
Glucosylceramide Synthase, a Key Enzyme in Sphingolipid Metabolism, Regulates Expression of Genes Accounting for Cancer Drug Resistance

 , ,
, , 
Abstract
1. Introduction
2. Results
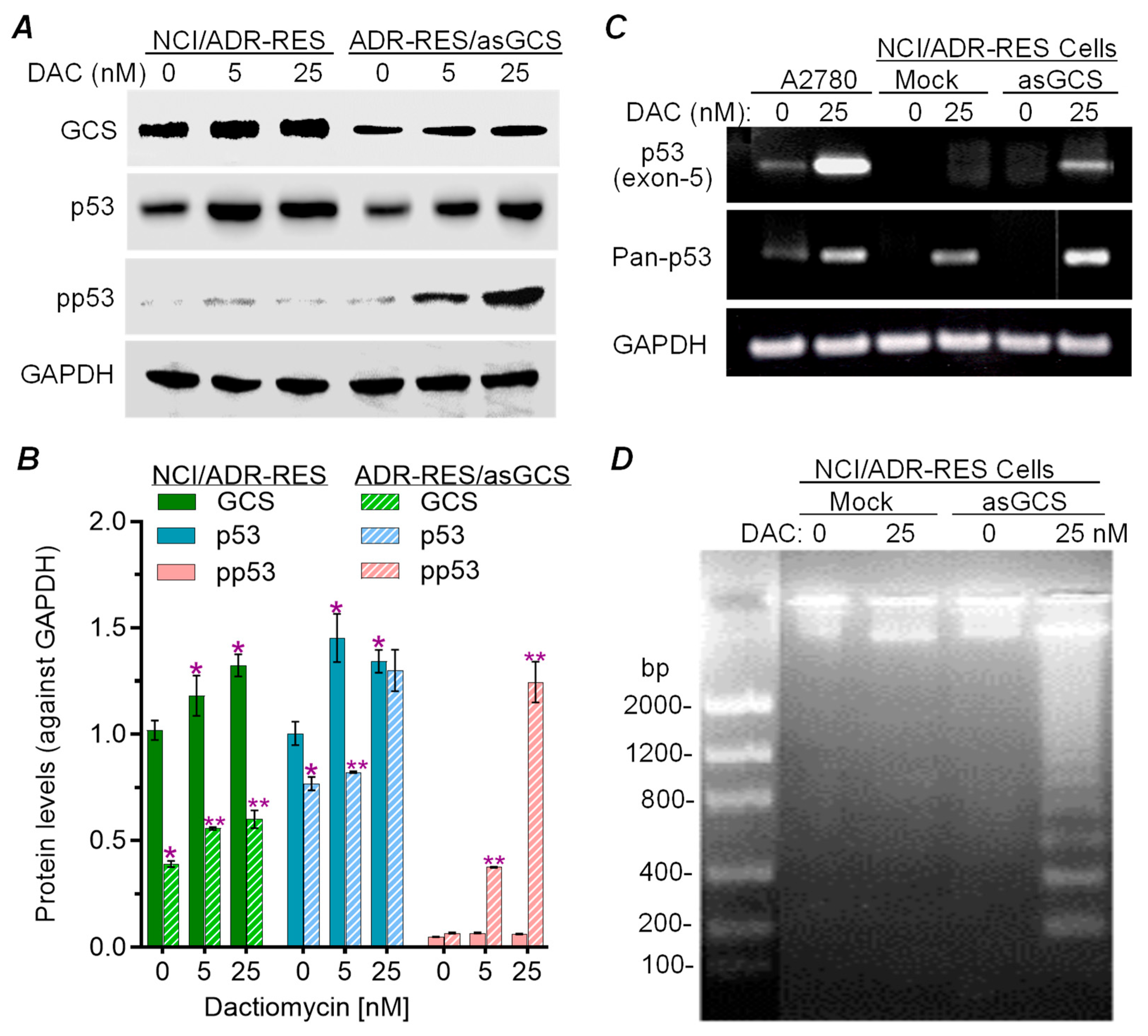
2.1. Silencing GCS Expression Sensitized NCI-ADR-RES Cells to Dactinomycin-Induced Apoptosis
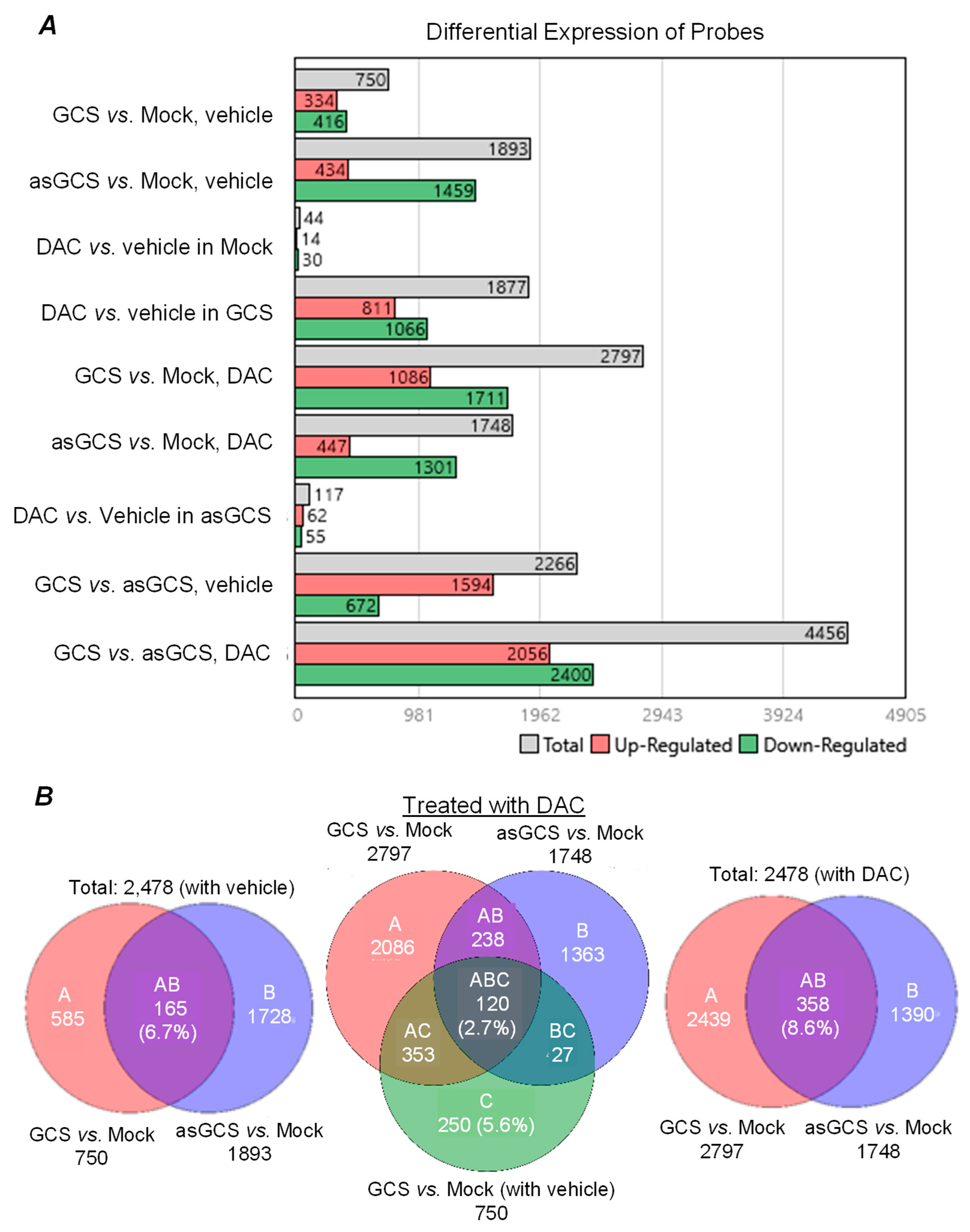
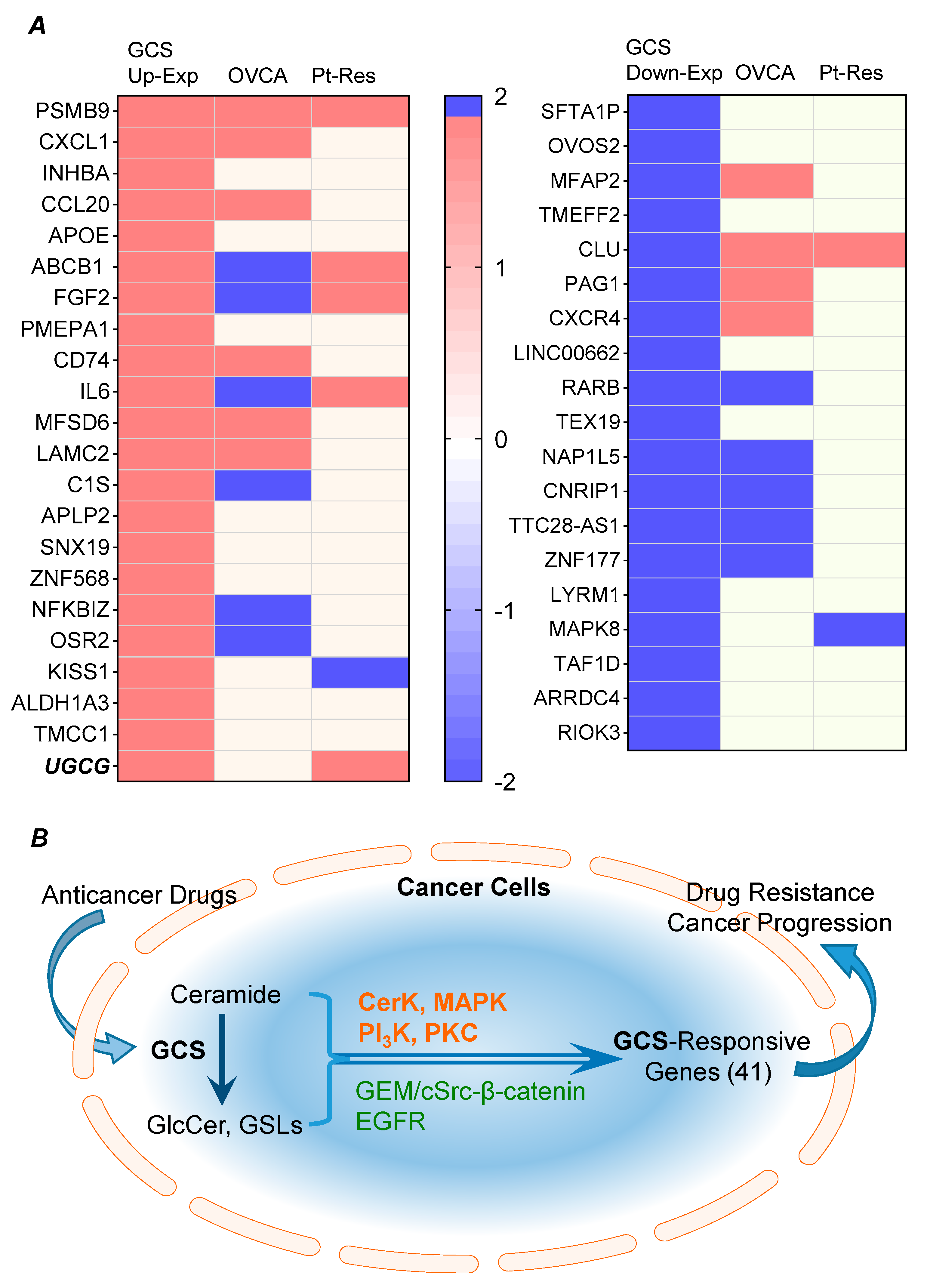
2.2. GCS Is Involved in Regulating Expression of Genes Protecting Cancer Cells
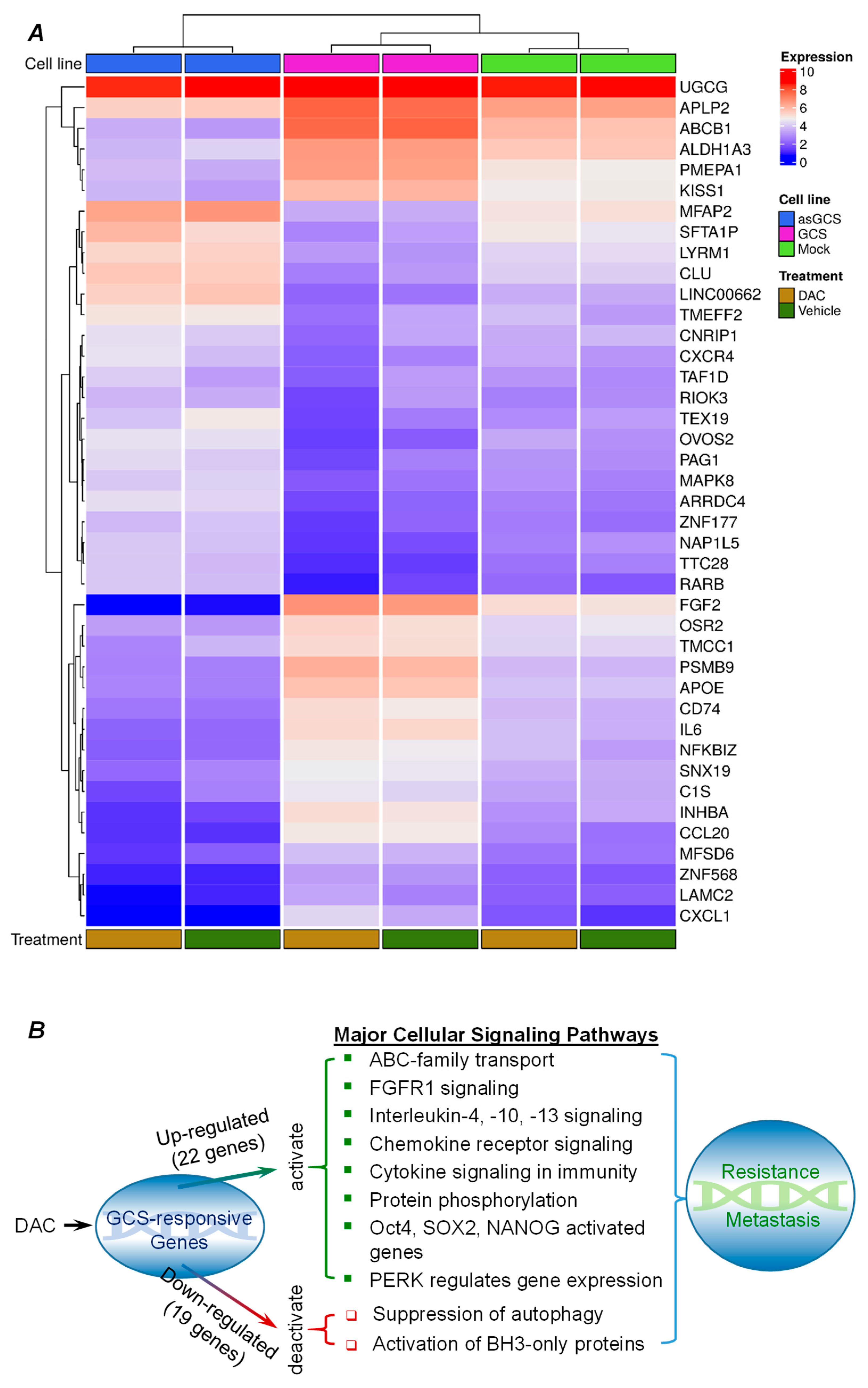
2.3. GCS-Responsive Genes Are Involved in Modulating Cancer Drug Resistance and Metastasis
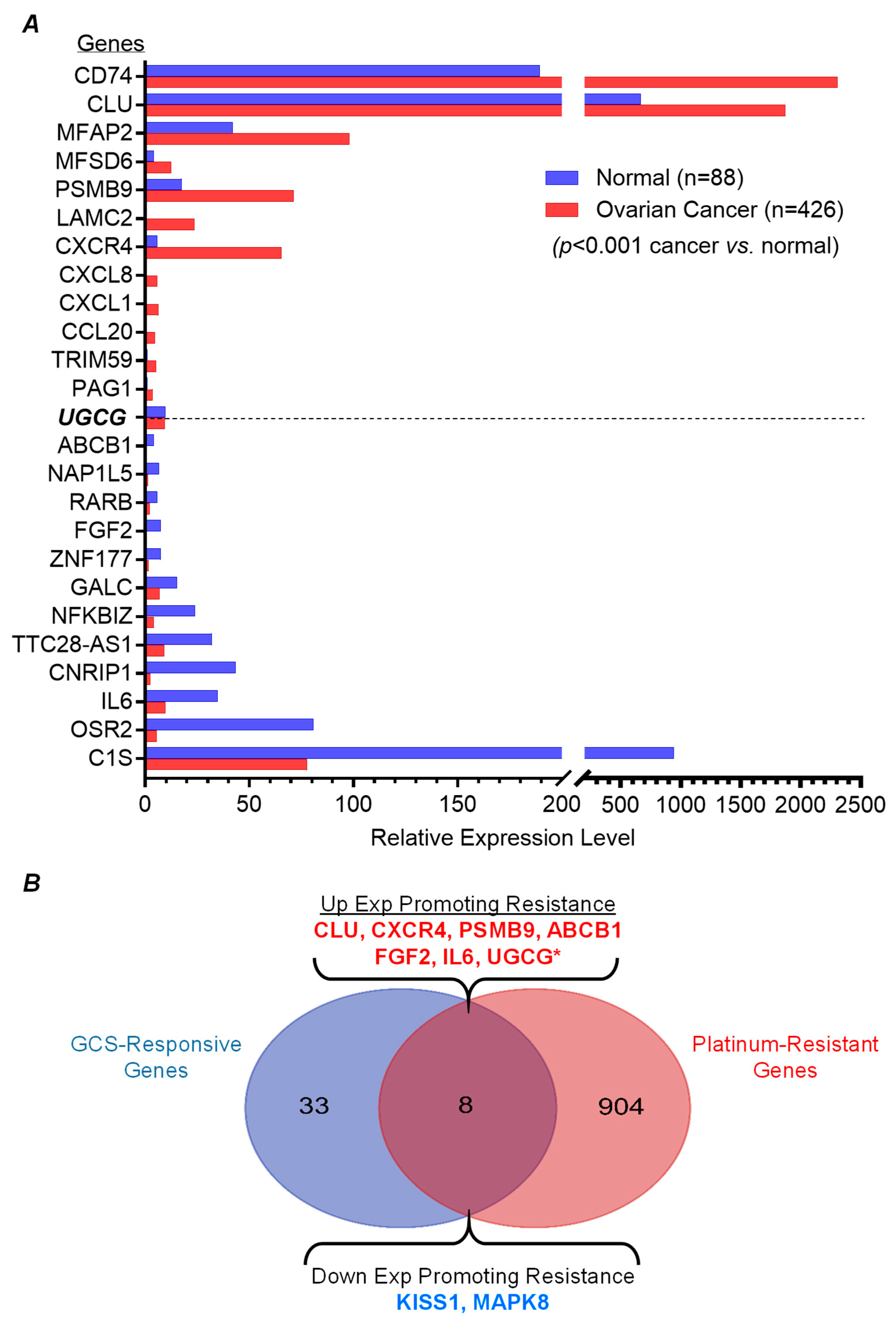
2.4. GCS-Responsive Genes Are Highly Expressed in Ovarian Cancer and Correlated with Platinum-Resistance
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. RT-PCR Analysis of p53 mRNA
4.3. DNA Fragmentation and Apoptosis Assay
4.4. Western Blot Analysis
4.5. Microarray Analysis
4.6. Database Submission of Microarray Data and Further Analysis
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Ichikawa, S.; Hirabayashi, Y.; Cabot, M.C. Glycosylation of ceramide potentiates cellular resistance to tumor necrosis factor-alpha-induced apoptosis. Exp. Cell Res. 1999, 252, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef]
- Russo, D.; Capolupo, L.; Loomba, J.S.; Sticco, L.; D’Angelo, G. Glycosphingolipid metabolism in cell fate specification. J. Cell Sci. 2018, 131, 219204. [Google Scholar] [CrossRef] [PubMed]
- Stuermer, C.A.; Lang, D.M.; Kirsch, F.; Wiechers, M.; Deininger, S.O.; Plattner, H. Glycosylphosphatidyl inositol-anchored proteins and fyn kinase assemble in noncaveolar plasma membrane microdomains defined by reggie-1 and -2. Mol. Biol. Cell 2001, 12, 3031–3045. [Google Scholar] [CrossRef]
- Roy, K.R.; Uddin, M.B.; Roy, S.C.; Hill, R.A.; Marshall, J.; Li, Y.T.; Chamcheu, J.C.; Lu, H.; Liu, Y.Y. Gb3-cSrc complex in glycosphingolipid-enriched microdomains contributes to the expression of p53 mutant protein and cancer drug resistance via beta-catenin-activated RNA methylation. FASEB Bioadv. 2020, 2, 653–667. [Google Scholar] [CrossRef]
- Kajiwara, K.; Yamada, T.; Bamba, T.; Fukusaki, E.; Imamoto, F.; Okada, M.; Oneyama, C. c-Src-induced activation of ceramide metabolism impairs membrane microdomains and promotes malignant progression by facilitating the translocation of c-Src to focal adhesions. Biochem. J. 2014, 458, 81–93. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol. Cancer 2010, 9, 145. [Google Scholar] [CrossRef]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.Y.; Khoo, K.H.; Li, S.C.; Li, Y.T.; et al. Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar] [CrossRef]
- Hosain, S.B.; Khiste, S.K.; Uddin, M.B.; Vorubindi, V.; Ingram, C.; Zhang, S.; Hill, R.A.; Gu, X.; Liu, Y.Y. Inhibition of glucosylceramide synthase eliminates the oncogenic function of p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells. Oncotarget 2016, 7, 60575–60592. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Seidman, J.D.; Horkayne-Szakaly, I.; Haiba, M.; Boice, C.R.; Kurman, R.J.; Ronnett, B.M. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int. J. Gynecol. Pathol. 2004, 23, 41–44. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. P53 and prognosis: New insights and further complexity. Cell 2005, 120, 7–10. [Google Scholar] [CrossRef]
- Uddin, M.B.; Roy, K.R.; Hosain, S.B.; Khiste, S.K.; Hill, R.A.; Jois, S.D.; Zhao, Y.; Tackett, A.J.; Liu, Y.Y. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem. Pharmacol. 2019, 160, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Vikhanskaya, F.; Siddique, M.M.; Kei Lee, M.; Broggini, M.; Sabapathy, K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Clin. Cancer Res. 2005, 11, 4348–4356. [Google Scholar] [CrossRef]
- Wong, R.P.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 2007, 6, 1054–1061. [Google Scholar] [CrossRef]
- Miller, L.D.; Long, P.M.; Wong, L.; Mukherjee, S.; McShane, L.M.; Liu, E.T. Optimal gene expression analysis by microarrays. Cancer Cell 2002, 2, 353–361. [Google Scholar] [CrossRef]
- Kavak, E.; Najafov, A.; Ozturk, N.; Seker, T.; Cavusoglu, K.; Aslan, T.; Duru, A.D.; Saygili, T.; Hoxhaj, G.; Hiz, M.C.; et al. Analysis of the Wnt/B-catenin/TCF4 pathway using SAGE, genome-wide microarray and promoter analysis: Identification of BRI3 and HSF2 as novel targets. Cell Signal 2010, 22, 1523–1535. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Hansen, N.; Cabot, M.C. Uncoupling ceramide glycosylation by transfection of glucosylceramide synthase antisense reverses adriamycin resistance. J. Biol. Chem. 2000, 275, 7138–7143. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Han, T.Y.; Yu, J.Y.; Bitterman, A.; Le, A.; Giuliano, A.E.; Cabot, M.C. Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells. J. Lipid Res. 2004, 45, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Kitano, T.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Iwai, K.; Tashima, M.; Uchiyama, T.; Okazaki, T. Possible role of ceramide as an indicator of chemoresistance: Decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin. Cancer Res. 2003, 9, 415–423. [Google Scholar]
- Madigan, J.P.; Robey, R.W.; Poprawski, J.E.; Huang, H.; Clarke, C.J.; Gottesman, M.M.; Cabot, M.C.; Rosenberg, D.W. A role for ceramide glycosylation in resistance to oxaliplatin in colorectal cancer. Exp. Cell Res. 2020, 388, 111860. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. Faseb J. 2001, 15, 719–730. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Patwardhan, G.A.; Bhinge, K.; Gupta, V.; Gu, X.; Jazwinski, S.M. Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells. Cancer Res. 2011, 71, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Vert, A.; Castro, J.; Ribo, M.; Vilanova, M.; Benito, A. Transcriptional profiling of NCI/ADR-RES cells unveils a complex network of signaling pathways and molecular mechanisms of drug resistance. Onco Targets Ther. 2018, 11, 221–237. [Google Scholar] [CrossRef]
- Ogretmen, B.; Safa, A.R. Expression of the mutated p53 tumor suppressor protein and its molecular and biochemical characterization in multidrug resistant MCF-7/Adr human breast cancer cells. Oncogene 1997, 14, 499–506. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of platinum resistance in ovarian cancer: What can we use to improve treatment. Endocr. Relat. Cancer 2018, 25, R303–R318. [Google Scholar] [CrossRef]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Piulats, J.M.; Vidal, A.; Garcia-Rodriguez, F.J.; Munoz, C.; Nadal, M.; Moutinho, C.; Martinez-Iniesta, M.; Mora, J.; Figueras, A.; Guino, E.; et al. Orthoxenografts of Testicular Germ Cell Tumors Demonstrate Genomic Changes Associated with Cisplatin Resistance and Identify PDMP as a Resensitizing Agent. Clin. Cancer Res. 2018, 24, 3755–3766. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.L. Mifepristone modulates glucosylceramide synthase expression and reverse multidrug resistance in ovarian cancer cells. Nan Fang. Yi Ke Da Xue Xue Bao 2008, 28, 1727–1730. [Google Scholar] [PubMed]
- Tyler, A.; Johansson, A.; Karlsson, T.; Gudey, S.K.; Brannstrom, T.; Grankvist, K.; Behnam-Motlagh, P. Targeting glucosylceramide synthase induction of cell surface globotriaosylceramide (Gb3) in acquired cisplatin-resistance of lung cancer and malignant pleural mesothelioma cells. Exp. Cell Res. 2015, 336, 23–32. [Google Scholar] [CrossRef]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Salustiano, E.J.; da Costa, K.M.; Freire-de-Lima, L.; Mendonca-Previato, L.; Previato, J.O. Inhibition of glycosphingolipid biosynthesis reverts multidrug resistance by differentially modulating ABC transporters in chronic myeloid leukemias. J. Biol. Chem. 2020, 295, 6457–6471. [Google Scholar] [CrossRef]
- Gan, J.; Gao, Q.; Wang, L.L.; Tian, A.P.; Zhu, L.D.; Zhang, L.T.; Zhou, W.; Mao, X.R.; Li, J.F. Glucosylceramide synthase regulates hepatocyte repair after concanavalin A-induced immune-mediated liver injury. PeerJ 2021, 9, e12138. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Sait, K.H.W.; Alam, Q.; Alam, M.Z.; Anfinan, N.; Wali, A.W.N.; Rasool, M. MDR1 Gene Polymorphisms and Its Association With Expression as a Clinical Relevance in Terms of Response to Chemotherapy and Prognosis in Ovarian Cancer. Front. Genet. 2020, 11, 516. [Google Scholar] [CrossRef]
- Holzmayer, T.A.; Hilsenbeck, S.; Von Hoff, D.D.; Roninson, I.B. Clinical correlates of MDR1 (P-glycoprotein) gene expression in ovarian and small-cell lung carcinomas. J. Natl. Cancer Inst. 1992, 84, 1486–1491. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, L.; Guo, Q.; Zhu, J.; Deng, Y.; Wu, X. Identification of Chemoresistance-Associated Key Genes and Pathways in High-Grade Serous Ovarian Cancer by Bioinformatics Analyses. Cancer Manag. Res. 2020, 12, 5213–5223. [Google Scholar] [CrossRef]
- Gan, Y.; Wientjes, M.G.; Au, J.L. Expression of basic fibroblast growth factor correlates with resistance to paclitaxel in human patient tumors. Pharm. Res. 2006, 23, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Mlynska, A.; Salciuniene, G.; Zilionyte, K.; Garberyte, S.; Strioga, M.; Intaite, B.; Barakauskiene, A.; Lazzari, G.; Dobrovolskiene, N.; Krasko, J.A.; et al. Chemokine profiling in serum from patients with ovarian cancer reveals candidate biomarkers for recurrence and immune infiltration. Oncol. Rep. 2019, 41, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, R.M.; Kabir, S.M.; Lee, E.S.; Adunyah, S.E.; Son, D.S. NF-kappaB-Mediated CCL20 Reigns Dominantly in CXCR2-Driven Ovarian Cancer Progression. PLoS ONE 2016, 11, e0164189. [Google Scholar] [CrossRef]
- Jin, Y.; Lin, Q.; Fei, H.; Xue, L.; Li, L.; Xi, Q.; Jiang, H. Bioinformatics Analysis of Potential Therapeutic Targets and Prognostic Biomarkers amid CXC Chemokines in Ovarian Carcinoma Microenvironment. J. Oncol. 2021, 2021, 8859554. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, C.; Hahn, M.A.; Baxter, R.C.; Marsh, D.J. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr. Relat. Cancer 2010, 17, 929–940. [Google Scholar] [CrossRef]
- Jordan, K.R.; Sikora, M.J.; Slansky, J.E.; Minic, A.; Richer, J.K.; Moroney, M.R.; Hu, J.; Wolsky, R.J.; Watson, Z.L.; Yamamoto, T.M.; et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin. Cancer Res. 2020, 26, 6362–6373. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, I.S.S.; Martins-Filho, A.; Micheli, D.C.; Lima, C.A.; Tavares-Murta, B.M.; Murta, E.F.C.; Nomelini, R.S. IL-6 and IL-8 as Prognostic Factors in Peritoneal Fluid of Ovarian Cancer. Immunol. Invest. 2020, 49, 510–521. [Google Scholar] [CrossRef]
- Dalal, V.; Kumar, R.; Kumar, S.; Sharma, A.; Kumar, L.; Sharma, J.B.; Roy, K.K.; Singh, N.; Vanamail, P. Biomarker potential of IL-6 and VEGF-A in ascitic fluid of epithelial ovarian cancer patients. Clin. Chim. Acta 2018, 482, 27–32. [Google Scholar] [CrossRef]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef]
- Wang, C.; Cicek, M.S.; Charbonneau, B.; Kalli, K.R.; Armasu, S.M.; Larson, M.C.; Konecny, G.E.; Winterhoff, B.; Fan, J.B.; Bibikova, M.; et al. Tumor hypomethylation at 6p21.3 associates with longer time to recurrence of high-grade serous epithelial ovarian cancer. Cancer Res. 2014, 74, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.U.; Kim, A.; Kim, J.Y.; Kim, K.H.; Hwang, C.; Lee, S.J.; Park, W.Y.; Jung, S.; Choi, H.J.; Kim, K. Differences in immune-related gene expressions and tumor-infiltrating lymphocytes according to chemotherapeutic response in ovarian high-grade serous carcinoma. J. Ovarian Res. 2020, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Jomary, C. Clusterin. Int. J. Biochem. Cell Biol. 2002, 34, 427–431. [Google Scholar] [CrossRef]
- Koltai, T. Clusterin: A key player in cancer chemoresistance and its inhibition. Onco. Targets Ther. 2014, 7, 447–456. [Google Scholar] [CrossRef]
- Hassan, M.K.; Watari, H.; Han, Y.; Mitamura, T.; Hosaka, M.; Wang, L.; Tanaka, S.; Sakuragi, N. Clusterin is a potential molecular predictor for ovarian cancer patient’s survival: Targeting clusterin improves response to paclitaxel. J. Exp. Clin. Cancer Res. 2011, 30, 113. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Lau, S.H.; Sham, J.S.; Wu, Q.L.; Fang, Y.; Liang, L.Z.; Che, L.H.; Zeng, Y.X.; Guan, X.Y. Up-regulated expression of cytoplasmic clusterin in human ovarian carcinoma. Cancer 2005, 103, 277–283. [Google Scholar] [CrossRef]
- Park, D.C.; Yeo, S.G.; Wilson, M.R.; Yerbury, J.J.; Kwong, J.; Welch, W.R.; Choi, Y.K.; Birrer, M.J.; Mok, S.C.; Wong, K.K. Clusterin interacts with Paclitaxel and confer Paclitaxel resistance in ovarian cancer. Neoplasia 2008, 10, 964–972. [Google Scholar] [CrossRef]
- Hassan, M.K.; Watari, H.; Christenson, L.; Bettuzzi, S.; Sakuragi, N. Intracellular clusterin negatively regulates ovarian chemoresistance: Compromised expression sensitizes ovarian cancer cells to paclitaxel. Tumour Biol. 2011, 32, 1031–1047. [Google Scholar] [CrossRef]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015, 22, 258–273. [Google Scholar] [CrossRef]
- Verheij, M.; Bose, R.; Lin, X.H.; Yao, B.; Jarvis, W.D.; Grant, S.; Birrer, M.J.; Szabo, E.; Zon, L.I.; Kyriakis, J.M.; et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 1996, 380, 75–79. [Google Scholar] [CrossRef]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Juin, S.K.; Bhattacharyya Majumdar, S.; Majumdar, S. Crucial role of glucosylceramide synthase in the regulation of stem cell-like cancer cells in B16F10 murine melanoma. Mol. Carcinog. 2021, 60, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Kaipio, K.; Chen, P.; Roering, P.; Huhtinen, K.; Mikkonen, P.; Ostling, P.; Lehtinen, L.; Mansuri, N.; Korpela, T.; Potdar, S.; et al. ALDH1A1-related stemness in high-grade serous ovarian cancer is a negative prognostic indicator but potentially targetable by EGFR/mTOR-PI3K/aurora kinase inhibitors. J. Pathol. 2020, 250, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Nagare, R.P.; Sneha, S.; Krishnapriya, S.; Ramachandran, B.; Murhekar, K.; Vasudevan, S.; Shabna, A.; Ganesan, T.S. ALDH1A1+ ovarian cancer stem cells co-expressing surface markers CD24, EPHA1 and CD9 form tumours in vivo. Exp. Cell Res. 2020, 392, 112009. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial-mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354. [Google Scholar] [CrossRef]
- Srivastava, M.; Ahlawat, N.; Srivastava, A. Ovarian Cancer Stem Cells: Newer Horizons. J. Obstet. Gynaecol. India 2021, 71, 115–117. [Google Scholar] [CrossRef]
- Wang, Y.; Zong, X.; Mitra, S.; Mitra, A.K.; Matei, D.; Nephew, K.P. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018, 3, 122360. [Google Scholar] [CrossRef]
- Cortes, M.; Sanchez-Moral, L.; de Barrios, O.; Fernandez-Acenero, M.J.; Martinez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Gyorffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J. 2017, 36, 3336–3355. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Liu, Y.Y. Sphingolipids and expression regulation of genes in cancer. Prog. Lipid Res. 2011, 50, 104–114. [Google Scholar] [CrossRef]
- Doumpas, N.; Lampart, F.; Robinson, M.D.; Lentini, A.; Nestor, C.E.; Cantu, C.; Basler, K. TCF/LEF dependent and independent transcriptional regulation of Wnt/beta-catenin target genes. EMBO J. 2019, 38, e98873. [Google Scholar] [CrossRef]
- Bremer, E.G.; Schlessinger, J.; Hakomori, S. Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J. Biol. Chem. 1986, 261, 2434–2440. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takahashi, K.; Shiozaki, K.; Yamaguchi, K.; Moriya, S.; Hosono, M.; Shima, H.; Miyagi, T. Potentiation of epidermal growth factor-mediated oncogenic transformation by sialidase NEU3 leading to Src activation. PLoS ONE 2015, 10, e0120578. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, H.; Hong, W.; Liu, L.; Wang, S.; Zhou, M.; Chen, B.; Bai, Y. Tyrphostin B42 attenuates trichostatin A-mediated resistance in pancreatic cancer cells by antagonizing IL-6/JAK2/STAT3 signaling. Oncol. Rep. 2018, 39, 1892–1900. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Hosain, S.B.; Liu, D.X.; Khiste, S.K.; Zhao, Y.; Bielawski, J.; Jazwinski, S.M.; Liu, Y.Y. Ceramide modulates pre-mRNA splicing to restore the expression of wild-type tumor suppressor p53 in deletion-mutant cancer cells. Biochim. Biophys. Acta 2014, 1841, 1571–1580. [Google Scholar] [CrossRef]
- Khiste, S.K.; Liu, Z.; Roy, K.R.; Uddin, M.B.; Hosain, S.B.; Gu, X.; Nazzal, S.; Hill, R.A.; Liu, Y.Y. Ceramide-Rubusoside Nanomicelles, a Potential Therapeutic Approach to Target Cancers Carrying p53 Missense Mutations. Mol. Cancer Ther. 2020, 19, 564–574. [Google Scholar] [CrossRef]
- Chalfant, C.E.; Rathman, K.; Pinkerman, R.L.; Wood, R.E.; Obeid, L.M.; Ogretmen, B.; Hannun, Y.A. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J. Biol. Chem. 2002, 277, 12587–12595. [Google Scholar] [CrossRef]
- Wooten-Blanks, L.G.; Song, P.; Senkal, C.E.; Ogretmen, B. Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 2007, 21, 3386–3397. [Google Scholar] [CrossRef]
- Perry, D.M.; Newcomb, B.; Adada, M.; Wu, B.X.; Roddy, P.; Kitatani, K.; Siskind, L.; Obeid, L.M.; Hannun, Y.A. Defining a role for acid sphingomyelinase in the p38/interleukin-6 pathway. J. Biol. Chem. 2014, 289, 22401–22412. [Google Scholar] [CrossRef]
- Mehta, K.; Devarajan, E.; Chen, J.; Multani, A.; Pathak, S. Multidrug-resistant MCF-7 cells: An identity crisis? J. Natl. Cancer Inst. 2002, 94, 1652–1654. [Google Scholar] [CrossRef]
- Fairchild, C.R.; Ivy, S.P.; Kao-Shan, C.S.; Whang-Peng, J.; Rosen, N.; Israel, M.A.; Melera, P.W.; Cowan, K.H.; Goldsmith, M.E. Isolation of amplified and overexpressed DNA sequences from adriamycin-resistant human breast cancer cells. Cancer Res. 1987, 47, 5141–5148. [Google Scholar]
- Lorenzi, P.L.; Reinhold, W.C.; Varma, S.; Hutchinson, A.A.; Pommier, Y.; Chanock, S.J.; Weinstein, J.N. DNA fingerprinting of the NCI-60 cell line panel. Mol. Cancer Ther. 2009, 8, 713–724. [Google Scholar] [CrossRef]
- Liu, Y.Y. Resuscitating wild-type p53 expression by disrupting ceramide glycosylation: A novel approach to target mutant p53 tumors. Cancer Res. 2011, 71, 6295–6299. [Google Scholar] [CrossRef]
- Kiewe, P.; Gueller, S.; Komor, M.; Stroux, A.; Thiel, E.; Hofmann, W.K. Prediction of qualitative outcome of oligonucleotide microarray hybridization by measurement of RNA integrity using the 2100 Bioanalyzer capillary electrophoresis system. Ann. Hematol. 2009, 88, 1177–1183. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Yu, J.Y.; Yin, D.; Patwardhan, G.A.; Gupta, V.; Hirabayashi, Y.; Holleran, W.M.; Giuliano, A.E.; Jazwinski, S.M.; Gouaze-Andersson, V.; et al. A role for ceramide in driving cancer cell resistance to doxorubicin. Faseb J. 2008, 22, 2541–2551. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef]
- Dozmorov, M.; Stone, R., 2nd; Clifford, J.L.; Sabichi, A.L.; Engles, C.D.; Hauser, P.J.; Culkin, D.J.; Hurst, R.E. System level changes in gene expression in maturing bladder mucosa. J. Urol. 2011, 185, 1952–1958. [Google Scholar] [CrossRef]
- Januchowski, R.; Zawierucha, P.; Rucinski, M.; Zabel, M. Microarray-based detection and expression analysis of extracellular matrix proteins in drugresistant ovarian cancer cell lines. Oncol. Rep. 2014, 32, 1981–1990. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Product (bp) | Positions |
|---|---|---|---|
| del-p53 (exon-5) forward reverse | 5′-TCACTGCCATGGAGGAG-3′ 5′-TTGAGGGCAGGGGAG-3′ | 400 | 113–512 |
| Pan-p53 forward reverse | 5′-TTGCCGTCCCAAGCAATG-3′ 5′-AAGTCACAGACTTGGCTGTCCCAGA-3′ | 268 | 223–490 |
| Gene Symbol | Description | GCS vs. Mock (Fold) | asGCS vs. Mock (Fold) |
|---|---|---|---|
| CXCL8 | chemokine (C-X-C motif) ligand 8 | 6.9 | −2.5 |
| PSMB9 | proteasome subunit beta 9 | 5.5 | −2 |
| CXCL1 | chemokine (C-X-C motif) ligand 1 | 5.3 | −2.8 |
| INHBA | inhibin beta A | 4.6 | −3.3 |
| CCL20 | chemokine (C-C motif) ligand 20 | 4.4 | −2.9 |
| APOE | apolipoprotein E | 3.6 | −2.3 |
| ABCB1 | ATP-binding cassette subfamily B member 1 | 3.2 | −5.5 |
| FGF2 | fibroblast growth factor 2 (basic) | 3.1 | −21.9 |
| PMEPA1 | prostate transmembrane protein, androgen induced 1 | 3 | −2.4 |
| CD74 | CD74 molecule | 2.8 | −2.4 |
| IL6 | interleukin 6 | 2.7 | −3.3 |
| MFSD6 | major facilitator superfamily domain containing 6 | 2.6 | −2.1 |
| LAMC2 | laminin, gamma 2 | 2.5 | −2.5 |
| C1S | complement component 1, s subcomponent | 2.4 | −3.3 |
| APLP2 | amyloid beta (A4) precursor-like protein 2 | 2.3 | −2.1 |
| SNX19 | sorting nexin 19 | 2.3 | −2.5 |
| ZNF568 | zinc finger protein 568 | 2.2 | −2.1 |
| NFKBIZ | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta | 2.2 | −3.5 |
| OSR2 | odd-skipped related transciption factor 2 | 2.2 | −2 |
| KISS1 | KiSS-1 metastasis-suppressor | 2.1 | −2.2 |
| ALDH1A3 | aldehyde dehydrogenase 1 family, member A3 | 2.1 | −3.9 |
| TMCC1 | transmembrane and coiled-coil domain family 1 | 2 | −2.8 |
| UGCG (GCS) | UDP-glucose ceramide glucosyltransferase | 1.3 | −1.1 |
| GALC | galactosylceramidase | −29.6 | −17.4 |
| RIOK3 | RIO kinase 3 | −2.1 | 2.1 |
| ARRDC4 | arrestin domain containing 4 | −2.1 | 3.3 |
| TAF1D | TATA box binding protein associated factor 1D | −2.1 | 2 |
| MAPK8 | mitogen-activated protein kinase 8 | −2.1 | 2.1 |
| LYRM1 | LYR motif containing 1 | −2.2 | 2.2 |
| ZNF177 | zinc finger protein 177 | −2.3 | 2.2 |
| TTC28-AS1 | TTC28 antisense RNA 1 | −2.4 | 3.1 |
| CNRIP1 | cannabinoid receptor interacting protein 1 | −2.4 | 2 |
| NAP1L5 | nucleosome assembly protein 1-like 5 | −2.5 | 2.6 |
| TEX19 | testis expressed 19 | −2.5 | 2.1 |
| RARB | retinoic acid receptor, beta | −2.5 | 3.4 |
| LINC00662 | long intergenic non-protein coding RNA 662 | −2.5 | 3.8 |
| CXCR4 | chemokine (C-X-C motif) receptor 4 | −2.6 | 2.2 |
| PAG1 | phosphoprotein membrane anchor with glycosphingolipid microdomains 1 | −2.7 | 2.4 |
| CLU | clusterin | −2.8 | 2.9 |
| TMEFF2 | transmembrane protein with EGF-like and two follistatin-like domains 2 | −2.8 | 2.3 |
| MFAP2 | microfibrillar associated protein 2 | −2.9 | 2.8 |
| OVOS2 | ovostatin 2 | −3.9 | 2.1 |
| SFTA1P | surfactant associated 1, pseudogene | −4.6 | 2.1 |
| Gene Symbol | Tumor (n = 426) | Normal (n = 88) | Fold | Adj p-Value |
|---|---|---|---|---|
| CXCL8 | 5.95 | 0.86 | 3.7 | 2.01 × 10−18 |
| PSMB9 | 71.24 | 17.55 | 3.9 | 6.24 × 10−23 |
| CXCL1 | 6.43 | 0.57 | 4.7 | 4.47 × 10−20 |
| MT1M | 2.73 | 5.89 | −1.9 | NS |
| INHBA | 2.23 | 1.79 | 1.2 | NS |
| CCL20 | 4.66 | 0.89 | 5.2 | 3.10 × 10−29 |
| APOE | 260.91 | 363.11 | −1.4 | NS |
| ABCB1 | 0.55 | 4.18 | −3.3 | 5.33 × 10−97 |
| FGF2 | 0.22 | 7.53 | −7.0 | 6.29 × 10−187 |
| PMEPA1 | 15.14 | 10.91 | 1.4 | NS |
| CD74 | 2309.00 | 189.54 | 12.1 | 5.89 × 10−52 |
| IL6 | 9.72 | 34.96 | −3.4 | 4.03 × 10−55 |
| MFSD6 | 12.42 | 4.34 | 2.5 | 1.56 × 10−29 |
| LAMC2 | 23.77 | 0.88 | 13.2 | 4.23 × 10−83 |
| C1S | 77.70 | 945.41 | −12.0 | 4.61 × 10−73 |
| APLP2 | 168.08 | 154.48 | 1.0 | NS |
| SNX19 | 16.92 | 21.60 | −1.3 | NS |
| LOC643201 | NS | |||
| NFKBIZ | 4.31 | 24.02 | −4.7 | 1.92 × 10−45 |
| OSR2 | 5.66 | 80.88 | −12.3 | 1.58 × 10−77 |
| ZNF568 | 4.03 | 6.07 | −1.4 | NS |
| ALDH1A3 | 2.30 | 2.84 | −1.2 | NS |
| IFIT1 | 40.64 | 8.15 | 4.6 | 1.96 × 10−24 |
| KISS1 | 0.43 | 0.00 | 1.4 | NS |
| TMCC1 | 11.42 | 8.98 | 1.3 | NS |
| UGCG (GCS) | 9.40 | 9.91 | −1.0 | NS |
| AKT3 | 1.80 | 27.46 | −10.2 | 5.57 × 10−86 |
| ARRDC4 | 2.37 | 5.44 | −1.9 | NS |
| CDK6 | 1.95 | 2.25 | −1.1 | NS |
| MAPK8 | 14.71 | 21.55 | −1.4 | NS |
| RIOK3 | 20.05 | 30.89 | −1.5 | NS |
| TAF1D | 61.50 | 113.83 | −1.8 | NS |
| LYRM1 | 31.50 | 33.43 | −1.1 | NS |
| IGFBP3 | 73.80 | 163.27 | −2.2 | 3.31 × 10−12 |
| ZNF177 | 1.66 | 7.52 | −3.2 | 1.05 × 10−51 |
| CNRIP1 | 2.58 | 43.46 | −12.4 | 7.14 × 10−125 |
| TTC28-AS1 | 9.09 | 32.05 | −3.3 | 1.37 × 10−58 |
| LINC00662 | 9.65 | 8.12 | 1.2 | NS |
| NAP1L5 | 1.39 | 6.78 | −3.3 | 2.06 × 10−67 |
| RARB | 2.12 | 5.89 | −2.2 | 2.37 × 10−20 |
| TEX19 | 0.04 | 0.00 | 1.0 | NS |
| CXCR4 | 65.44 | 5.78 | 9.7 | 2.42 × 10−63 |
| PAG1 | 3.63 | 1.17 | 2.1 | 6.33 × 10−22 |
| CLU | 1874.12 | 671.15 | 2.8 | 1.43 × 10−6 |
| TMEFF2 | 0.02 | 0.06 | −1.0 | NS |
| MFAP2 | 98.05 | 42.01 | 2.3 | 4.58 × 10−5 |
| TFRC | 30.48 | 67.49 | −2.2 | 5.51 × 10−15 |
| OVOS2 | 1.59 | 1.02 | 1.3 | NS |
| GJA1 | 19.68 | 28.36 | −1.4 | NS |
| SFTA1P | 0.27 | 0.34 | −1.1 | NS |
| PLEKHA8 | 1.82 | 3.83 | −1.7 | NS |
| PDAP1 | 81.42 | 85.53 | −1.1 | NS |
| MATR3 | 6.00 | 6.62 | −1.1 | NS |
| TRIM59 | 5.41 | 1.00 | 3.2 | 2.43 × 10−51 |
| ARMCX4 | 2.48 | 35.09 | −10.3 | 8.83 × 10−151 |
| MALAT1 | 577.01 | 714.13 | −2.2 | NS |
| GALC | 6.88 | 15.41 | 1.3 | 4.89 × 10−27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostaq, M.S.; Kang, L.; Patwardhan, G.A.; Zhao, Y.; Shi, R.; Liu, Y.-Y. Glucosylceramide Synthase, a Key Enzyme in Sphingolipid Metabolism, Regulates Expression of Genes Accounting for Cancer Drug Resistance. Int. J. Mol. Sci. 2025, 26, 5112. https://doi.org/10.3390/ijms26115112
Mostaq MS, Kang L, Patwardhan GA, Zhao Y, Shi R, Liu Y-Y. Glucosylceramide Synthase, a Key Enzyme in Sphingolipid Metabolism, Regulates Expression of Genes Accounting for Cancer Drug Resistance. International Journal of Molecular Sciences. 2025; 26(11):5112. https://doi.org/10.3390/ijms26115112
Chicago/Turabian StyleMostaq, Md Saqline, Lin Kang, Gauri A. Patwardhan, Yunfeng Zhao, Runhua Shi, and Yong-Yu Liu. 2025. "Glucosylceramide Synthase, a Key Enzyme in Sphingolipid Metabolism, Regulates Expression of Genes Accounting for Cancer Drug Resistance" International Journal of Molecular Sciences 26, no. 11: 5112. https://doi.org/10.3390/ijms26115112
APA StyleMostaq, M. S., Kang, L., Patwardhan, G. A., Zhao, Y., Shi, R., & Liu, Y.-Y. (2025). Glucosylceramide Synthase, a Key Enzyme in Sphingolipid Metabolism, Regulates Expression of Genes Accounting for Cancer Drug Resistance. International Journal of Molecular Sciences, 26(11), 5112. https://doi.org/10.3390/ijms26115112