
Mutational Patterns in Colorectal Cancer: Do PDX Models Retain the Heterogeneity of the Original Tumor?


Abstract
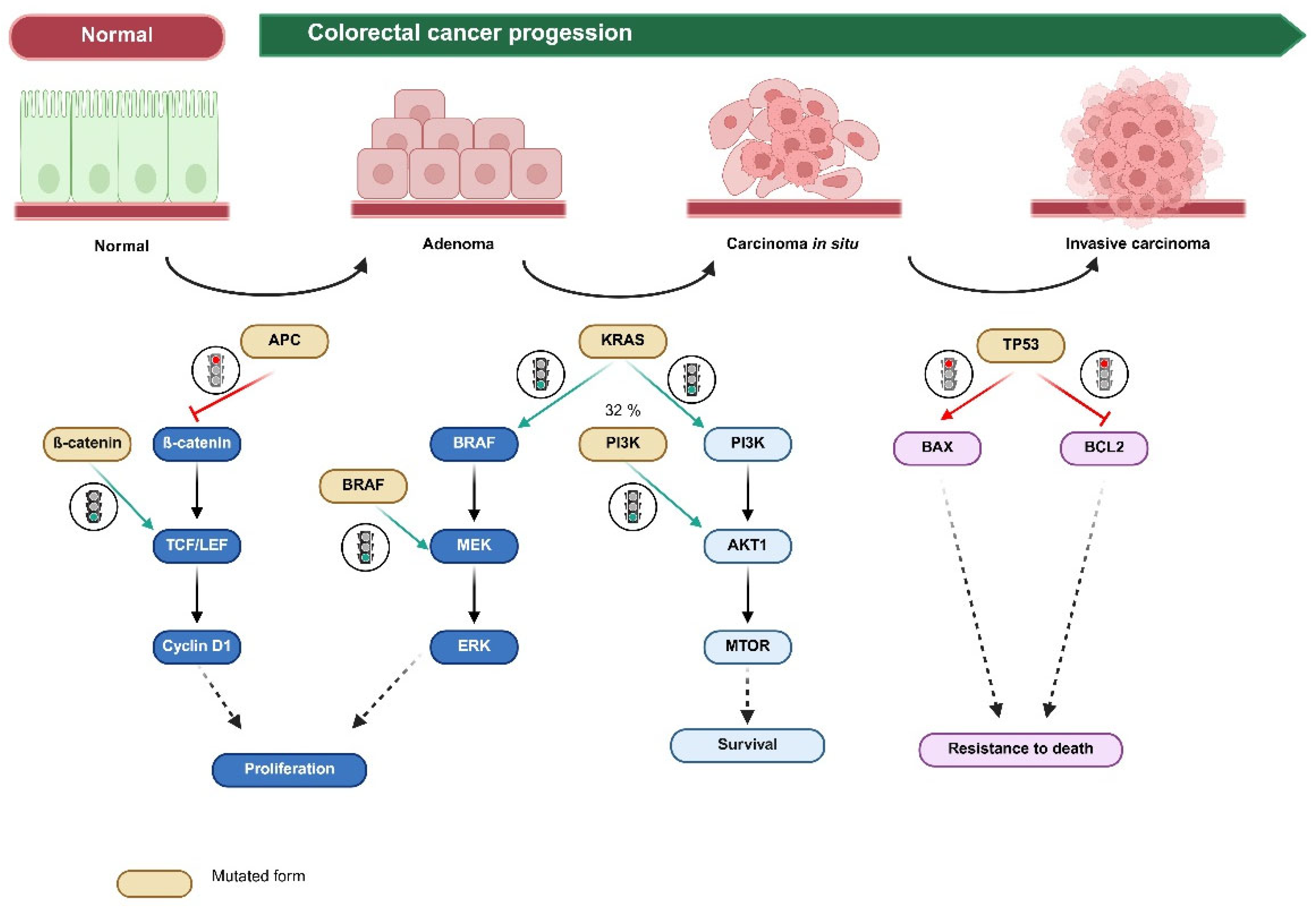
1. Introduction
2. Results
2.1. Single-Mutation Frequency Comparison Between the Clinical Cohort and the PDX Cohort
2.2. Comparison of Concordance in Mutational Patterns Between the Clinical Cohort and the PDX Cohort
2.3. Comparison of the Concordance in Mutational Patterns in the Clinically Significant Mutations (KRAS, BRAF, NRAS, and ERBB2) Between the Clinical Cohort and the PDX Cohort
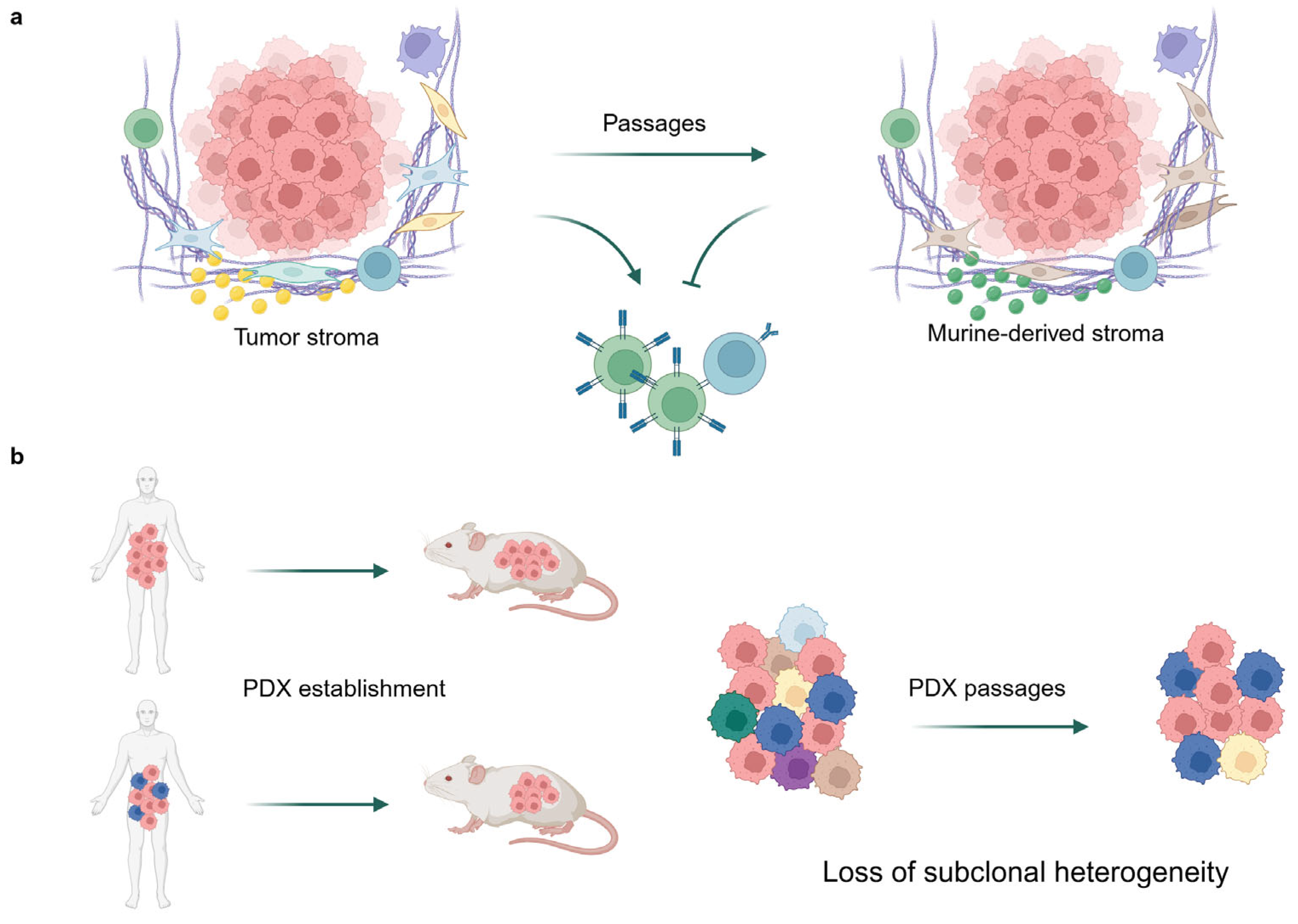
3. Discussion
4. Materials and Methods
4.1. PDX Cohort Establishment
4.2. Clinical Cohort Establishment
4.3. Mutation Analysis of the PDX Cohort
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| PDX | Patient-derived xenograft |
| CMS | Consensus molecular subtype |
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Dariya, B.; Aliya, S.; Merchant, N.; Alam, A.; Nagaraju, G.P. Colorectal cancer biology, diagnosis, and therapeutic approaches. Crit. Rev. Oncog. 2020, 25, 71–94. [Google Scholar] [CrossRef]
- Jungwirth, J.; Urbanova, M.; Boot, A.; Preusser, M.; Jakubecova, J.; Kiss, I.; Mayerova, D.; Vogel, A.; Stros, P.; Brabec, P.; et al. Mutational analysis of driver genes defines the colorectal adenoma: In situ carcinoma transition. Sci. Rep. 2022, 12, 2570. [Google Scholar] [CrossRef]
- Afrăsânie, V.A.; Marinca, M.V.; Alexa-Stratulat, T.; Gafton, B.; Păduraru, M.; Adavidoaiei, A.M.; Miron, L.; Rusu, C. KRAS, NRAS, BRAF, HER2 and microsatellite instability in metastatic colorectal cancer—Practical implications for the clinician. Radiol. Oncol. 2019, 53, 265–274. [Google Scholar] [CrossRef]
- Luo, X.J.; Zhao, Q.; Liu, J.; Zheng, J.B.; Qiu, M.Z.; Ju, H.Q.; Xu, R.H. Novel genetic and epigenetic biomarkers of prognostic and predictive significance in stage II/III colorectal cancer. Mol. Ther. 2021, 29, 587–596. [Google Scholar] [CrossRef]
- Harada, S.; Morlote, D. Molecular pathology of colorectal cancer. Adv. Anat. Pathol. 2020, 27, 20–26. [Google Scholar] [CrossRef]
- Jia He, C.; Zhang, C.; Ozkan, A.; Feng, T.; Duan, P.; Wang, S.; Yang, X.; Xie, J.; Liu, X. Patient-derived tumor models and their distinctive applications in personalized drug therapy. Mechanobiol. Med. 2023, 1, 100014. [Google Scholar] [CrossRef]
- Jin, J.; Yoshimura, K.; Sewastjanow-Silva, M.; Song, S.; Ajani, J.A. Challenges and Prospects of Patient-Derived Xenografts for Cancer Research. Cancers 2023, 15, 4352. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hou, X.; Du, C.; Lu, L.; Yuan, S.; Zhan, M.; You, P.; Du, H. Opportunities and challenges of patient-derived models in cancer research: Patient-derived xenografts, patient-derived organoid and patient-derived cells. World J. Surg. Oncol. 2022, 20, 37. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thota, R.; Yang, M.; Pflieger, L.; Schell, M.J.; Rajan, M.; Davis, T.B.; Wang, H.; Presson, A.; Pledger, W.J.; Yeatman, T.J. APC and TP53 mutations predict cetuximab sensitivity across consensus molecular subtypes. Cancers 2021, 13, 5394. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, W.; Wu, K.; Shi, L. The roles of KRAS in cancer metabolism, tumor microenvironment and clinical therapy. Mol. Cancer 2025, 24, 14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cassidy, J.W.; Caldas, C.; Bruna, A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015, 75, 2963–2968. [Google Scholar] [CrossRef]
- Liu, M.Q.; Yang, X. Patient-derived xenograft models: Current status, challenges, and innovations in cancer research. Genes. Dis. 2025, in press, 101520. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; San Román, M.; Pozas, J.; Alonso-Gordoa, T.; Pozas, M.; Conde, E.; Rosas, M.; Grande, E.; García-Bermejo, M.L.; Carrato, A. BRAF mutated colorectal cancer: New treatment approaches. Cancers 2020, 12, 1571. [Google Scholar] [CrossRef]
- Shan, K.S.; Rehman, T.U.; Ivanov, S.; Domingo, G.; Raez, L.E. Molecular Targeting of the BRAF Proto-Oncogene/Mitogen-Activated Protein Kinase (MAPK) Pathway across Cancers. Int. J. Mol. Sci. 2024, 25, 624. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prasetyanti, P.R.; van Hooff, S.R.; van Herwaarden, T.; de Vries, N.; Kalloe, K.; Rodermond, H.; van Leersum, R.; de Jong, J.H.; Franitza, M.; Nürnberg, P.; et al. Capturing colorectal cancer inter-tumor heterogeneity in patient-derived xenograft (PDX) models. Int. J. Cancer 2019, 144, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Linnekamp, J.F.; Hooff, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.A.S.T.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Suto, H. Microsatellite instability-high colorectal cancer patient-derived xenograft models for cancer immunity research. J. Cancer Res. Ther. 2021, 17, 1358–1369. [Google Scholar] [CrossRef]
- Kerstetter-Fogle, A.E.; Harris, P.L.R.; Brady-Kalnay, S.M.; Sloan, A.E. Generation of glioblastoma patient-derived intracranial xenografts for preclinical studies. Int. J. Mol. Sci. 2020, 21, 5113. [Google Scholar] [CrossRef]
- Sunil, H.S.; O’Donnell, K.A. Capturing heterogeneity in PDX models: Representation matters. Nat. Commun. 2024, 15, 4652. [Google Scholar] [CrossRef]
- Hoge, A.C.H. DNA-based copy number analysis confirms genomic evolution of PDX models. NPJ Precis. Oncol. 2022, 6, 30. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Michel, M.; Kaps, L.; Maderer, A.; Galle, P.R.; Moehler, M. The role of p53 dysfunction in colorectal cancer and its implication for therapy. Cancers 2021, 13, 2296. [Google Scholar] [CrossRef]
- Dang, H.X.; Krasnick, B.A.; White, B.S.; Grossman, J.G.; Strand, M.S.; Zhang, J.; Cabanski, C.R.; Miller, C.A.; Fulton, R.S.; Goedegebuure, S.P.; et al. The clonal evolution of metastatic colorectal cancer. Sci. Adv. 2020, 6, eaay9691. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hynds, R.E. Progress towards non-small-cell lung cancer models that represent clinical evolutionary trajectories. Open Biol. 2021, 11, 200247. [Google Scholar] [CrossRef]
- Mullins, C.S.; Micheel, B.; Matschos, S.; Leuchter, M.; Bürtin, F.; Krohn, M.; Hühns, M.; Klar, E.; Prall, F.; Linnebacher, M. Integrated biobanking and tumor model establishment of human colorectal carcinoma provides excellent tools for preclinical research. Cancers 2019, 11, 1520. [Google Scholar] [CrossRef] [PubMed]
- Matschos, S.; Bürtin, F.; Kdimati, S.; Radefeldt, M.; Krake, S.; Prall, F.; Engel, N.; Krohn, M.; Micheel, B.; Kreutzer, M.; et al. The HROC-Xenobank—A high quality assured PDX biobank of >100 individual colorectal cancer models. Cancers 2021, 13, 5882. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Mutation In | Clinical Cohort | PDX Cohort | χ2 | p | Test Used | ||
|---|---|---|---|---|---|---|---|
| n = 7936 | (%) | n = 137 | (%) | ||||
| APC | 5366 | 67.6 | 59 | 43.1 | 35.72 | <0.001 | Chi-square |
| TP53 | 5179 | 65.3 | 5179 | 42.3 | 30.06 | <0.001 | Chi-square |
| KRAS | 3347 | 42.2 | 28 | 20.4 | 25.27 | <0.001 | Chi-square |
| BRAF | 750 | 9.5 | 28 | 20.4 | 17.43 | <0.001 | Chi-square |
| NRAS | 346 | 4.4 | 3 | 2.2 | NA | 0.288 | Fisher’s exact |
| ERBB2 | 195 | 2.5 | 2 | 1.5 | NA | 1.000 | Fisher’s exact |
| Mutation In | Clinical Cohort | PDX Cohort | χ2 | p | Test Used | ||
|---|---|---|---|---|---|---|---|
| n = 7936 | (%) | n = 137 | (%) | ||||
| TP53 + APC | 2332 | 29.4 | 24 | 17.5 | 8.61 | 0.003 | Chi-square |
| KRAS + TP53 + APC | 1439 | 18.1 | 6 | 4.4 | 16.41 | <0.001 | Chi-square |
| KRAS + APC | 1015 | 12.8 | 6 | 4.4 | 7.88 | 0.005 | Chi-square |
| TP53 | 911 | 11.5 | 21 | 15.3 | 1.60 | 0.207 | Chi-square |
| No mutation | 766 | 9.7 | 41 | 29.9 | 59.30 | <0.001 | Chi-square |
| APC | 580 | 7.3 | 23 | 16.8 | 16.17 | <0.001 | Chi-square |
| KRAS + TP53 | 497 | 6.3 | 7 | 5.1 | 0.14 | 0.708 | Chi-square |
| KRAS | 396 | 5.0 | 9 | 6.6 | 0.41 | 0.521 | Chi-square |
| Mutation In | Clinical Cohort | PDX Cohort | χ2 | p | Test Used | ||
|---|---|---|---|---|---|---|---|
| n = 7936 | % | n = 137 | % | ||||
| No mutation | 3474 | 43.7 | 76 | 55.4 | 7.01 | 0.008 | Chi-Square |
| KRAS | 3192 | 40.2 | 28 | 20.4 | 3142.43 | <0.001 | Chi-Square |
| BRAF | 689 | 8.6 | 28 | 20.4 | 655.33 | <0.001 | Chi-Square |
| NRAS | 303 | 3.8 | 3 | 2.19 | NA | 0.450 | Fisher’s Exact |
| ERBB2 | 104 | 1.3 | 2 | 1.46 | NA | 0.701 | Fisher’s Exact |
| ERBB2 + KRAS | 83 | 1.05 | 0 | NA | 0.406 | Fisher’s Exact | |
| KRAS + BRAF | 45 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| NRAS + KRAS | 26 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| NRAS + BRAF | 12 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| NRAS + ERBB2 | 4 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| BRAF + ERBB2 | 2 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| NRAS + BRAF + ERBB2 | 1 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| KRAS + BRAF + ERBB2 | 1 | <1 | 0 | NA | 1.000 | Fisher’s Exact | |
| A | B | Neither | A Not B | B Not A | Both | Log2 Odds Ratio | p-Value | q-Value | Tendency |
|---|---|---|---|---|---|---|---|---|---|
| KRAS | BRAF | 3885 | 3301 | 704 | 46 | <−3 | <0.001 | <0.001 | Mutual exclusivity |
| APC | BRAF | 2074 | 5112 | 496 | 254 | −2.267 | <0.001 | <0.001 | Mutual exclusivity |
| KRAS | NRAS | 4269 | 3321 | 320 | 26 | <−3 | <0.001 | <0.001 | Mutual exclusivity |
| APC | TP53 | 1162 | 1595 | 1408 | 3771 | 0.964 | <0.001 | <0.001 | Co-occurrence |
| TP53 | KRAS | 1346 | 3243 | 1411 | 1936 | −0.812 | <0.001 | <0.001 | Mutual exclusivity |
| APC | KRAS | 1677 | 2912 | 893 | 2454 | 0.662 | <0.001 | <0.001 | Co-occurrence |
| TP53 | BRAF | 2386 | 4800 | 371 | 379 | −0.978 | <0.001 | <0.001 | Mutual exclusivity |
| APC | NRAS | 2510 | 5080 | 60 | 286 | 1.236 | <0.001 | <0.001 | Co-occurrence |
| BRAF | ERBB2 | 6995 | 746 | 191 | 4 | −2.348 | <0.001 | <0.001 | Mutual exclusivity |
| BRAF | NRAS | 6853 | 737 | 333 | 13 | −1.462 | <0.001 | <0.001 | Mutual exclusivity |
| TP53 | ERBB2 | 2669 | 5072 | 88 | 107 | −0.644 | 0.003 | 0.004 | Mutual exclusivity |
| NRAS | ERBB2 | 7400 | 341 | 190 | 5 | −0.808 | 0.284 | 0.356 | |
| TP53 | NRAS | 2642 | 4948 | 115 | 231 | 0.101 | 0.564 | 0.63 | |
| APC | ERBB2 | 2503 | 5238 | 67 | 128 | −0.131 | 0.588 | 0.63 | |
| KRAS | ERBB2 | 4478 | 3263 | 111 | 84 | 0.055 | 0.826 | 0.826 |
| Project Name | Sample Count | Source | Year | Reference Genome | Cancer Type |
|---|---|---|---|---|---|
| Colorectal Adenocarcinoma (DFCI/Orion) | 74 | DFCI/Orion | 2024 | GRCh37/hg19 | CRC |
| Colorectal Adenocarcinoma (MSK, Nat Commun) | 179 | MSK | 2022 | GRCh37/hg19 | CRC |
| Colorectal Cancer (MSK, JNCI) | 1516 | MSK | 2021 | GRCh37/hg19 | CRC |
| Colorectal Adenocarcinoma (DFCI, Cell Reports) | 619 | DFCI | 2016 | GRCh37/hg19 | CRC |
| Colorectal Adenocarcinoma (Genentech, Nature) | 74 | Genentech | 2012 | GRCh37/hg19 | CRC |
| Colorectal Adenocarcinoma (TCGA, Firehose Legacy) | 640 | TCGA | GRCh37/hg19 | CRC | |
| Colorectal Adenocarcinoma (TCGA, Nature) | 276 | TCGA | 2012 | GRCh37/hg19 | CRC |
| Colorectal Adenocarcinoma (TCGA, PanCancer Atlas) | 594 | TCGA | GRCh37/hg19 | CRC | |
| Colorectal Adenocarcinoma Triplets (MSK, Genome Biol) | 138 | MSK | 2014 | GRCh37/hg19 | CRC |
| Colorectal Cancer (CAS Shanghai, Cancer Cell) | 146 | CAS Shanghai | 2020 | GRCh37/hg19 | CRC |
| Colorectal Cancer (MSK, Cancer Discovery) | 22 | MSK | 2022 | GRCh37/hg19 | CRC |
| Colorectal Cancer (MSK, Gastroenterology) | 471 | MSK | 2020 | GRCh37/hg19 | CRC |
| Colorectal Cancer (MSK, JCO Precis Oncol) | 47 | MSK | 2022 | GRCh37/hg19 | CRC |
| Disparities in metastatic colorectal cancer (MSK) | 64 | MSK | 2020 | GRCh37/hg19 | CRC |
| Metastatic Colorectal Cancer (MSK, Cancer Cell) | 1134 | MSK | 2018 | GRCh37/hg19 | CRC |
| Pre-cancer Colorectal Polyps (HTAN Vanderbilt, Cell) | 61 | HTAN Vanderbilt | 2021 | GRCh37/hg19 | CRC |
| Colon Adenocarcinoma (CPTAC, GDC) | 109 | CPTAC | GRCh38/hg38 | Colon Cancer | |
| Colon Adenocarcinoma (CaseCCC, PNAS) | 29 | CaseCCC | 2015 | GRCh37/hg19 | Colon Cancer |
| Colon Adenocarcinoma (TCGA, GDC) | 463 | TCGA | GRCh38/hg38 | Colon Cancer | |
| Colon Cancer (CPTAC-2 Prospective, Cell) | 110 | CPTAC-2 | 2019 | GRCh37/hg19 | Colon Cancer |
| Colon Cancer (Sidra-LUMC AC-ICAM, Nat Med) | 348 | Sidra-LUMC | 2023 | GRCh37/hg19 | Colon Cancer |
| Rectal Cancer (MSK, Nature Medicine) | 788 | MSK | 2022 | GRCh37/hg19 | Rectal Cancer |
| Rectal Cancer (MSK, Nature Medicine) | 339 | MSK | 2019 | GRCh37/hg19 | Rectal Cancer |
| Colorectal Cancer Radiation (MSK) | 48 | MSK | 2024 | GRCh37/hg19 | Rectal Cancer |
| Rectal Adenocarcinoma (TCGA, GDC) | 171 | TCGA | GRCh38/hg38 | Rectal Cancer | |
| Appendiceal Cancer (MSK, J Clin Oncol) | 273 | MSK | 2022 | GRCh37/hg19 | Appendiceal Cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hage, M.; Su, Z.; Linnebacher, M. Mutational Patterns in Colorectal Cancer: Do PDX Models Retain the Heterogeneity of the Original Tumor? Int. J. Mol. Sci. 2025, 26, 5111. https://doi.org/10.3390/ijms26115111
El Hage M, Su Z, Linnebacher M. Mutational Patterns in Colorectal Cancer: Do PDX Models Retain the Heterogeneity of the Original Tumor? International Journal of Molecular Sciences. 2025; 26(11):5111. https://doi.org/10.3390/ijms26115111
Chicago/Turabian StyleEl Hage, Maria, Zhaoran Su, and Michael Linnebacher. 2025. "Mutational Patterns in Colorectal Cancer: Do PDX Models Retain the Heterogeneity of the Original Tumor?" International Journal of Molecular Sciences 26, no. 11: 5111. https://doi.org/10.3390/ijms26115111
APA StyleEl Hage, M., Su, Z., & Linnebacher, M. (2025). Mutational Patterns in Colorectal Cancer: Do PDX Models Retain the Heterogeneity of the Original Tumor? International Journal of Molecular Sciences, 26(11), 5111. https://doi.org/10.3390/ijms26115111