
Integrative Single-Cell and Bulk RNA Sequencing Identifies a Glycolysis-Related Prognostic Signature for Predicting Prognosis in Pancreatic Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
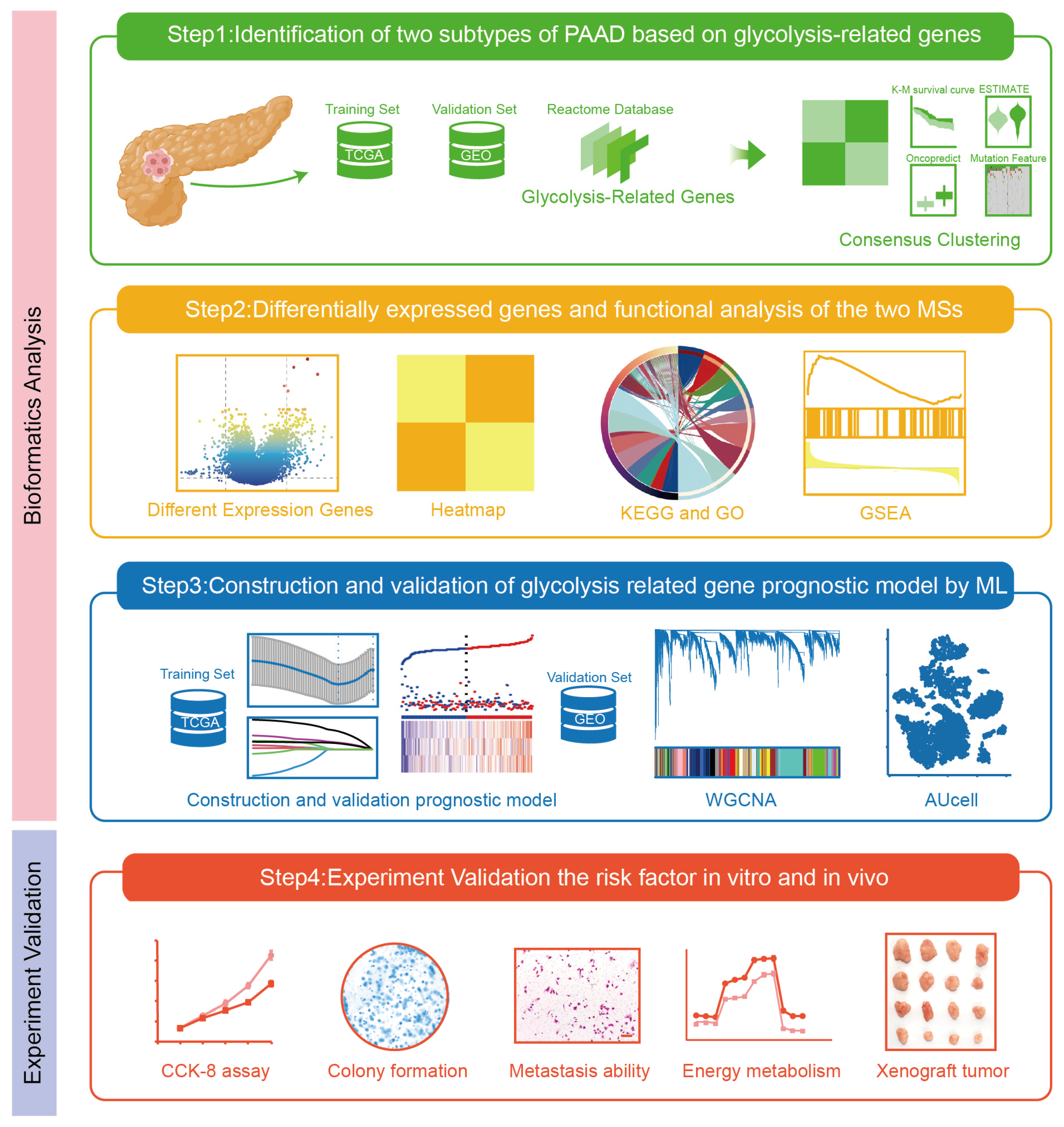
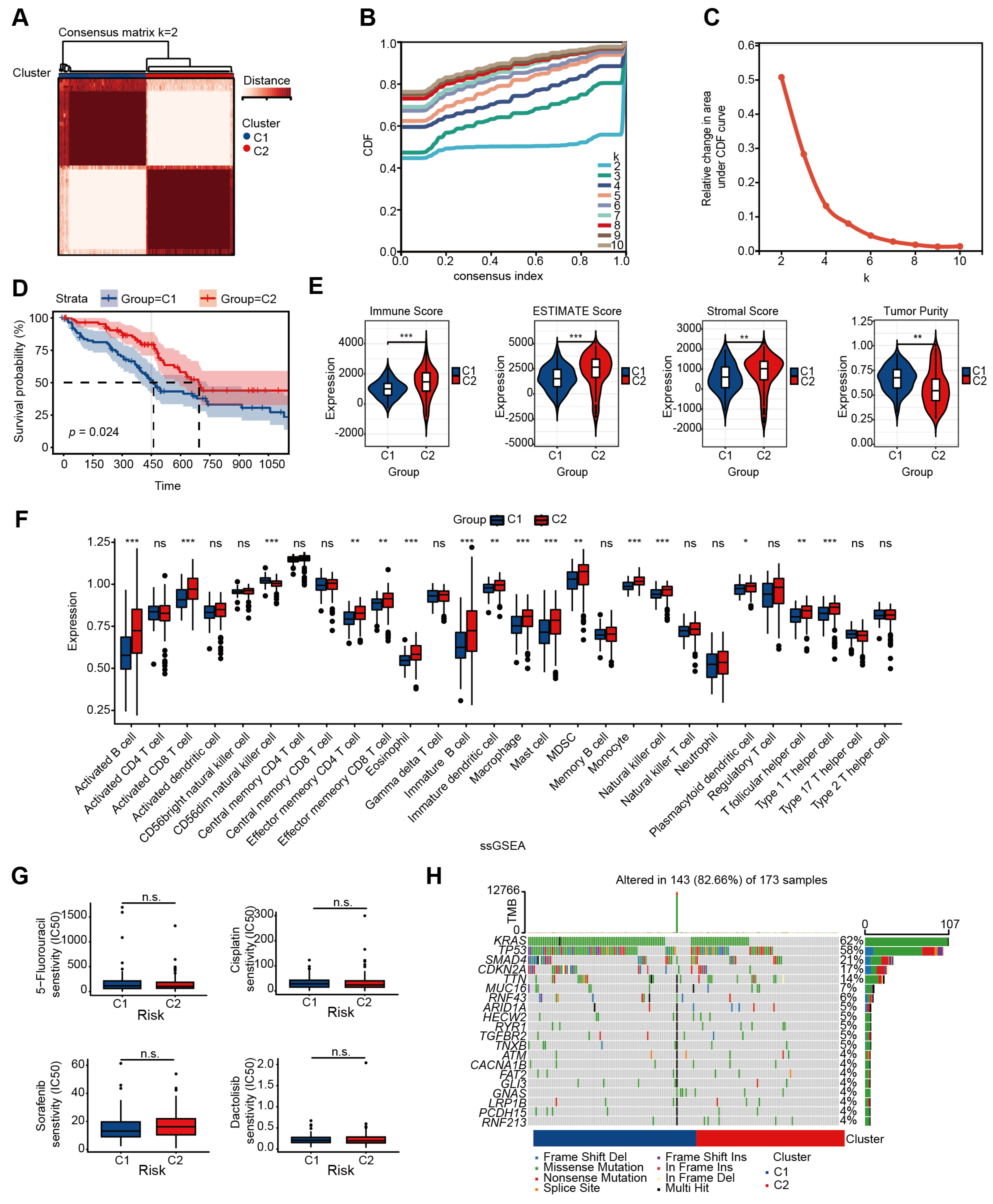
2.1. Identification of Two Molecular Subtypes of PAAD Depending on Glycolysis-Related Genes
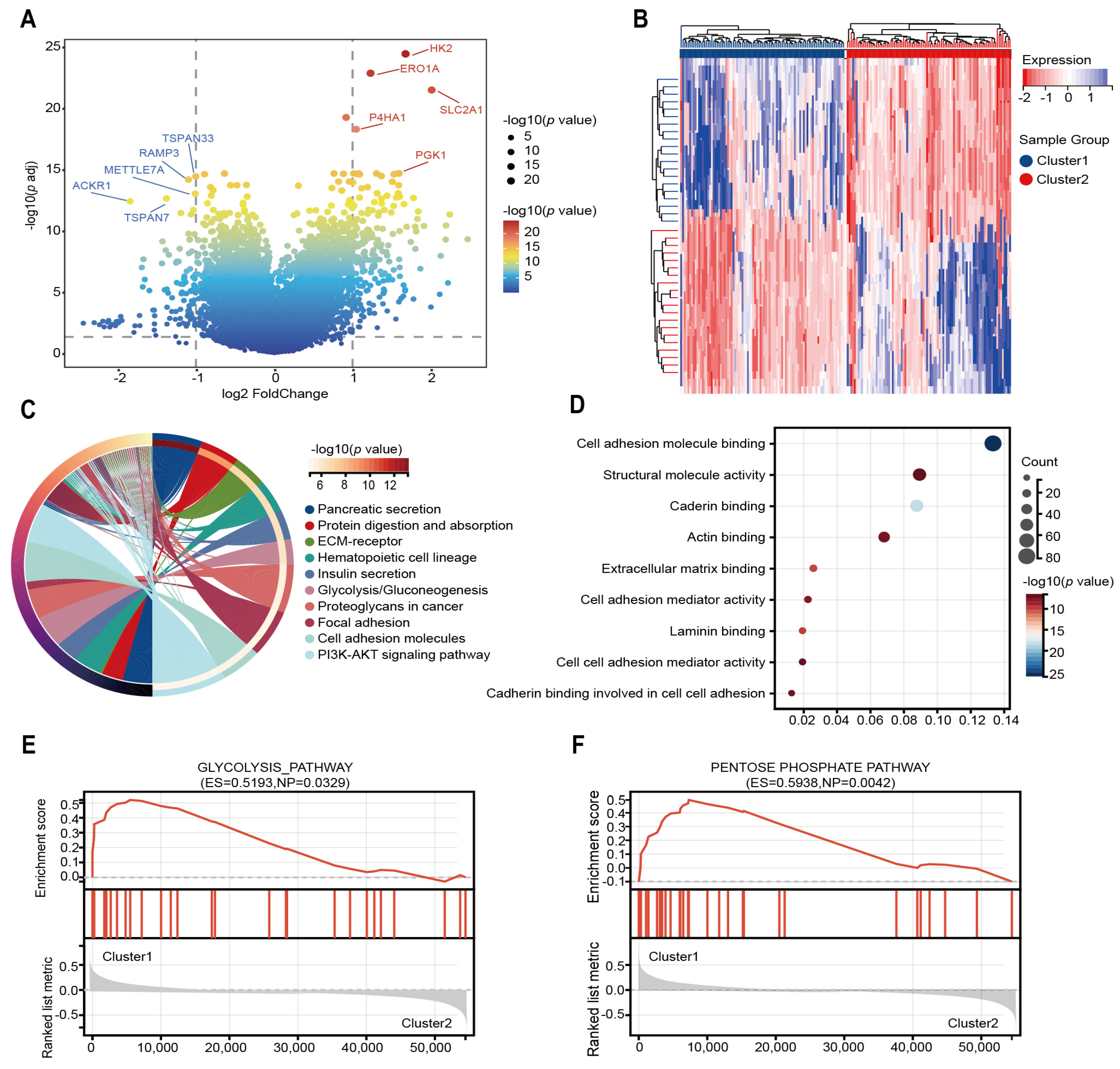
2.2. The Different Expressed Genes and Enrichment Analysis of Two Subtypes
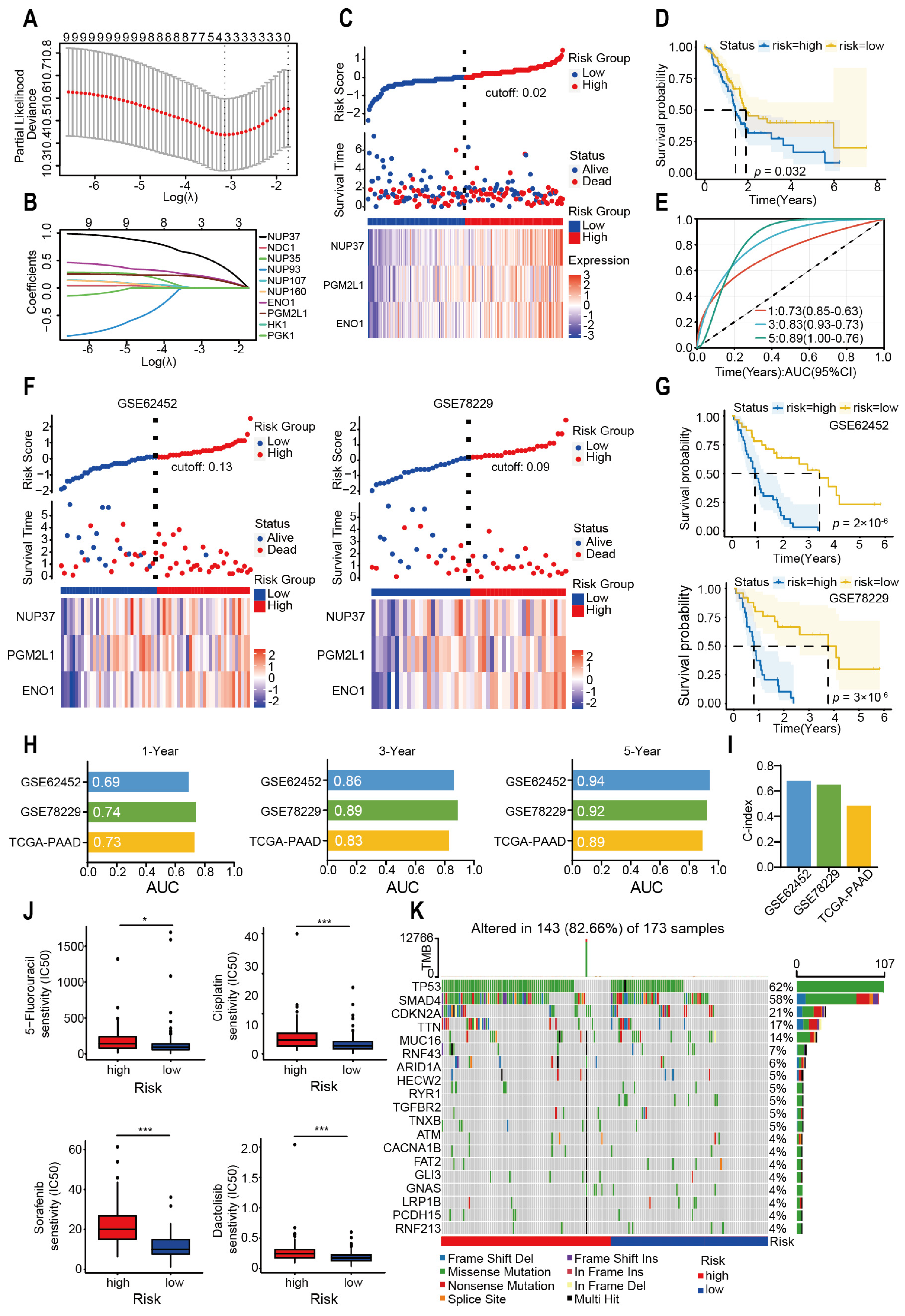
2.3. Glycolysis-Related Gene’s Prognostic Risk Model Was Constructed by Machine Learning
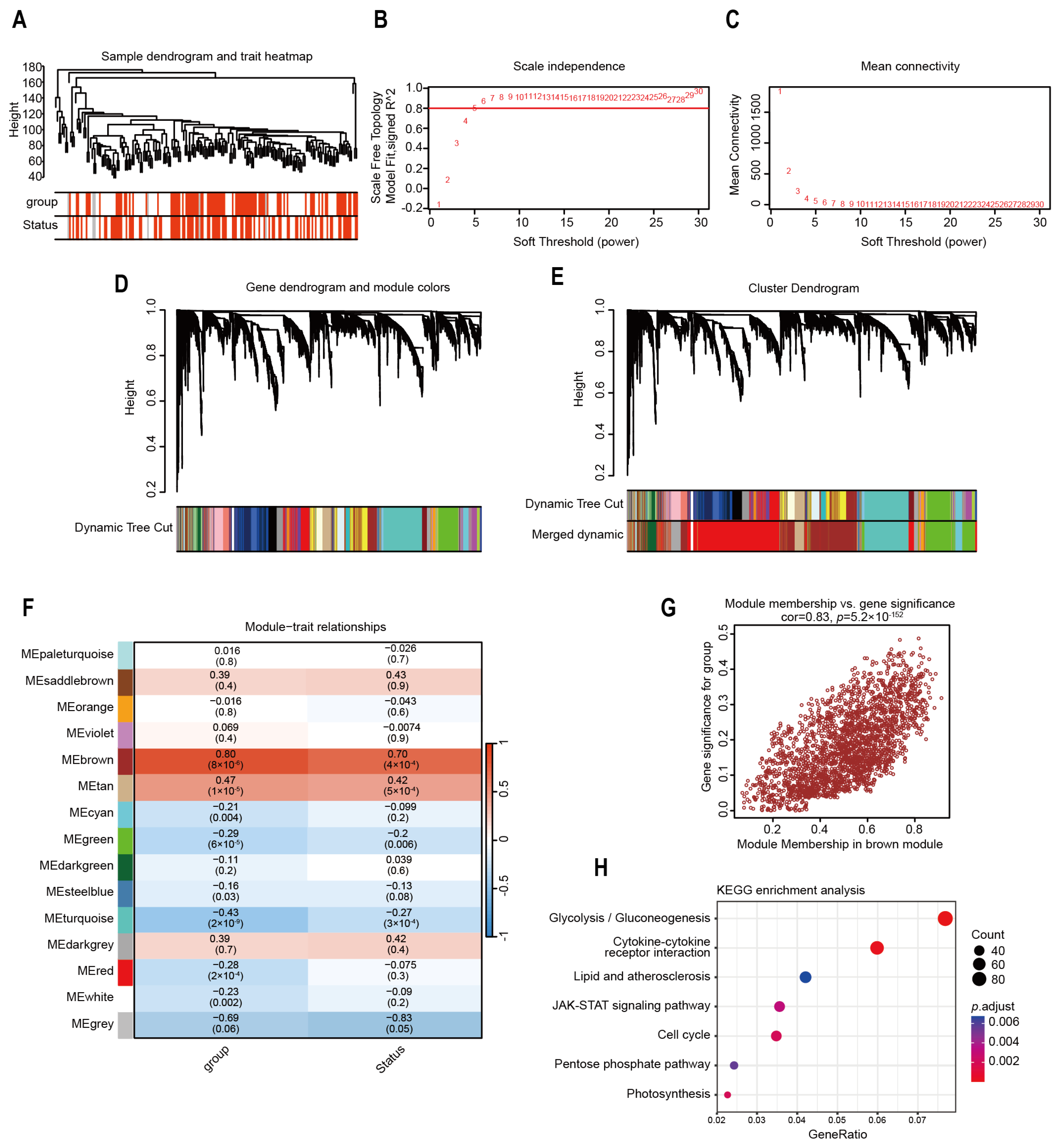
2.4. Identification of Highly Correlated Gene Modules in Risk Model
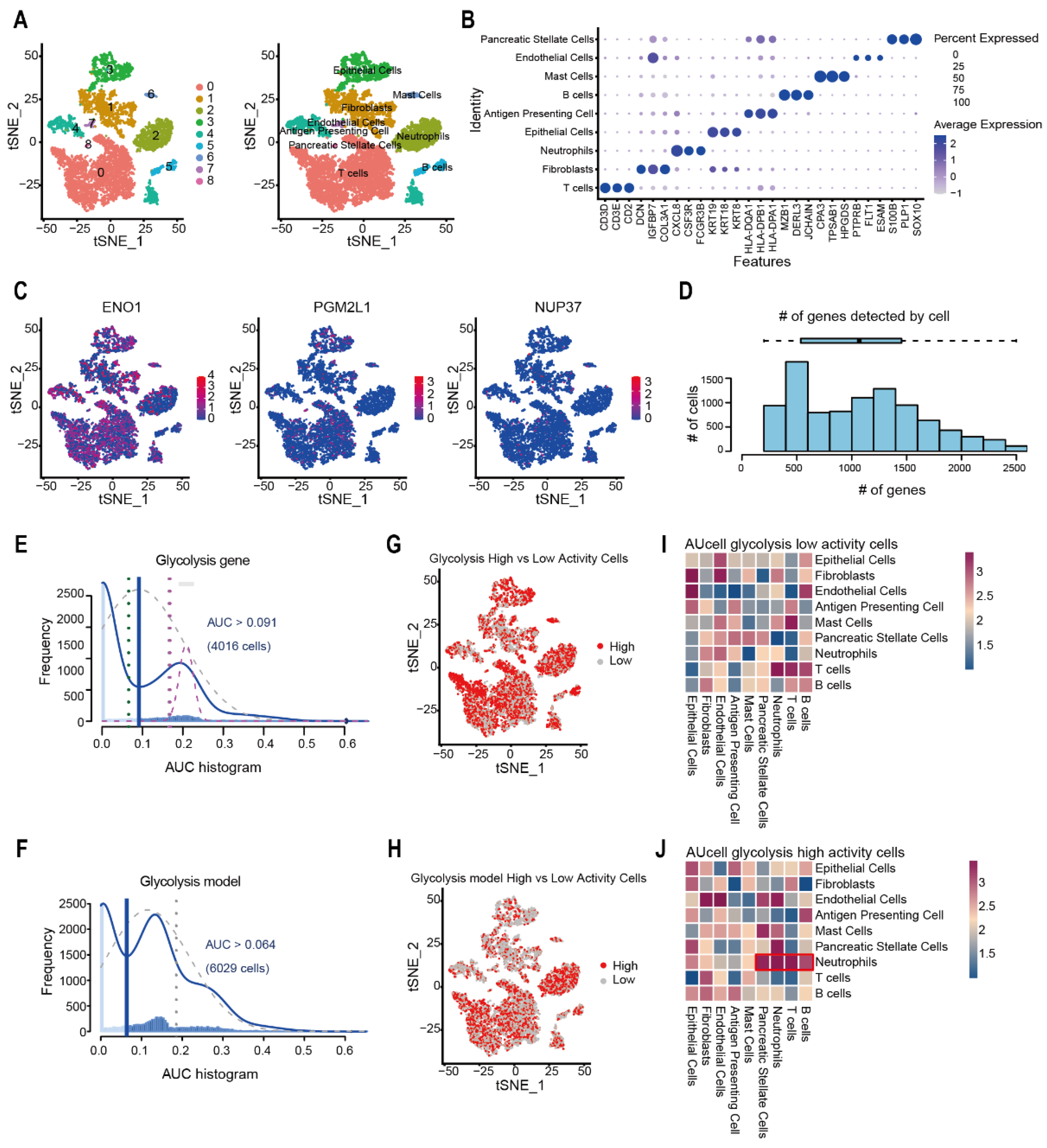
2.5. Evaluation of Glycolysis Prognosis Model in Pancreatic Cancer Through scRNA-Seq Analysis
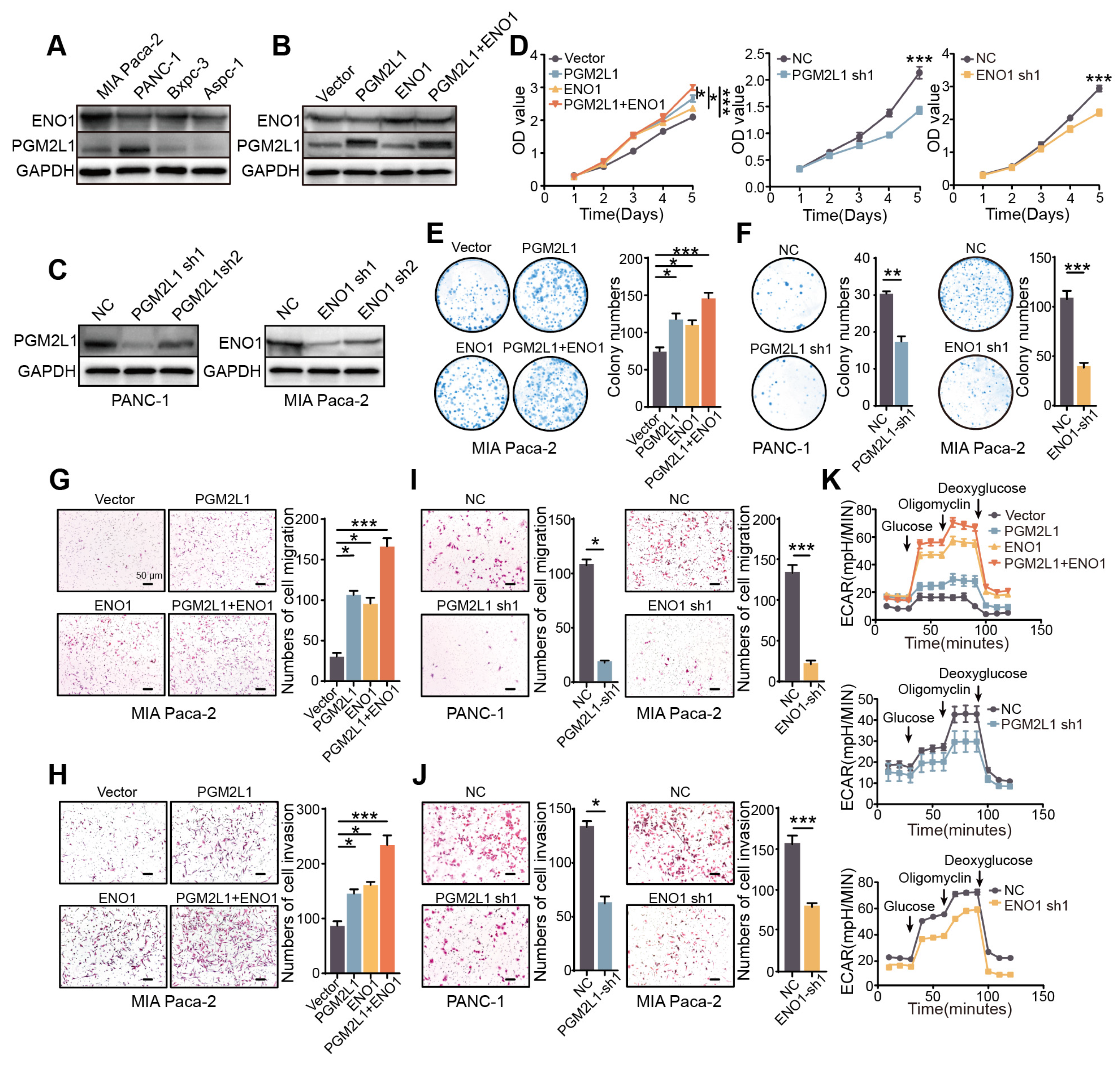
2.6. PGM2L1 and ENO1 Promote Proliferation, Migration, Invasion, and Glycolysis of PAAD
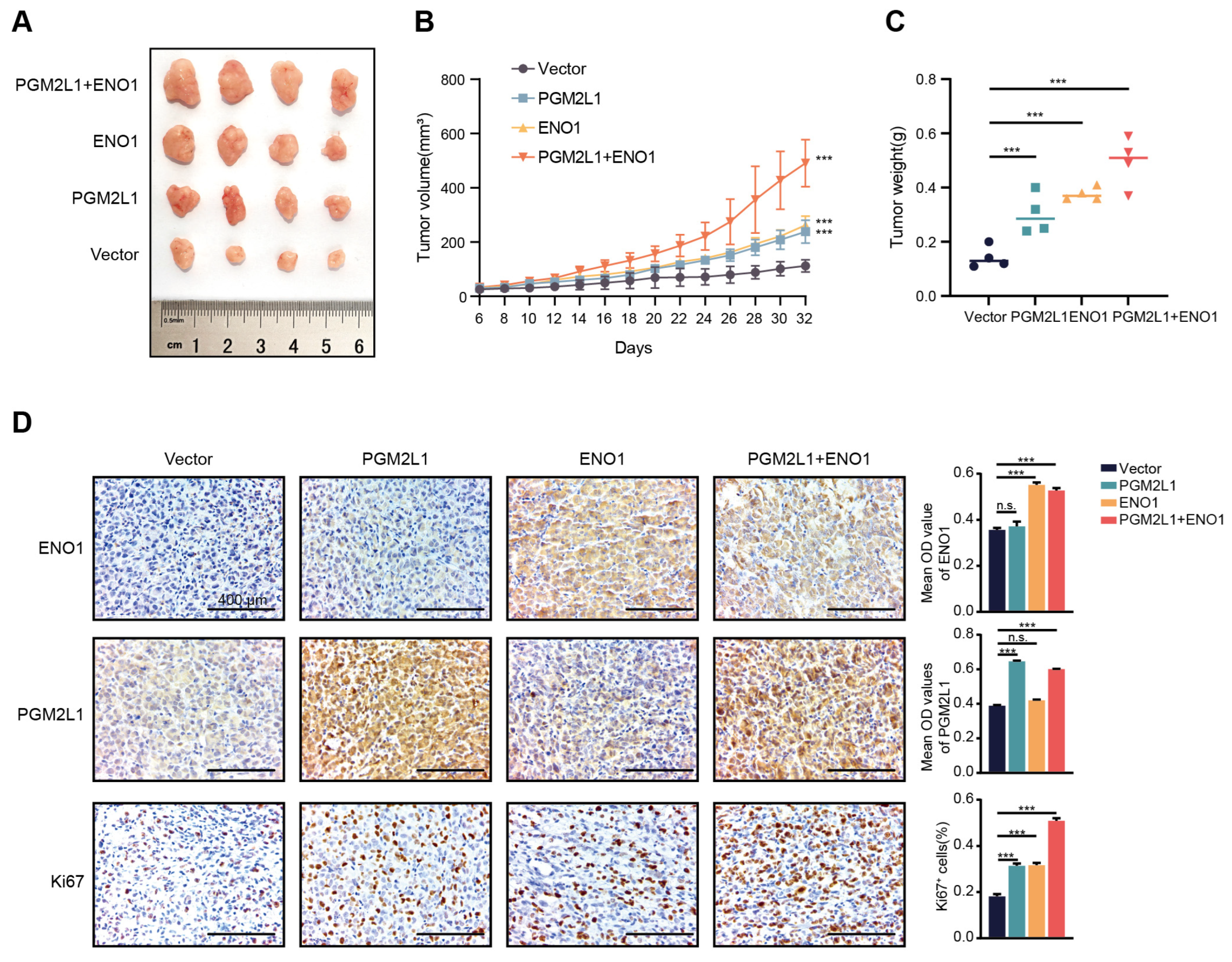
2.7. PGM2L1 and ENO1 Promoted Xenograft Tumor Growth in Mouse Models
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Identification of Molecular Subgroups
4.3. Analysis of Immune Cell Infiltration
4.4. Identifying Differentially Expressed Genes and Functional Enrichment
4.5. Genomic Mutation and Drug Sensitivity Analysis
4.6. WGCNA
4.7. Single-Cell Sequencing Data Processing
4.8. AUCell Scoring
4.9. CCK-8 and Colony Formation
4.10. Transwell Assay
4.11. Western Blot
4.12. Glycolysis Stress Assay
4.13. Xenograft Formation Assay
4.14. Immunohistochemistry (IHC) Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PAAD | Pancreatic adenocarcinoma |
| PDAC | Pancreatic ductal adenocarcinoma |
| OS | Overall survival rate |
| DEG | Differently expressed genes |
| GO | Gene Ontology |
| MF | Molecular function |
| BP | Biological processes |
| CC | Cellular component |
| GSEA | Gene Set Enrichment Analysis |
| LASSO | Least Absolute Shrinkage and Selection Operato |
| WGCNA | Weighted correlation network analysis |
| shRNA | Short hairpin RNA |
| ECAR | Extracellular acidification rate |
| PGM | Phosphoglucomutase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| IHC | Immunohistochemistry |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Liu, Y.H.; Hu, C.M.; Hsu, Y.S.; Lee, W.H. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 817. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Tu, Y.K.; Wu, M.S.; Lin, J.T.; Wang, H.P.; Chien, K.L. Blood glucose concentration and risk of pancreatic cancer: Systematic review and dose-response meta-analysis. BMJ 2015, 350, g7371. [Google Scholar] [CrossRef]
- Shegay, P.V.; Shatova, O.P.; Zabolotneva, A.A.; Shestopalov, A.V.; Kaprin, A.D. Moonlight functions of glycolytic enzymes in cancer. Front. Mol. Biosci. 2023, 10, 1076138. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Huang, S.; Chen, J.; Ding, P.; Wang, Q.; Li, L.; Lv, X.; Li, L.; Zhang, P.; et al. FOXM1D potentiates PKM2-mediated tumor glycolysis and angiogenesis. Mol. Oncol. 2021, 15, 1466–1485. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Glycolysis-induced drug resistance in tumors-A response to danger signals? Neoplasia 2021, 23, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Fan, G.; Liu, Y.; Liu, L.; Zhang, T.; Liu, P.; Tu, Q.; Zhang, X.; Luo, S.; Yao, L.; et al. The transcription factor KLF14 regulates macrophage glycolysis and immune function by inhibiting HK2 in sepsis. Cell. Mol. Immunol. 2022, 19, 504–515. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e4. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Beijing, China. Available online: http://gepia2.cancer-pku.cn/ (accessed on 6 March 2025).
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An Update to the Integrated Cancer Data Analysis Platform. LA, USA. Available online: https://ualcan.path.uab.edu/ (accessed on 5 March 2025).
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Mehla, K.; Singh, P.K. Metabolic Subtyping for Novel Personalized Therapies Against Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 6–8. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Nigam, M.; Mishra, A.P.; Deb, V.K.; Dimri, D.B.; Tiwari, V.; Bungau, S.G.; Bungau, A.F.; Radu, A.F. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomed. Pharmacother. 2023, 164, 115015. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Zhou, C.; Qian, W.; Li, J.; Ma, J.; Chen, X.; Jiang, Z.; Cheng, L.; Duan, W.; Wang, Z.; Wu, Z.; et al. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 302. [Google Scholar] [CrossRef]
- Levink, I.J.M.; Jansen, M.; Azmani, Z.; van IJcken, W.; van Marion, R.; Peppelenbosch, M.P.; Cahen, D.L.; Fuhler, G.M.; Bruno, M.J. Mutation Analysis of Pancreatic Juice and Plasma for the Detection of Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 13116. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yu, Y.; Zong, K.; Lv, P.; Gu, Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 497. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Y.; Gao, Z.; Li, X.; Weng, M.; Shi, C.; Wang, C.; Sun, L. Fisetin inhibits the proliferation, migration and invasion of pancreatic cancer by targeting PI3K/AKT/mTOR signaling. Aging 2021, 13, 24753–24767. [Google Scholar] [CrossRef]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.S.; Cai, C.Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol. Cancer 2022, 21, 112. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.K.; Sun, Y.; Lv, L.; Ping, Y. ENO1 and Cancer. Mol. Ther. Oncolytics 2022, 24, 288–298. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Luo, J.; Fu, X.; Liu, P.; Liu, S.; Pan, Y. FAM126A interacted with ENO1 mediates proliferation and metastasis in pancreatic cancer via PI3K/AKT signaling pathway. Cell Death Discov. 2022, 8, 248. [Google Scholar] [CrossRef]
- Li, H.J.; Ke, F.Y.; Lin, C.C.; Lu, M.Y.; Kuo, Y.H.; Wang, Y.P.; Liang, K.H.; Lin, S.C.; Chang, Y.H.; Chen, H.Y.; et al. ENO1 Promotes Lung Cancer Metastasis via HGFR and WNT Signaling-Driven Epithelial-to-Mesenchymal Transition. Cancer Res. 2021, 81, 4094–4109. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Satani, N.; Hammoudi, N.; Yan, V.C.; Barekatain, Y.; Khadka, S.; Ackroyd, J.J.; Georgiou, D.K.; Pham, C.D.; Arthur, K.; et al. An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat. Metab. 2020, 2, 1413–1426. [Google Scholar] [CrossRef]
- Wang, C.; Huang, M.; Lin, Y.; Zhang, Y.; Pan, J.; Jiang, C.; Cheng, M.; Li, S.; He, W.; Li, Z.; et al. ENO2-derived phosphoenolpyruvate functions as an endogenous inhibitor of HDAC1 and confers resistance to antiangiogenic therapy. Nat. Metab. 2023, 5, 1765–1786. [Google Scholar] [CrossRef]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.H.; Hammoudi, N.; Peng, Z.; Pisaneschi, F.; Link, T.M.; Lee, G.R.t.; Sun, D.; et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, N.; Zhou, C.; Yan, X.; Liu, Z.; Jiang, R.; Luo, Y.; Jiang, P.; Mu, Y.; Xiao, S.; Huang, X.; et al. Integrative Single-Cell and Bulk RNA Sequencing Identifies a Glycolysis-Related Prognostic Signature for Predicting Prognosis in Pancreatic Cancer. Int. J. Mol. Sci. 2025, 26, 5105. https://doi.org/10.3390/ijms26115105
Wu N, Zhou C, Yan X, Liu Z, Jiang R, Luo Y, Jiang P, Mu Y, Xiao S, Huang X, et al. Integrative Single-Cell and Bulk RNA Sequencing Identifies a Glycolysis-Related Prognostic Signature for Predicting Prognosis in Pancreatic Cancer. International Journal of Molecular Sciences. 2025; 26(11):5105. https://doi.org/10.3390/ijms26115105
Chicago/Turabian StyleWu, Nan, Chong Zhou, Xu Yan, Ziang Liu, Ruohan Jiang, Yuzhou Luo, Ping Jiang, Yu Mu, Shan Xiao, Xien Huang, and et al. 2025. "Integrative Single-Cell and Bulk RNA Sequencing Identifies a Glycolysis-Related Prognostic Signature for Predicting Prognosis in Pancreatic Cancer" International Journal of Molecular Sciences 26, no. 11: 5105. https://doi.org/10.3390/ijms26115105
APA StyleWu, N., Zhou, C., Yan, X., Liu, Z., Jiang, R., Luo, Y., Jiang, P., Mu, Y., Xiao, S., Huang, X., Zhou, Y., Sun, D., & Jin, Y. (2025). Integrative Single-Cell and Bulk RNA Sequencing Identifies a Glycolysis-Related Prognostic Signature for Predicting Prognosis in Pancreatic Cancer. International Journal of Molecular Sciences, 26(11), 5105. https://doi.org/10.3390/ijms26115105