | 29 | 6.89 | 7.01 | 39 ★ | 7.63 | 8.04 |
| 10 | 7.85 | 7.66 | 20 ★ | 5.97 | 6.39 | 30 | 6.97 | 7.22 | 40 ★ | 7.37 | 7.22 |
| Ligand | MolDock Score kcal/mol | H-Bond kcal/mol |
|---|---|---|
| X1 | −153.5 | −4.9 |
| X2 | −152.8 | −7.1 |
| X3 | −152.1 | −4.2 |
| X4 | −150.7 | −5.5 |
| X5 | −150.7 | −2.5 |
| X6 | −150.3 | −7.2 |
| X7 | −149.9 | −6.2 |
| X8 | −148.7 | −5.6 |
| X9 | −148.2 | −2.5 |
| X10 | −148.1 | −3.4 |
| X11 | −146.5 | −2.5 |
| X12 | −146.3 | −3.8 |
| X13 | −146.2 | −0.3 |
| X14 | −146.0 | −3.8 |
| X15 | −146.0 | −6.9 |
| X16 | −145.2 | −6.5 |
| X17 | −145.1 | −1.5 |
| X18 | −143.1 | −2.5 |
| X19 | −142.0 | −3.5 |
| X20 | −140.3 | −2.4 |
| Native | −131.9 | −2.5 |
| Compounds | ΔGbind kcal/mol | vdW Energy kcal/mol | Electrostatic Energy kcal/mol |
|---|---|---|---|
| X1 | +1.1 | −37.4 | −24.5 |
| X2 | −1.4 | −38.6 | −17.1 |
| X3 | −8.2 | −48.1 | −15.9 |
| X4 | −15.3 | −40.0 | −23.8 |
| X5 | +22.8 | −25.9 | −10.5 |
| X6 | −12.0 | −51.4 | −14.0 |
| C1 | C2 | C3 * |
 | 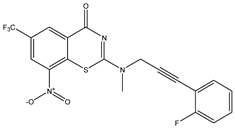 | 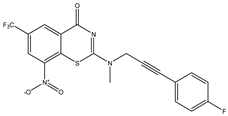 |
| C4 | C5 * | C6 |
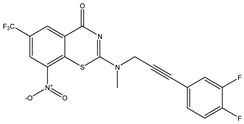 | 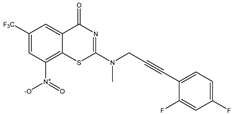 |  |
| C7 | C8 | C9 |
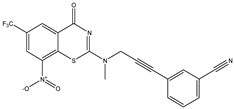 | 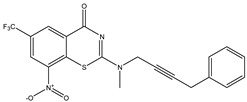 |  |
| C10 | C11 * | C12 |
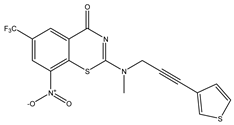 | 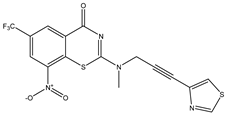 | 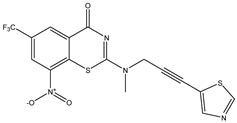 |
| C13 | C14 | C15 |
 | 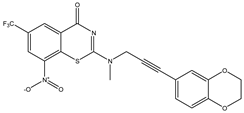 | 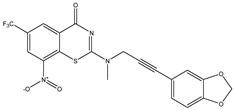 |
| C16 | C17 | C18 |
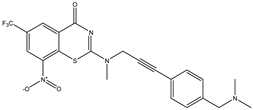 |  | 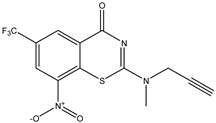 |
| C19 | C20 * | C21 |
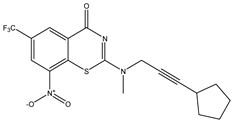 | 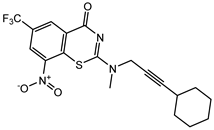 |  |
| C22 | C23 | C24 * |
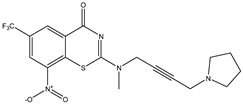 | 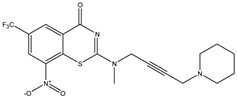 | 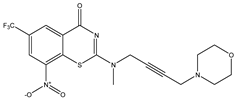 |
| C25 | C26 | C27 * |
 | 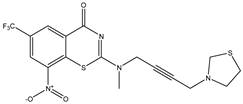 | 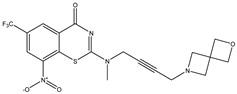 |
| C28 | C29 | C30 |
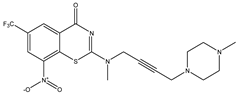 | 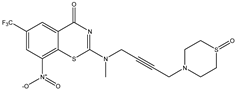 | 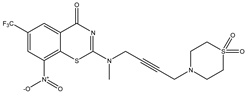 |
| C31 | C32 | C33 |
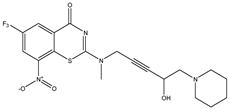 |  | 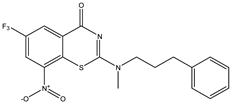 |
| C34 * | C35 * | C36 |
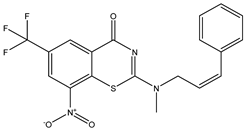 | 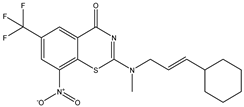 |  |
| C37 | C38 | C39 * |
 |  | 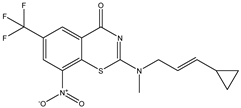 |
| C40 * | ||
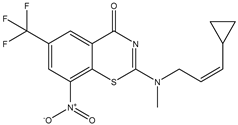 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guendouzi, A.; Belkhiri, L.; Slimani, Z.; Guendouzi, A.; Moroy, G. Virtual Screening of Novel Benzothiozinone Derivatives to Predict Potential Inhibitors of Mycobacterium Tuberculosis Kinases 2D-QSAR, Molecular Docking, MM-PBSA Dynamics Simulations, and ADMET Properties. Int. J. Mol. Sci. 2025, 26, 5129. https://doi.org/10.3390/ijms26115129
Guendouzi A, Belkhiri L, Slimani Z, Guendouzi A, Moroy G. Virtual Screening of Novel Benzothiozinone Derivatives to Predict Potential Inhibitors of Mycobacterium Tuberculosis Kinases 2D-QSAR, Molecular Docking, MM-PBSA Dynamics Simulations, and ADMET Properties. International Journal of Molecular Sciences. 2025; 26(11):5129. https://doi.org/10.3390/ijms26115129
Chicago/Turabian StyleGuendouzi, Abdelmadjid, Lotfi Belkhiri, Zakaria Slimani, Abdelkrim Guendouzi, and Gautier Moroy. 2025. "Virtual Screening of Novel Benzothiozinone Derivatives to Predict Potential Inhibitors of Mycobacterium Tuberculosis Kinases 2D-QSAR, Molecular Docking, MM-PBSA Dynamics Simulations, and ADMET Properties" International Journal of Molecular Sciences 26, no. 11: 5129. https://doi.org/10.3390/ijms26115129
APA StyleGuendouzi, A., Belkhiri, L., Slimani, Z., Guendouzi, A., & Moroy, G. (2025). Virtual Screening of Novel Benzothiozinone Derivatives to Predict Potential Inhibitors of Mycobacterium Tuberculosis Kinases 2D-QSAR, Molecular Docking, MM-PBSA Dynamics Simulations, and ADMET Properties. International Journal of Molecular Sciences, 26(11), 5129. https://doi.org/10.3390/ijms26115129